
Abstract
Aldosterone is a mineralocorticoid hormone, as its main renal effect has been considered as electrolyte and water homeostasis in the distal tubule, thus maintaining blood pressure and extracellular fluid homeostasis through the activation of mineralocorticoid receptor (MR) in epithelial cells. However, over the past decade, numerous studies have documented the significant role of aldosterone in the progression of chronic kidney disease (CKD) which has become a subject of interest. It is being studied that aldosterone can affect cardiovascular and renal system, thereby contributing to tissue inflammation, injury, glomerulosclerosis, and interstitial fibrosis. Aldosterone acts on renal vessels, renal cells (glomerular mesangial cells, podocytes, vascular smooth muscle cells, tubular epithelial cells, and interstitial fibroblasts), and infiltrating inflammatory cells, inducing reactive oxygen species (ROS) production, upregulated epithelial growth factor receptor (EGFR), and type 1 angiotensin (AT1) receptor expressions, and activating nuclear factor kappa B (NF-κB), activator protein-1 (AP-1), and EGFR to further promote cell proliferation, apoptosis, and proliferation. Phenotypic transformation of epithelial cells stimulates the expression of transforming growth factor-β (TGF-β), connective tissue growth factor (CTGF), osteopontin (OPN), and plasminogen activator inhibitor-1 (PAI-1), eventually leading to renal fibrosis. MR antagonisms are related to inhibition of aldosterone-mediated pro-inflammatory and pro-fibrotic effect. In this review, we will summarize the important role of aldosterone in the pathogenesis of renal injury and fibrosis, emphasizing on its multiple underlying mechanisms and advances in aldosterone research along with the potential therapeutics for targeting MR in a renal fibrosis.
Aanchal Shrestha and Ruo-Chen Che contributed equally to this work.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
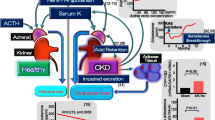
In 1953, Simpson and Tait were the first to isolate a mineralocorticoid hormone, aldosterone (Simpson 1953). Aldosterone was initially found to have a physiological effect in regulating the electrolyte and water balance in the distal tubule, thus maintaining blood pressure and extracellular fluid homeostasis (Simpson 1953; Williams and Williams 2003) that is acted principally via cytosolic MR in epithelial cells (Rogerson and Fuller 2000). However, over the past two decades, aldosterone has also shown to act on the blood vessels, heart, and kidney. MRs are found in different tissues, such as vascular smooth muscle cells and endothelial cells, cardiomyocytes in the heart, mesangial cells and podocytes in the kidney, fibroblasts, adipocytes, hypothalamus in the brain, and macrophages expanding the distribution of aldosterone actions among non-epithelial tissues (Jaisser and Farman 2016; Funder 2010; Shibata et al. 2007; Brown 2013). Thus, there has been a shift of paradigm in our understanding of the more complex role of aldosterone, apart from the physiological role in renal sodium reabsorption. There are numerous studies which demonstrated aldosterone as a major deleterious hormone in the cardiovascular and renal system, thereby contributing to tissue inflammation, injury, glomerulosclerosis, and interstitial fibrosis (Epstein 2006; Hostetter and Ibrahim 2003). A study done by Greene et al. in 1996 first showed the role of aldosterone as an independent mediator in the progression of CKD, where aldosterone is infused intravenously in unilateral nephrectomized rat model which diminished the renoprotective effect of angiotensin-converting enzyme (ACE) inhibitor and angiotensin receptor blocker (ARB) (Greene et al. 1996). In the pathogenesis of CKD, the activation of renin–angiotensin–aldosterone system (RAAS) is an important event (Gonzalez et al. 2004). In RAAS, angiotensin II (Ang II) is one of the key elements which augments adrenal production of aldosterone and has appeared as a helper for renal injury by increasing the intraglomerular capillary pressure and ultrafiltration of plasma proteins and by promoting pro-inflammatory effects, pro-fibrogenic and growth stimulatory action (Rüster and Wolf 2006). CKD has become a massive public health concern which has increased the overall mortality by 31.7% over the last 10 years (Wang et al. 2016). Renal fibrosis, as the hallmark feature of CKD, is characterized by pathomorphologic changes comprising of glomerulosclerosis and tubulointerstitial fibrosis along with the process of excessive production and progressive extracellular matrix (ECM) protein accumulation such as collagen I and fibronectin leading to scar formation.
Herein, we will summarize the pivotal role of aldosterone in the pathogenesis of renal injury and fibrosis, emphasizing on its multiple underlying mechanisms and advances in aldosterone research along with the potential therapeutics for targeting MR in a renal fibrosis.
2 Aldosterone: Its Synthesis, Secretion, and Action
Aldosterone is synthesized in the body from cholesterol mainly in the cells of the zona glomerulosa layer of the adrenal cortex. However, extra-adrenal sites of aldosterone synthesis have also been identified in the heart, blood vessels, and brain (Bonvalet et al. 1995; Coirini et al. 1985; Kornel 1994). The enzyme aldosterone synthase, encoded by CYP11B2 gene, can convert deoxycorticosterone to aldosterone in a series of steps, including molecular arrangement and enzymatic reactions. Likewise, similar enzyme β-hydroxylase, encoded by the CYP11B1 gene, catalyzes the conversion of deoxycortisol to cortisol (Lenzini et al. 2007). There are many hormonal and paracrine factors that modify the regulation of aldosterone secretion. However, under physiological conditions, the important ones are Ang II, extracellular potassium, and adrenocorticotropic hormone (ACTH). Ang II, an octapeptide, is the main precursor of RAAS which is generated by the action of ACE on the inactive precursor Ang I. Ang II acts on aldosterone secretion mainly by binding to the G-protein-coupled receptor type 1 angiotensin (AT1), whereas type 2 angiotensin (AT2) receptor counteracts many of the AT1 receptor-mediated processes (Unger et al. 2011). An increased extracellular potassium concentration also augments aldosterone secretion leading to potassium excretion in the kidney. Both aldosterone and Ang II stimulate aldosterone secretion independently and equipotently (Williams 2005). The action of ACTH in the zona glomerulosa cells is mediated by cAMP which induces aldosterone secretion. In severe sodium or fluid loss, ACTH is also secreted and synergizes with Ang II or potassium in order to stimulate glomerulosa cells (Spat and Hunyady 2004). In the alteration of aldosterone synthesis, inhibitory factors such as atrial natriuretic peptides (ANP), dopamine, somatostatin, and extracellular calcium also participate. ANP is an effective inhibitor of aldosterone secretion. ANP secretion is increased in response to sodium and/or water load, thereby inhibiting the aldosterone secretion (Spat and Hunyady 2004).
At present, the varied actions of aldosterone seem to be related to two different pathways, such as genomic pathway and non-genomic pathway. In genomic pathway, aldosterone shows its physiological effects by binding to classical MR, a ligand-activated transcription factor which belongs to the nuclear receptor family with equal affinity for both mineralocorticoids and glucocorticoids (Arriza et al. 1987). It then results in a series of intracellular events beginning with the translocation of receptor ligand into the nucleus to promote protein synthesis and finally mediates the insertion of sodium channels into the epithelial cells of the collecting duct [epithelial sodium channel (ENaC)] to allow sodium reabsorption and potassium and hydrogen ions secretion (Porter and Edelman 1964; Porter et al. 1964). This process takes hours to produce the epithelial effect which can be blocked by protein synthesis inhibitor. The presence of isoenzymes 11β-hydroxysteroid dehydrogenase (11β-HSD) types 1 and 2, along with other factors independent of steroid receptor binding, seems to account for much of the mineralocorticoid selectivity seen biologically (Funder et al. 1988; Funder and Myles 1996). The specificity of the action of aldosterone in the cells containing MR (distal tubules and collecting ducts) is ensured by the expression and activity of 11β-HSD type 2 enzyme, which induces physiological responses by metabolizing circulating glucocorticoid hormones (cortisol in humans, corticosterone in rodents) into inactive 11-dehydro-derivatives (cortisone, 11-dehydrocorticosterone) with very low affinity for the MR (Farman and Rafestin-Oblin 2001). During the hypovolemic and hypoperfusion state of the kidney, RAAS activation leads to vasoconstriction and volume expansion.
Over the ensuing years, the non-genomic aldosterone effects have been demonstrated. In this, a non-classical type of MR is expressed mostly in nonelectrolyte transporting cells and the effects can be observed within minutes as nuclear binding or protein synthesis does not appear to take place (Christ and Wehling 1999; Boldyreff and Wehling 2003; Mihailidou and Funder 2005). The cell signaling pathway involves activation of protein kinase C along with release of intracellular calcium (Mihailidou and Funder 2005). Studies have shown that an alternative G-protein-coupled estrogen receptor, GPR30, may promote rapid, non-genomic effects in vascular endothelial cells (Gros et al. 2013) and vascular smooth cells (Gros et al. 2011).
3 Mechanism of Aldosterone-Mediated Renal Fibrosis
There has been a curious history regarding the relationship between aldosterone and renal disease which led to much researches conducted in the past. In 1964, Conn described the first 125 proven cases of primary hyperaldosteronism with hypertension in patients, among which 85% had gross proteinuria (Conn et al. 1964). The proteinuria was considered as a result of hypertension that accompanies the Conn syndrome, until 1996 when the experiment done by Greene et al. on a rat kidney model showed that mineralocorticoid hormones, particularly aldosterone, can induce proteinuria in the absence of hypertension (Greene et al. 1996). They showed that the treatment with ARB (losartan) and ACE inhibitor (enalapril) in remnant rat kidneys reduced proteinuria, hypertension, and glomerulosclerosis. They also demonstrated that rats injected with exogenous aldosterone in order to maintain very high aldosterone levels were subjected to losartan and enalapril, experienced proteinuria, hypertension, and glomerulosclerosis to the same level as the untreated rats with remnant kidneys did. Another study done on rats with complete unilateral ureteral obstruction (UUO) showed that the renal fibrosis was significantly reduced after the treatment with MR antagonist spironolactone with no changes in serum potassium, aldosterone, and urine sodium (Trachtman et al. 2004). In response to deoxycorticosterone acetate (DOCA) salt treatment, p47phox−/− and gp91phox−/Y mice were found to reduce blood pressure only within the first 2–3 days of treatment and later no significant difference was observed between these mice and wild-type (WT) control group (Zhang et al. 2011). On the contrary, four different kinds of mitochondrial ROS inhibitors (rotenone: mitochondrial respiratory chain complex I inhibitor, 5-hydroxydecanoate: specific mitochondrial ATP-sensitive potassium channel blocker, benzylguanidine: mitochondrial complexes I and III, and the cell-permeable manganese tetrakis (4-benzoic acid), and porphyrin: mitochondrial superoxide dismutase analogues) are found to lower the blood pressure and ROS production in renal tissue, indicating that mitochondria mediates aldosterone-induced ROS production (Zhang et al. 2011). This provides direct evidence that mitochondrial dysfunction contributes to aldosterone-dependent or MR-dependent ROS formation. Juknevicius and colleagues demonstrated that aldosterone infusion for 3 days in normal rats causes a more than twofold increase in the urinary excretion of TGF-β without changes in blood pressure or evidence of kidney damage through an MR-dependent posttranscriptional effect (Juknevicius et al. 2004).
Taken together, these data clearly indicate that aldosterone was involved in the pathogenesis of renal damage and has a direct histological mechanism besides mediating renal damage through hemodynamic pathways. In the following section, we will illustrate the cells, principal molecules, and other mediators involved in the contribution of aldosterone to the pathogenesis of renal fibrosis.
3.1 Induced Mesangial Injury
Mesangial cells are active intrinsic kidney cells which play a crucial role in maintaining the glomerular structural integrity of the microvascular bed which provides mesangial matrix homeostasis and modifies glomerular filtration (Abboud 2012; Brunskill and Potter 2012). Chronic aldosterone infusion increases ROS production through the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nishiyama et al. 2004; Huang et al. 2009). Superoxide anion and hydrogen peroxide are ROS that implicates as important mediators in the mechanisms of aldosterone-mediated renal damage. There are a number of oxidant-generating systems that participate in ROS production such as NADPH oxidase, xanthine oxidase, mitochondrial respiratory chain, cyclooxygenase, lipoxygenase, cytochrome P-450, and nitric oxide synthase.
In rat mesangial cells, aldosterone could directly promote superoxide anion (O2-) generation through the activation of NADPH oxidase and translocation of the cytosolic components of NADPH enzyme p47phox and p67phox to the cell membrane (Miyata et al. 2005). Experiment done in cultured human mesangial cells shows that aldosterone stimulates mesangial cell apoptosis in a dose- and time-dependent manner. The administration of MR antagonist or antioxidant attenuates the proapoptotic effects of aldosterone. Aldosterone also induces dephosphorylation of Bcl-2-associated death promoter (BAD), a protein, which after dephosphorylation is linked to mitochondrial dysfunction leading to accumulation of cytochrome c into the cytosol and mesangial cell apoptosis (Mathew et al. 2008). Aldosterone-infused mesangial cells exhibited increased ROS production, which was blocked by MR antagonist, mitochondrial respiratory chain complex I inhibitor, or an NADPH oxidase inhibitor. These results indicate that mitochondria and NADPH oxidase mediate aldosterone-induced renal ROS production (Huang et al. 2009). Evidence points to aldosterone has a role in mesangial cell proliferation, which involved in MR expression and extracellular signal-regulated kinase 1 and 2 (ERK1/2) activation. Similarly, we have shown that aldosterone-induced mesangial cell proliferation was mediated by Ki-RasA:GTP/c-Raf/MEK/ERK and PI3 K/Akt/mTOR/p70S6K1, an EGF-activated signaling pathway downstream from the EGFR (Huang et al. 2009). Recently, Zhang et al. revealed that Connexin 43 (Cx43), an important regulator of cell growth, showed decreased expression in mesangial cell proliferation and treatment with MR antagonist spironolactone, ERK1/2 inhibitor PD98059, and PKC inhibitor GF109203X attenuated the downregulation of Cx43 expression which suggests that this process might be mediated through (ERK1/2) and PKC pathways (Zhang et al. 2015). TGF-β is a pro-fibrogenic cytokine that has been extensively studied as a responsible factor for renal fibrosis. Aldosterone induces TGF-β1 expression via ERK1/2, c-Jun N-terminal kinase (JNK) and AP-1 pathways (Han et al. 2009).
Aldosterone also exhibited a pro-fibrotic effect by rapidly inducing mRNA and protein expression of CTGF in a time- and concentration-dependent manner (Gauer et al. 2007). The use of MR antagonist spironolactone, canrenoate, or eplerenone could not inhibit the rapid CTGF induction which suggests an MR-independent mechanism for this effect. However, selective inhibitor of glucocorticoid receptor (GR), RU-486, inhibited aldosterone-induced CTGF expression, which indicates that GR is important in aldosterone-induced regulation of CTGF (Gauer et al. 2007). CTGF can promote migration, hypertrophy, fibronectin production, and actin disassembly in mesangial cells and also increases type I, type III, and type IV collagen production by mesangial cells (Phanish et al. 2010). Aldosterone could upregulate CTGF expression in two ways: TGF-β dependent and TGF-β independent. In vivo study done by Terada et al. (2012) showed that in cultured rat mesangial cells, aldosterone promoted the expression of CTGF in TGF-β-dependent manner. In this study, aldosterone stimulates serum- and glucocorticoid-regulated kinase-1 (SGK1) expression and promotes expressions of CTGF and intercellular adhesion molecule-1 (ICAM-1) via NF-κB. However, in a mouse model of diabetic nephropathy, aldosterone significantly increases CTGF gene expression and protein synthesis even in the presence of TGF-β1 neutralizing antibody, which shows the TGF-β-independent pathway for aldosterone-induced CTGF production in cultured mesangial cells (Han et al. 2006). Moreover, treatment with spironolactone markedly decreases renal CTGF and collagen synthesis. Another possible mechanism through which aldosterone may involve in the development of fibrosis is by activating PAI-1 (Huang et al. 2008; Yuan et al. 2007). Huang et al. (2008) observed the increment in aldosterone-induced PAI-1 is partially facilitated by TGF-β1. TGF-β1 and aldosterone acts synergistically to stimulate the PAI-1 expression and decrease extracellular matrix degradation (Huang et al. 2008). Another study done by Yuan et al. (2007) also demonstrated upregulation of PAI-1 mRNA and protein expression by aldosterone, however, was independent of aldosterone-induced increases in TGF-β1 expression and intracellular ROS production. In a high glucose media, aldosterone produced an inflammatory response by upregulating NF-κB and monocyte chemoattractant protein-1 (MCP-1) via AT1a and AT2 pathways (Hao et al. 2015).
Evidences point to show that EGFR may take part in aldosterone-induced mesangial cell proliferation (Huang et al. 2009; Sheng et al. 2016). EGFR is a transmembrane protein with intrinsic tyrosine kinase activity. Upon ligand binding to the receptor induces dimerization and phosphorylation of tyrosine residues in its cytosolic domains. These phosphorylated residues act as docking sites for adaptor proteins and activate downstream signaling cascades (Forrester et al. 2016). The most widely studied signaling pathways are phosphatidylinositol-3 kinase (PI3 K)/AKT and Ras/mitogen-activated protein kinase (MAPK). Aldosterone, after binding to their receptor, can transactivate EGFR, via “a disintegrin and metalloproteases” (ADAMs), thereby regulating several processes including cellular functions, proliferation, hypertrophy, and migration. We have shown that aldosterone transactivates EGFR in cultured mesangial cells, because pretreatment with the EGFR antagonist AG1478 blocked mesangial cell proliferation as well as the activation of Ras/MAPK and PI3 K/Akt (Huang et al. 2009).
In addition, the mitogenic effect of aldosterone was shown to be mediated by Ki-RasA/c-Raf/MEK/ERK and PI3 K/Akt/mTOR/p70S6K1 signaling pathways (Huang et al. 2009). Similarly, Sheng et al. highlighted a role of EGFR in aldosterone-mediated renal fibrosis which relies on ROS-induced EGFR/ERK activation (Sheng et al. 2016).
3.2 Induced Podocyte Injury
The glomerular filtration barrier is composed of three layers: the capillary endothelium, the glomerular basement membrane (GBM), and the podocytes (specialized epithelial cells). Podocytes extend long primary processes from its cell body, the end of which contains foot processes (Wolf et al. 2005). The foot processes of adjacent podocytes are connected by a continuous membrane-like structure called the slit diaphragm and form an interdigitating pattern adhering to the glomerular basement membrane (Wolf et al. 2005; Pavenstadt et al. 2003). The proteins of the slit diaphragm such as nephrin, podocin, and CD2-associated protein (CD2AP) are not only structural, but also serve as a survival function for podocytes by interacting with each other within the body of the podocytes (Wolf et al. 2005). Podocytes thus act as a final filtration barrier against urinary protein loss (Wolf et al. 2005; Shankland 2006). Podocyte injury is the basic pathogenic mechanism that leads to the development of proteinuria and progression of glomerular sclerosis. Several studies have shown that podocytes express MR, which is the target site of aldosterone (Shibata et al. 2007; Kiyomoto et al. 2008; Nagase et al. 2006).
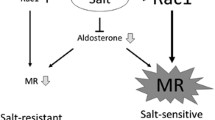
Our study provided direct evidence supporting the role of ROS in contributing to aldosterone-induced podocyte injury (Zhu et al. 2011). Aldosterone-infused mice showed podocyte injury with reduced glomerular expression of nephrin and increased ROS production in renal glomeruli. We examined the important role of mitochondria dysfunction in aldosterone-induced podocyte injury as pronounced by decreased mitochondrial membrane potential, ATP levels, and mitochondrial DNA copy number seen in podocytes and glomeruli. The study done by Nishiyama et al. (2004) showed that aldosterone/salt treatment induced hypertension and renal injury, as characterized by proteinuria, glomerular changes, and collagen accumulation. The renal injury was also associated with increase in renal cortical thiobarbituric acid reactive substance (TBARS) contents, a marker of ROS production, and mRNA expression of NADPH oxidase components, p22phox, Nox-4, and gp91phox. The use of MR antagonist eplerenone prevented the increases in TBARS levels and NADPH oxidase expression in the kidney, and tempol also reduced ROS levels and ameliorated renal injury (Nishiyama et al. 2004). Shibata et al. (2007) showed that continuous infusion of aldosterone in uninephrectomized rats induced massive proteinuria with hypertension and glomerular podocyte injury and decreased gene expressions of nephrin and podocin were closely related to podocyte injury. In the same study, NADPH oxidase activity and TBARS contents were enhanced and podocyte damage was ameliorated by tempol further supporting an important role for ROS in this rat model. It is found that aldosterone can induce the activation of SGK1 signaling pathway through oxidative stress and damage the podocyte (Shibata et al. 2007). Previously conducted studies have found that aldosterone promotes apoptosis of podocytes by inducing mitochondrial dysfunction (Su et al. 2013; Yuan, et al. 2012a). We showed that under normal conditions, endogenous peroxisome proliferator-activated receptor-g coactivator-1α (PGC-1α), a transcriptional coactivator of peroxisome proliferator-activated receptor-γ, and other nuclear hormone receptors are important for podocyte maintenance of mitochondrial function (Yuan, et al. 2012a). SIRT1/PGC-1α axis activation protects against aldosterone-induced podocyte injury likely by preventing mitochondrial dysfunction and SIRT1 agonist, resveratrol-blocked mitochondrial dysfunction, and podocyte injury by activating SIRT1/PGC-1 alpha (Yuan et al. 2012a). Aldosterone-induced podocyte apoptosis seems to be mediated through the activation of p38 MAPK signaling pathway because pretreatment with inhibitor of p38 MAPK suppressed apoptosis (Chen et al. 2009). We recently demonstrated the possible signaling pathways involved in mitochondrial dynamics (Yuan et al. 2017). In this study, aldosterone induced podocyte injury and increased the expression of p53, leading to mitochondrial dysfunction through activation of dynamin-related GTPase dynamin-related protein 1 (Drp1)-mediated mitochondrial fission. Pathogenic role of NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome in podocyte injury, has been identified in our another study (Bai et al. 2017). Herein, exposure of podocytes to aldosterone enhanced NLRP3, indicating an activation of NLRP3 inflammasome. NLRP3 deletion abolished aldosterone-induced NLRP3 inflammasome activation and also ameliorated aldosterone-induced podocyte injury evidenced by the improved proteinuria and silt diaphragm protein loss. In the ARHGDIA gene (encoding RhoGDI 1 alpha) knockout mice, the damage of foot processes is positively correlated with the intensity of Rac1 activation, and the Rac1 inhibitor NSC23766 can inhibit the activation of the SGK1 signaling pathway and reduce the proteinuria and the foot processes damage (Shibata et al. 2008).
3.3 Regulation of Vessels and Vascular Smooth Muscle Cells (VSMCs)
Aldosterone increases the VSMCs proliferation, increases the expression of vascular collagen components, decreases vascular compliance, and constricts the glomerular afferent and efferent arterioles. It increases the glomeruli pressure, perfusion, and filtration which stimulates vasculitis and hence results in renal fibrosis. The main mechanism of aldosterone in stimulation of VSMCs proliferation is as follows: (1) The combination of aldosterone/MR complex with the hormone response element (HRE) of AT1 receptor gene promoter upregulates the AT1 receptor expression which can enhance the effect of Ang II-induced vascular smooth muscle hypertrophy, hence stimulating the proliferation of VSMCs (Yamada et al. 2008) and (2) Aldosterone activates ERK1/2 signaling pathway and promotes the proliferation of VSMCs (Min et al. 2005). A study showed the synergistic signaling interaction between aldosterone and Ang II on the mitogenic action in VSMCs (Min et al. 2005). Here, aldosterone and Ang II increased the early activation of ERK1/2 involving the transactivation of the EGFR, whereas delayed activation involved increased MAPK-1 and Ki-ras2A expression. Other investigators used small interference RNA for AT1aR, AT1bR, and MR to explain the cross talk between aldosterone and angiotensin II in mouse VSMCs. Aldosterone and Ang II induced ERK1/2, JNK, and NF-κB phosphorylation (Bomback and Klemmer 2007). This study showed ERK1/2 activation through an AT1-dependent, MR-independent mechanism, and JNK and NF-κB activation required both AT1- and MR-dependent mechanisms after aldosterone stimulation.
Aldosterone increases expression of PAI-1 in VSMCs and endothelial cells (Brown et al. 2000a, b). PAI-1 is a member of the serine protease inhibitor (serpin) family, and it can promote fibrosis by preventing matrix metalloproteinases (MMPs) activation and ECM degradation by plasminogen activators and plasmin. PAI-1 is the major physiological inhibitor of tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA). It inhibits the activation of plasminogen to plasmin by tPA and also can also exert anti-fibrotic effects by retarding cellular infiltration and interfering with uPA or plasmin-mediated activation and the release of latent growth factors. By interacting with Ang II, aldosterone increases PAI-1 expression in a dose-dependent manner both in vitro cultured vascular smooth muscle cells and in vivo vascular endothelial cells (Brown et al. 2000b). This effect was blocked by MR antagonist spironolactone, indicating that aldosterone promotes PAI-1 expression through the classical salt mineralocorticoid receptor mechanism. In this study, plasma PAI-1 levels in patients with primary hyperaldosteronism increased significantly. In a radiation model of renal damage, inhibition of aldosterone also downregulates PAI-1 expression in vivo, and local PAI-1 expression is closely associated with sclerosis (Brown et al. 2000a). These collective evidences support the hypothesis that aldosterone induces renal fibrosis at least in part through its effects on PAI-1 expression. Grossmann et al. (2007) showed the aldosterone increases the expression of EGFR in vascular smooth muscle cells via an interaction with the EGFR promoter, which is MR specific.
Nitric oxide (NO) has several functions in the kidney including dilatation of the renal blood vessels, regulation of renal hemodynamics, blunting of tubuloglomerular feedback, modulation of renal sympathetic nerve activity, and inhibition of matrix protein aggregation, smooth muscle cell proliferation, and fibroblast proliferation. Nω-nitro-L-arginine methyl ester (L-NAME) is a potent inhibitor of NO synthase which can cause systemic hypertension, vascular injury, and vascular endothelial cell dysfunction. It thus plays an important role in renal fibrosis. Ikeda et al. (2009) demonstrated significant increase in blood pressure, proteinuria, renal inflammation, and fibrosis in L-NAME-treated rats. These effects were prevented significantly after the use of aldosterone antagonist, spironolactone, which may suggest the role of aldosterone in contributing to renal inflammation and fibrosis by inhibiting the formation of NO. Besides, Arima et al. (2003) examined the vascular action of aldosterone that can cause dose-dependent constriction in both afferent and efferent glomerular arterioles with more susceptibility in efferent arterioles. These effects are via non-genomic pathway as the mineralocorticoid receptor antagonist spironolactone was not able to block the vasoconstriction effect of aldosterone. Instead, the effect of aldosterone on both the arterioles was due to activation of phospholipase C and thus promoting calcium transport through L- and T-type calcium channels. In addition, aldosterone-induced vasculitis may also be involved in the process of renal fibrosis (Blasi et al. 2003). It was demonstrated that aldosterone/salt administration induces elevated expression of inflammatory components including MCP-1, IL-1β, and IL-6 in the blood vessels of rats. It was also accompanied with severe albuminuria, renal vascular injury, and histopathological changes in the kidney. The author further observed that eplerenone attenuated these inflammatory components, improved vasculitis, and reduced renal inflammation and fibrosis (Blasi et al. 2003).
3.4 Phenotypic Transition of Renal Tubular Epithelial Cells
Epithelial–mesenchymal transition (EMT) is defined as phenotypic conversion in epithelial cells which leads to loss of contacts between epithelial cell-to-cell basement membrane and hence gives rise to matrix-producing fibroblasts and myofibroblasts (Acloque et al. 2009; Thiery 2002; Kalluri and Weinberg 2009). EMT is known to play a pivotal role in the mechanism of tubulointerstitial fibrosis (Burns and Thomas 2011). We have previously demonstrated that the aldosterone induces EMT in renal epithelial cells through MR-mediated, mitochondria-derived ROS, and activation of ERK1/2 signaling pathway (Zhang et al. 2007). This effect was significantly blocked by MR antagonist eplerenone and mitochondrial respiratory chain complex I inhibitor rotenone. In the DOCA/salt mice model, EMT was evidenced by exhibiting the tubular epithelial cells as elongated and fibroblast-like morphology, expression of alpha-smooth muscle actin (fibroblast markers) was increased, and E-cadherin (epithelial markers) was decreased. We further extended our investigation demonstrating the role of mitochondrial function in aldosterone-induced EMT (Yuan et al. 2012b). PGC-1α and silent mating-type information regulation 2 homolog 1 (SIRT1) function together to control the mitochondrial biogenesis and function. In this study, aldosterone-infused human kidney cells showed decreased PGC1 expression and induced mitochondrial dysfunction leading to promoting EMT, whereas overexpression of PGC-1α prevented mitochondrial dysfunction and EMT. SIRT1/PGC-1α axis activation protected against aldosterone-induced EMT by inhibiting mitochondrial dysfunction (Yuan et al. 2012b). Aldosterone has been reported to contribute to normal renal tubular epithelial cell differentiation and proliferation from isolated renal stem cells (Minuth et al. 2005), and in vivo experiment done on juvenile animals can also cause dysregulation of renal cell growth and promote renal fibrosis (Heber et al. 2007). Human tubular epithelial cells treated with aldosterone activate EGFR via TGF-α/ADAM17 (Morgado-Pascual et al. 2015). Blockade of ADAM-17/TGF-α/EGFR pathway using the specific inhibitors, the ADAM17 inhibitor TAPI-2, an anti-TGF-α neutralizing antibody, and the EGFR kinase inhibitor AG1478, significantly diminished aldosterone-induced pro-inflammatory gene upregulation which supports the involvement of ADAM-17/TGF-α/EGFR axis in the regulation of pro-inflammatory factors by aldosterone (Morgado-Pascual et al. 2015).
3.5 Fibroblasts
In rat kidney fibroblast cells, aldosterone induces proliferation via MR-mediated kinase activity of the growth factor receptors and downstream cell signaling through P13 K/Akt, ERK, and JNK pathways (Huang et al. 2012). Aldosterone induced uninephrectomized rats in the presence of AT1 receptor blockade, increased the expression of TGF-β1 and collagen mRNAs, and diffused renal medullary and cortical fibrosis accompanied by the accumulation of abundant myofibroblasts at the sites of fibrosis. This suggests the independent role of aldosterone in renal fibrosis through TGF-β1 signaling pathways, thereby upregulating the collagen synthesis, downregulating the release of the extracellular matrix metalloproteinase collagenase, and hence promoting fibroblast proliferation (Sun et al. 2000). CTGF is recognized as a pro-fibrotic protein and plays an important role in the pathogenesis of chronic fibrotic disorders (Phanish et al. 2010). The CTGF gene promoter contains a TGF-β1-responsive element as well as other regulatory sequences and thus acts as an early responder during the onset of fibrotic diseases. In addition to its own pro-fibrotic effect, it is a downstream mediator of at least some of the pro-fibrotic effects of TGF-β1, in particular proliferation of fibroblasts and secretion of ECM proteins by fibroblasts (Phanish et al. 2010). OPN is a key mediator of aldosterone-induced renal fibrosis. It is a glycosylated phosphoprotein that is produced by osteoblasts, macrophages, endothelial cells, and epithelial cells and acts to facilitate cell adhesion and migration. In the normal kidney, it is mainly expressed in the loop of Henle and distal nephron and is thought to play a role in renal fibroblast proliferation and ECM synthesis. Aldosterone significantly increased OPN mRNA expression and that OPN-small inference RNA completely blocked aldosterone-induced collagen synthesis and renal fibroblast proliferation in renal fibroblasts (Irita et al. 2008). This study also showed the use of MR antagonist spironolactone abolished aldosterone-induced OPN expression through AP-1 and NF-κB binding activities, which suggests that OPN may be a downstream factor of aldosterone in renal fibrosis.
3.6 Infiltration of Inflammatory Cells
Infiltration of inflammatory cells is one of the major cellular events in renal fibrosis. In response to injury, inflammation is an essential part of host defense mechanism. However, inflammation which is non-resolving tends to develop fibrotic diseases (Liu 2011). Following renal fibrosis, the inflammatory cells become activated, which produce molecules such as ROS that damage tissues; hence, growth factors and fibrogenic cytokines are produced. Macrophage, a member of the mononuclear phagocyte family, has a key role in renal injury, inflammation, and fibrosis. They are pleiotropic inflammatory cells that take part in inflammatory reactions (Han et al. 2019). Blasi et al. (2003) showed the possible role of aldosterone in mediating renal fibrosis which is preceded by inflammatory components infiltration including macrophage, and increased the expression of pro-inflammatory cytokines OPN, interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), MCP-1, suggesting that inflammation is an essential contributor of renal fibrosis. The heterogeneity of macrophage polarization has been recognized as an important feature of renal disease (Li et al. 2015). Aldosterone plus salt treated in rat kidney model contributes to classical macrophage activation to the inflammatory M1 phenotype, along with hypertension, renal damage, and fibrosis. These effects were effectively blocked by MR antagonism spironolactone, thereby showing the beneficial effect of MR antagonist in hypertensive renal disease (Martín-Fernández et al. 2016). NF-κB, a eukaryotic transcription factor, is one of the most important pro-inflammatory signal pathways which has been intensively investigated over the past 30 years because of its involvement of several biological programs. NF-κB can be activated by various molecules, such as lipopolysaccharide, cytokines, and ROS, and influence the transcription of several genes involved in the inflammatory response, cell growth, and adhesion (Baeuerle and Henkel 1994). Ding et al. conducted an experiment showing the role of NF-κB in the pathogenesis of aldosterone-induced renal injury (Ding et al. 2012). It was found that the activity of NF-κB and inflammation in the renal tissue of aldosterone-/salt-treated rats increased significantly along with the upregulated expression of ICAM-1, TGF-β, CTGF, and type IV collagen. Treatment with pyrrolidine dithiocarbamate (PDTC), a NF-κB inhibitor, significantly reduced the pathological damage of kidney tissue and reduced the expression of factors including CTGF, ICAM-1, TGF-β, and collagen IV (Ding et al. 2012).
4 Aldosterone Breakthrough
We have already mentioned studies that showed proteinuria induced by aldosterone is one of the important signs in the progression of renal disease and renal fibrosis. The renin–angiotensin system (RAS) plays a key role in the development of proteinuria in kidney diseases. Blockade of RAS with the use of ACE inhibitors decreases the levels of circulating Ang II, and ARBs inhibit the action of AT1R significantly reducing the urinary protein excretion which further delays the chronic progression of renal disease. However, with the use of ACE inhibitors or ARBs, Ang II inhibits release of renin and leads to increased plasma renal activity. Besides, after the initiation of ACE inhibitors or ARBs therapy for several weeks, plasma aldosterone concentration is increased to pretreatment level, following a decrease of aldosterone levels in the initial treatment phase. This phenomenon is known as aldosterone breakthrough, which also suggests the role of aldosterone in mediating proteinuria (Bomback and Klemmer 2007). Up to 10–53% CKD patients on ACE inhibitors or ARBs were found to have aldosterone breakthrough in previous studies using various definitions (Bomback and Klemmer 2007). It was shown that patients who experienced aldosterone breakthrough had enhanced decline in GFR compared with those without breakthrough (Schjoedt et al. 2004). Aldosterone breakthrough provides a theoretical basis for clinical application of aldosterone receptor antagonists. Many observational studies have confirmed that the combination of MR antagonist and ACE inhibitors or ARBs can effectively reduce proteinuria and delay renal fibrosis in patients with CKD (Schjoedt et al. 2004; Furumatsu et al. 2008; Schjoedt et al. 2006; Chrysostomou et al. 2006; Bianchi et al. 2006). Sato and Fukuda (2013) reported that aldosterone breakthrough occurred in 55% of hypertensive patients after the use of direct renin inhibitor (DRI) aliskiren, which acts by directly blocking the enzymatic action of renin without interfering with its production and interaction with renin receptors. Adding MR receptor antagonist eplerenone to DRI to those patients showed decrease in albuminuria and may benefit hypertensive patients by exerting a cardiovascular and/or renal protective effect.
The mechanism of aldosterone breakthrough is not fully elucidated. It may be related to the following factors: (1) Ang II is produced by alternative pathway and non-renin pathway. ACE inhibitor can block the classical Ang II generation pathway, but it does not play a role in the alternative pathway and non-renin pathway of Ang II generation (Urata et al. 1994a, b; Cicoira et al. 2001); (2) Long-term blockade of AT1 receptor leads to increase in Ang II which may mediate aldosterone synthesis through AT2 receptor (Naruse et al. 2002); (3) Some aldosterone stimulating factors that do not depend upon Ang II are endothelin, vasopressin, catecholamine, ACTH, glucocorticoid, hyperkalemia, hyperlipidemia, and reduced high-density lipoprotein.
5 Treatment
5.1 Clinical Trials
5.1.1 MR Antagonists
The two clinically approved and commonly used MR antagonists are spironolactone and eplerenone, both of which are steroidal compounds. They share similar molecular structure of aldosterone, natural MR ligands, and cortisol. Searle Laboratories (Skokie, Ill) introduced spironolactone as the first MR antagonist in 1959 (Jaisser and Farman 2016). It was recommended for the treatment of edema, hypertension, primary aldosteronism, and heart failure. It is commonly used as a diuretic. Spironolactone is a potent MR antagonist; however, due to its poorly selective nature toward MR, it inhibits the androgen and progesterone receptors (Kolkhof and Borden 2012). Therapeutic use of this drug has led to several side effects such as feminization, gynecomastia, impotence, and menstrual irregularities, which reflect its anti-androgenic and progestogenic activity. In the Randomized Aldactone Evaluation Study (RALES), 10% of men in the spironolactone-treated group had gynecomastia and breast pain and the incidence rate of hyperkalemia was only slightly higher than the placebo group (Pitt et al. 1999). Other side effects of spironolactone are hyperkalemia, hyponatremia, dizziness, abnormal renal function, and high creatinine concentration. These side effects limit the patients with spironolactone therapy. In recent years, spironolactone has shown to prevent renal fibrosis. In 2002, a second-generation MR antagonist eplerenone was launched for the treatment of hypertension and congestive heart failure. In comparison with spironolactone, eplerenone has more selectivity for MR but less potency (40-fold less affinity) and interacts less with other steroid receptors (Kolkhof and Borden 2012). Therefore, there are fewer hormonal-related side effects with eplerenone. In the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS), 15.6% of the patients with eplerenone therapy had serum potassium (K+) >5.5 mEq/L in comparison with 11.2% with the placebo group (Pitt et al. 2008). Essentially, patients who are taking spironolactone and eplerenone need to monitor their serum potassium level and renal function carefully. The ratio of sodium/potassium in the urine is an accurate indicator of the appropriate dose of spironolactone.
Nonsteroidal compound dihydropyridine, which is also known as L-type calcium channel antagonists, can also act as MR antagonist (Kolkhof and Borden 2012). A potent and selective dihydropyridine-based MR antagonist, BR-4628, was generated due to optimization of antagonists without L-type calcium channel activation. Finerenone (BAY 94–8862) is a third-generation, novel dihydronaphthyridine class of MR antagonist formed by the optimization of BR-4628 which has shown better selectivity nature for MR over spironolactone and eplerenone (Jaisser and Farman 2016). The use of finerenone was investigated in a phase 2 clinical trial of Mineralocorticoid Receptor Antagonist Tolerability Study (ARTS), which showed equal effectiveness as spironolactone in reducing the levels of B-type natriuretic peptide (BNP), amino-terminal proBNP, and albuminuria (Pitt et al. 2013). However, the rate of hyperkalemia and renal dysfunction was lowered in finerenone group demonstrating its safety profile than spironolactone in patients with chronic heart failure and kidney impairment. Another clinical trial, the ARTS-diabetic nephropathy (ARTS-DN), was launched in 823 patients suffering from type II diabetic mellitus or diabetic nephropathy in order to testify the efficacy of different oral doses of finerenone compared to placebo, who were initially receiving ACE inhibitors or ARBs (Bakris et al. 2015). At day 90 follow-up, finerenone (7.5–20 mg once daily) reduced the albuminuria (17.2–40.2%) in a dose-dependent manner as compared with placebo (13.6%). There was no significant correlation seen between changes in urinary albumin–creatinine ratio and changes in systolic blood pressure which suggests that lowering of blood pressure does not lower albumin excretion, and intraglomerular pressure is also independent of urinary albumin–creatinine ratio changes.
5.1.2 Aldosterone Synthase Inhibitor
Inhibition of aldosterone synthase is an alternative approach to antagonize the harmful aldosterone actions (both MR dependent and MR independent) by directly attenuating its production in cardiac, vascular, and renal target organs (Azizi et al. 2012). Therefore, enzymatic activity of the aldosterone synthase is inactivated resulting in decreased aldosterone concentrations in plasma and tissue. Aldosterone synthase inhibitor has been studied as a new treatment option for management of hypertension, cardiac failure, and renal diseases. LCI699 is the first orally active aldosterone synthase inhibitor drug that was discovered for humans. LCI699, when used in 14 patients with primary aldosteronism observed dose-dependent reduction in plasma and urinary aldosterone concentrations by 70–80%, decreased blood pressure, and plasma renin activity was increased mildly. Notably, LCI699 was shown to have limited selectivity to aldosterone synthase CYP11B2 (Azizi et al. 2012).
5.2 Experimental Studies
As mentioned earlier, various experiments were carried out in animal models which have shown a potential role of MR antagonists, along with other pathways inhibitors in delaying the progression of renal fibrosis through different mechanisms. In this section, we will briefly mention a few of them. We have examined the role of MR antagonist, mitochondrial respiratory chain complex I inhibitor, or an NADPH oxidase inhibitor in significantly mitigating the mesangial cell proliferation and ROS production which is induced by aldosterone (Huang et al. 2009). In this, eplerenone also inhibited EGFR transactivation which is a downstream effector of PI3 K/Akt and Ras/MAPK signaling pathway. Aldosterone-induced ERK1/2 phosphorylation in mesangial cells was significantly attenuated when using eplerenone as pretreatment (Nishiyama et al. 2005). Spironolactone can block the role of NO synthase inhibitor L-NAME in renal tissue which showed that reduced expression of TGF-beta 1, CTGF, OPN, and PAI-1 in renal tissue, proteinuria, and blood pressure was significantly decreased, hence alleviating renal fibrosis and renal function deterioration (Ikeda et al. 2009). In another study, we also showed that aldosterone-induced EMT was significantly blocked with the use of eplerenone and rotenone (Zhang et al. 2007). Treatment with eplerenone significantly improved aldosterone-induced renal injury and increased serum- and glucocorticoid-inducible protein kinase-1 (SGK1), ICAM-1, and CTGF expressions in the rat mesangial cells which may be considered as another therapeutic target in the treatment of renal fibrosis (Terada et al. 2012). Blockade of ADAM-17/TGF-α/EGFR pathway diminished aldosterone-induced pro-inflammatory gene upregulation (Morgado-Pascual et al. 2015) which may be another treatment option for preventing the progression of renal fibrosis. When treated with eplerenone, podocyte damage and proteinuria were markedly attenuated in the aldosterone-treated rats (Shibata et al. 2007). Besides, treatment with eplerenone or tempol also significantly reduced podocyte injury and proteinuria in other rat models of hypertensive glomerulosclerosis (Nagase et al. 2006, 2007) and type 2 diabetic rats (Nishiyama et al. 2010). Our study has shown activator of SIRT1, resveratrol-blocked mitochondrial dysfunction, and podocyte injury by activating SIRT1/PGC-1 alpha which may be therapeutically useful in renal diseases (Yuan et al. 2012a). Blockade of Drp1 inhibited mitochondrial fission and dysfunction, and podocyte apoptosis in an animal model of aldosterone-induced nephropathy which may provide another promising therapeutics for podocyte injury (Yuan et al. 2017). In order to discover highly selective aldosterone synthase inhibitor, an experimental study was conducted in cynomolgus monkey-based models which showed BI 689648 as a highly selective one (Weldon et al. 2016). It revealed an in vitro IC50 of 2 nM against aldosterone synthase and cortisol synthase and 150-fold selectivity. For in vivo selectivity profiling, an adrenocorticotropin-challenge model was used which exhibited BI 689648 to be more than 20-fold selective compared with FAD286 and LCI699. This study may highlight an important step forward in the identification of effective and high potential aldosterone synthase inhibitors for clinical setup in cardiometabolic diseases, diabetic nephropathy, and CKD.
6 Summary
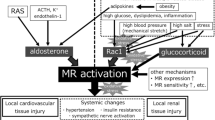
Apart from its physiological action on salt and water homeostasis, herein, we have discussed aldosterone as a potent factor in the pathogenesis of renal fibrosis. The prevailing in vivo and in vitro experiments suggest the independent detrimental role of aldosterone along with its MR, eventually contributing to the development of renal injury and fibrosis. As shown in Fig. 15.1, aldosterone has multiple mechanisms underlying the pathogenic role in kidney ultimately promoting renal injury and fibrosis. Some small clinical studies have shown that the use of aldosterone antagonists in patients with CKD can reduce proteinuria and delay the progression of renal fibrosis. However, large-scale clinical studies are needed to confirm the safety and effectiveness of aldosterone receptor antagonists in CKD patients.

Aldosterone stimulates the increase in ROS production which induces mitochondrial dysfunction. ROS also activates EGFR. EGFR transactivation activates PI3 K/Akt/mTOR/p70S6K1 and Ki-RasA/c-Raf/MEK/ERK and signaling pathways. Activation of p38 MAPK could trigger the Caspase 3 death pathway. Aldosterone induces SGK1 phosphorylation and promotes expressions of CTGF and ICAM-1 via NF-κB. In kidney, these changes lead to activation of pro-inflammatory and pro-fibrotic pathways ultimately promoting glomerulosclerosis and tubulointerstitial fibrosis
References
Abboud HE (2012) Mesangial cell biology. Exp Cell Res 318:979–985
Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA (2009) Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest 119:1438–1449
Arima S, Kohagura K, Xu H-L, Sugawara A, Abe T, Satoh F et al (2003) Nongenomic vascular action of aldosterone in the glomerular microcirculation. J Am Soc Nephrol 14:2255–2263
Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE et al (1987) Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 237:268–275
Azizi M, Amar L, Menard J (2012) Aldosterone synthase inhibition in humans. Nephrol Dial Transplant 28:36–43
Baeuerle PA, Henkel T (1994) Function and activation of NF-kappaB in the immune system. Annu Rev Immunol 12:141–179
Bai M, Chen Y, Zhao M, Zhang Y, He JC-J, Huang S et al (2017) NLRP3 inflammasome activation contributes to aldosterone-induced podocyte injury. Am J Physiol Renal Physiol 312:F556–F564
Bakris GL, Agarwal R, Chan JC, Cooper ME, Gansevoort RT, Haller H et al (2015) Effect of finerenone on albuminuria in patients with diabetic nephropathy: a randomized clinical trial. JAMA 314:884–894
Bianchi S, Bigazzi R, Campese VM (2006) Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 70:2116–2123
Blasi ER, Rocha R, Rudolph AE, Blomme EA, Polly ML, McMahon EG (2003) Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int 63:1791–1800
Boldyreff B, Wehling M (2003) Non-genomic actions of aldosterone: mechanisms and consequences in kidney cells. Nephrol Dial Transplant 18:1693–1695
Bomback AS, Klemmer PJ (2007) The incidence and implications of aldosterone breakthrough. Nat Clin Pract Nephrol 3:486–492
Bonvalet J, Alfaidy N, Farman N, Lombes M (1995) Aldosterone: intracellular receptors in human heart. Eur Heart J 16:92–97
Brown NJ (2013) Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat Rev Nephrol 9:459–469
Brown NJ, Kim KS, Chen YQ, Blevins LS, Nadeau JH, Meranze SG et al (2000a) Synergistic effect of adrenal steroids and angiotensin II on plasminogen activator inhibitor-1 production. J Clin Endocrinol Metab 85:336–344
Brown NJ, Nakamura S, Ma L, Nakamura I, Donnert E, Freeman M et al (2000b) Aldosterone modulates plasminogen activator inhibitor-1 and glomerulosclerosis in vivo. Kidney Int 58:1219–1227
Brunskill EW, Potter SS (2012) Changes in the gene expression programs of renal mesangial cells during diabetic nephropathy. BMC Nephrol 13:70
Burns W, Thomas M (2011) Angiotensin II and its role in tubular epithelial to mesenchymal transition associated with chronic kidney disease. Cells Tissues Organs 193:74–84
Chen C, Liang W, Jia J, Van Goor H, Singhal PC, Ding G (2009) Aldosterone induces apoptosis in rat podocytes: role of PI3-K/Akt and p38MAPK signaling pathways. Nephron Exp Nephrol 113:e26–e34
Christ M, Wehling M (1999) Rapid actions of aldosterone: lymphocytes, vascular smooth muscle and endothelial cells. Steroids 64:35–41
Chrysostomou A, Pedagogos E, MacGregor L, Becker GJ (2006) Double-blind, placebo-controlled study on the effect of the aldosterone receptor antagonist spironolactone in patients who have persistent proteinuria and are on long-term angiotensin-converting enzyme inhibitor therapy, with or without an angiotensin II receptor blocker. Clin J Am Soc Nephrol 1:256–262
Cicoira M, Zanolla L, Rossi A, Golia G, Franceschini L, Cabrini G et al (2001) Failure of aldosterone suppression despite angiotensin-converting enzyme (ACE) inhibitor administration in chronic heart failure is associated with ACE DD genotype. J Am Coll Cardiol 37:1808–1812
Coirini H, Mari A, De Nicola AF, Rainbow TC, McEwen BS (1985) Further studies of brain aldosterone binding sites employing new mineralocorticoid and glucocorticoid receptor markers in vitro. Brain Res 361:212–216
Conn JW, Knopf RF, Nesbit RM (1964) Clinical characteristics of primary aldosteronism from an analysis of 145 cases. Am J Surg 107:159–172
Ding W, Yang L, Zhang M, Gu Y (2012) Chronic inhibition of nuclear factor kappa B attenuates aldosterone/salt-induced renal injury. Life Sci 90:600–666
Epstein M (2006) Aldosterone blockade: an emerging strategy for abrogating progressive renal disease. Am J Med 119:912–919
Farman N, Rafestin-Oblin M-E (2001) Multiple aspects of mineralocorticoid selectivity. Am J Physiol Renal Physiol 280:F181–F192
Forrester SJ, Kawai T, O’Brien S, Thomas W, Harris RC, Eguchi S (2016) Epidermal growth factor receptor transactivation: mechanisms, pathophysiology, and potential therapies in the cardiovascular system. Annu Rev Pharmacol Toxicol 56:627–653
Funder J, Myles K (1996) Exclusion of corticosterone from epithelial mineralocorticoid receptors is insufficient for selectivity of aldosterone action: in vivo binding studies. Endocrinology 137:5264–5268
Funder JW (2010) Aldosterone and mineralocorticoid receptors in the cardiovascular system. Prog Cardiovasc Dis 52:393–400
Funder JW, Pearce PT, Smith R, Smith AI (1988) Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science 242:583–585
Furumatsu Y, Nagasawa Y, Tomida K, Mikami S, Kaneko T, Okada N et al (2008) Effect of renin-angiotensin-aldosterone system triple blockade on non-diabetic renal disease: addition of an aldosterone blocker, spironolactone, to combination treatment with an angiotensin-converting enzyme inhibitor and angiotensin II receptor blocker. Hypertens Res 31:59–67
Gauer S, Segitz V, Goppelt-Struebe M (2007) Aldosterone induces CTGF in mesangial cells by activation of the glucocorticoid receptor. Nephrol Dial Transplant 22:3154–3159
Gonzalez A, López B, Dı́ez J (2004) Fibrosis in hypertensive heart disease: role of the renin-angiotensin-aldosterone system. Med Clin North Am 88:83–97
Greene E, Kren S, Hostetter T (1996) Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 98:1063–1068
Gros R, Ding Q, Sklar LA, Prossnitz EE, Arterburn JB, Chorazyczewski J et al (2011) GPR30 expression is required for the mineralocorticoid receptor–independent rapid vascular effects of aldosterone. Hypertension 57:442–451
Gros R, Ding Q, Liu B, Chorazyczewski J, Feldman RD (2013) Aldosterone mediates its rapid effects in vascular endothelial cells through GPER activation. Am J Physiol Cell Physiol 304:C532–C540
Grossmann C, Krug AW, Freudinger R, Mildenberger S, Voelker K, Gekle M (2007) Aldosterone-induced EGFR expression: interaction between the human mineralocorticoid receptor and the human EGFR promoter. Am J Physiol Endocrinol Metab 292:E1790–E1800
Han K, Kang Y, Han SY, Jee Y, Lee M, Han J et al (2006) Spironolactone ameliorates renal injury and connective tissue growth factor expression in type II diabetic rats. Kidney Int 70:111–120
Han JS, Choi BS, Yang CW, Kim YS (2009) Aldosterone-induced TGF-β1 expression is regulated by mitogen-activated protein kinases and activator protein-1 in mesangial cells. J Korean Med Sci 24:S195–S203
Han HI, Skvarca LB, Espiritu EB, Davidson AJ, Hukriede NA (2019) The role of macrophages during acute kidney injury: destruction and repair. Pediatr Nephrol 34:1–9
Hao J, Ren L, Zhang L, Kong D, Hao L (2015) Aldosterone-induced inflammatory response of mesangial cells via angiotension II receptors. J Renin Angiotensin Aldosterone Syst 16:739–748
Heber S, Denk L, Hu K, Minuth WW (2007) Modulating the development of renal tubules growing in serum-free culture medium at an artificial interstitium. Tissue Eng 13:281–292
Hostetter TH, Ibrahim HN (2003) Aldosterone in chronic kidney and cardiac disease. J Am Soc Nephrol 14:2395–2401
Huang W, Xu C, Kahng KW, Noble NA, Border WA, Huang Y (2008) Aldosterone and TGF-β1 synergistically increase PAI-1 and decrease matrix degradation in rat renal mesangial and fibroblast cells. Am J Physiol Renal Physiol 294:F1287–F1295
Huang S, Zhang A, Ding G, Chen R (2009) Aldosterone-induced mesangial cell proliferation is mediated by EGF receptor transactivation. Am J Physiol Renal Physiol 296:F1323–F1333
Huang L, Nikolic-Paterson D, Ma F, Tesch G (2012) Aldosterone induces kidney fibroblast proliferation via activation of growth factor receptors and PI3K/MAPK signalling. Nephron Exp Nephrol 120:e115–e122
Ikeda H, Tsuruya K, Toyonaga J, Masutani K, Hayashida H, Hirakata H et al (2009) Spironolactone suppresses inflammation and prevents L-NAME–induced renal injury in rats. Kidney Int 75:147–155
Irita J, Okura T, Kurata M, Miyoshi K-i, Fukuoka T, Higaki J (2008) Osteopontin in rat renal fibroblasts: functional properties and transcriptional regulation by aldosterone. Hypertension 51:507–513
Jaisser F, Farman N (2016) Emerging roles of the mineralocorticoid receptor in pathology: toward new paradigms in clinical pharmacology. Pharmacol Rev 68:49–75
Juknevicius I, Segal Y, Kren S, Lee R, Hostetter TH (2004) Effect of aldosterone on renal transforming growth factor-β. Am J Physiol Renal Physiol 286:F1059–F1062
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119:1420–1428
Kiyomoto H, Rafiq K, Mostofa M, Nishiyama A (2008) Possible underlying mechanisms responsible for aldosterone and mineralocorticoid receptor-dependent renal injury. J Pharmacol Sci 108:399–405
Kolkhof P, Borden SA (2012) Molecular pharmacology of the mineralocorticoid receptor: prospects for novel therapeutics. Mol Cell Endocrinol 350:310–317
Kornel L (1994) Colocalization of 11β-hydroxysteroid dehydrogenase and mineralocorticoid receptors in cultured vascular smooth muscle cells. Am J Hypertens 7:100–103
Lenzini L, Seccia TM, Aldighieri E, Belloni AS, Bernante P, Giuliani L et al (2007) Heterogeneity of aldosterone-producing adenomas revealed by a whole transcriptome analysis. Hypertension 50:1106–1113
Li C, Ding XY, Xiang DM, Xu J, Huang XL, Hou FF et al (2015) Enhanced M1 and impaired M2 macrophage polarization and reduced mitochondrial biogenesis via inhibition of AMP kinase in chronic kidney disease. Cell Physiol Biochem 36:358–372
Liu Y (2011) Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol 7:684–696
Martín-Fernández B, Rubio-Navarro A, Cortegano I, Ballesteros S, Alía M, Cannata-Ortiz P et al (2016) Aldosterone induces renal fibrosis and inflammatory M1-macrophage subtype via mineralocorticoid receptor in rats. PLoS ONE 11:e0145946
Mathew JT, Patni H, Chaudhary AN, Liang W, Gupta A, Chander PN et al (2008) Aldosterone induces mesangial cell apoptosis both in vivo and in vitro. Am J Physiol Renal Physiol 295:F73–F81
Mihailidou AS, Funder JW (2005) Nongenomic effects of mineralocorticoid receptor activation in the cardiovascular system. Steroids 70:347–351
Min LJ, Mogi M, Li JM, Iwanami J, Iwai M, Horiuchi M (2005) Aldosterone and angiotensin II synergistically induce mitogenic response in vascular smooth muscle cells. Circ Res 97:434–442
Minuth WW, Denk L, Heber S (2005) Growth of embryonic renal parenchyme at the interphase of a polyester artificial interstitium. Biomaterials 26:6588–6598
Miyata K, Rahman M, Shokoji T, Nagai Y, Zhang GX, Sun GP et al (2005) Aldosterone stimulates reactive oxygen species production through activation of NADPH oxidase in rat mesangial cells. J Am Soc Nephrol 16:2906–2912
Morgado-Pascual JL, Rayego-Mateos S, Valdivielso JM, Ortiz A, Egido J, Ruiz-Ortega M (2015) Paricalcitol inhibits aldosterone-induced proinflammatory factors by modulating epidermal growth factor receptor pathway in cultured tubular epithelial cells. Biomed Res Int 2015:783538
Nagase M, Shibata S, Yoshida S, Nagase T, Gotoda T, Fujita T (2006) Podocyte injury underlies the glomerulopathy of Dahl salt-hypertensive rats and is reversed by aldosterone blocker. Hypertension 47:1084–1093
Nagase M, Matsui H, Shibata S, Gotoda T, Fujita T (2007) Salt-induced nephropathy in obese spontaneously hypertensive rats via paradoxical activation of the mineralocorticoid receptor: role of oxidative stress. Hypertension 50:877–883
Naruse M, Tanabe A, Sato A, Takagi S, Tsuchiya K, Imaki T et al (2002) Aldosterone breakthrough during angiotensin II receptor antagonist therapy in stroke-prone spontaneously hypertensive rats. Hypertension 40(1):28–33
Nishiyama A, Yao L, Nagai Y, Miyata K, Yoshizumi M, Kagami S et al (2004) Possible contributions of reactive oxygen species and mitogen-activated protein kinase to renal injury in aldosterone/salt-induced hypertensive rats. Hypertension 43:841–848
Nishiyama A, Yao L, Fan Y, Kyaw M, Kataoka N, Hashimoto K et al (2005) Involvement of aldosterone and mineralocorticoid receptors in rat mesangial cell proliferation and deformability. Hypertension 45:710–716
Nishiyama A, Kobori H, Konishi Y, Morikawa T, Maeda I, Okumura M et al (2010) Mineralocorticoid receptor blockade enhances the antiproteinuric effect of an angiotensin II blocker through inhibiting podocyte injury in type 2 diabetic rats. J Pharmacol Exp Ther 332:1072–1080
Pavenstadt H, Kriz W, Kretzler M (2003) Cell biology of the glomerular podocyte. Physiol Rev 83:253–307
Phanish MK, Winn S, Dockrell M (2010) Connective tissue growth factor-(CTGF, CCN2)–a marker, mediator and therapeutic target for renal fibrosis. Nephron Exp Nephrol 114:e83–e92
Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A et al (1999) The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med 341:709–717
Pitt B, Bakris G, Ruilope LM, DiCarlo L, Mukherjee R (2008) Serum potassium and clinical outcomes in the eplerenone post-acute myocardial infarction heart failure efficacy and survival study (EPHESUS). Circulation 118:1643–1650
Pitt B, Kober L, Ponikowski P, Gheorghiade M, Filippatos G, Krum H et al (2013) Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94-8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: a randomized, double-blind trial. Eur Heart J 34:2453–2463
Porter GA, Edelman IS (1964) The action of aldosterone and related corticosteroids on sodium transport across the toad bladder. J Clin Invest 43:611–620
Porter GA, Bogoroch R, Edelman IS (1964) On the mechanism of action of aldosterone on sodium transport: the role of RNA synthesis. Proc Natl Acad Sci U S A 52:1326–1333
Rogerson FM, Fuller PJ (2000) Mineralocorticoid action. Steroids 65:61–73
Rüster C, Wolf G (2006) Renin-angiotensin-aldosterone system and progression of renal disease. J Am Soc Nephrol 17:2985–2991
Sato A, Fukuda S (2013) Effect of aldosterone breakthrough on albuminuria during treatment with a direct renin inhibitor and combined effect with a mineralocorticoid receptor antagonist. Hypertens Res 36:879–884
Schjoedt K, Andersen S, Rossing P, Tarnow L, Parving H-H (2004) Aldosterone escape during blockade of the renin–angiotensin–aldosterone system in diabetic nephropathy is associated with enhanced decline in glomerular filtration rate. Diabetologia 47:1936–1939
Schjoedt K, Rossing K, Juhl T, Boomsma F, Tarnow L, Rossing P et al (2006) Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 70:536–542
Shankland S (2006) The podocyte’s response to injury: role in proteinuria and glomerulosclerosis. Kidney Int 69:2131–2147
Sheng L, Yang M, Ding W, Zhang M, Niu J, Qiao Z et al (2016) Epidermal growth factor receptor signaling mediates aldosterone-induced profibrotic responses in kidney. Exp Cell Res 346:99–110
Shibata S, Nagase M, Yoshida S, Kawachi H, Fujita T (2007) Podocyte as the target for aldosterone: roles of oxidative stress and Sgk1. Hypertension 49:355–364
Shibata S, Nagase M, Yoshida S, Kawarazaki W, Kurihara H, Tanaka H et al (2008) Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med 14:1370–1376
Simpson S (1953) Isolation from the adrenals of a new crystalline hormone with especially high effectiveness on mineral metabolism. Experientia 9(333–335):3
Spat A, Hunyady L (2004) Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev 84:489–539
Su M, Dhoopun A-R, Yuan Y, Huang S, Zhu C, Ding G et al (2013) Mitochondrial dysfunction is an early event in aldosterone-induced podocyte injury. Am J Physiol Renal Physiol 305:F520–F531
Sun Y, Zhang J, Zhang JQ, Ramires FJ (2000) Local angiotensin II and transforming growth factor-β1 in renal fibrosis of rats. Hypertension 35:1078–1084
Terada Y, Ueda S, Hamada K, Shimamura Y, Ogata K, Inoue K et al (2012) Aldosterone stimulates nuclear factor-kappa B activity and transcription of intercellular adhesion molecule-1 and connective tissue growth factor in rat mesangial cells via serum-and glucocorticoid-inducible protein kinase-1. Clin Exp Nephrol 16:81–88
Thiery JP (2002) Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer 2:442–454
Trachtman H, Weiser AC, Valderrama E, Morgado M, Palmer LS (2004) Prevention of renal fibrosis by spironolactone in mice with complete unilateral ureteral obstruction. J Urol 172:1590–1594
Unger T, Paulis L, Sica DA (2011) Therapeutic perspectives in hypertension: novel means for renin–angiotensin–aldosterone system modulation and emerging device-based approaches. Eur Heart J 32:2739–2747
Urata H, Hoffmann S, Ganten D (1994a) Tissue angiotensin II system in the human heart. Eur Heart J 15:68–78
Urata H, Strobel F, Ganten D (1994b) Widespread tissue distribution of human chymase. J Hypertens Suppl 12:S17–S22
Wang H, Naghavi M, Allen C, Barber RM, Bhutta ZA, Carter A et al (2016) Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 388:1459–1544
Weldon SM, Cerny MA, Gueneva-Boucheva K, Cogan D, Guo X, Moss N et al (2016) Selectivity of BI 689648, a novel, highly selective aldosterone synthase inhibitor: comparison with FAD286 and LCI699 in nonhuman primates. J Pharmacol Exp Ther 359:142–150
Williams GH (2005) Aldosterone biosynthesis, regulation, and classical mechanism of action. Heart Fail Rev 10:7–13
Williams JS, Williams GH (2003) 50th anniversary of aldosterone. J Clin Endocrinol Metab 88:2364–2372
Wolf G, Chen S, Ziyadeh FN (2005) From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes 54:1626–1634
Yamada M, Kushibiki M, Osanai T, Tomita H, Okumura K (2008) Vasoconstrictor effect of aldosterone via angiotensin II type 1 (AT1) receptor: possible role of AT1 receptor dimerization. Cardiovasc Res 79:169–178
Yuan J, Jia R, Bao Y (2007) Aldosterone up-regulates production of plasminogen activator inhibitor-1 by renal mesangial cells. J Biochem Mol Biol 40:180–188
Yuan Y, Huang S, Wang W, Wang Y, Zhang P, Zhu C et al (2012a) Activation of peroxisome proliferator-activated receptor-γ coactivator 1α ameliorates mitochondrial dysfunction and protects podocytes from aldosterone-induced injury. Kidney Int 82:771–789
Yuan Y, Chen Y, Zhang P, Huang S, Zhu C, Ding G et al (2012b) Mitochondrial dysfunction accounts for aldosterone-induced epithelial-to-mesenchymal transition of renal proximal tubular epithelial cells. Free Radic Biol Med 53:30–43
Yuan Y, Zhang A, Qi J, Wang H, Liu X, Zhao M et al (2017) P53/Drp1-dependent mitochondrial fission mediates aldosterone-induced podocyte injury and mitochondrial dysfunction. Am J Physiol Renal Physiol 314:F798–F808
Zhang A, Jia Z, Guo X, Yang T (2007) Aldosterone induces epithelial-mesenchymal transition via ROS of mitochondrial origin. Am J Physiol Renal Physiol 293:F723–F731
Zhang A, Jia Z, Wang N, Tidwell TJ, Yang T (2011) Relative contributions of mitochondria and NADPH oxidase to deoxycorticosterone acetate-salt hypertension in mice. Kidney Int 80:51–60
Zhang A, Han Y, Wang B, Li S, Gan W (2015) Beyond gap junction channel function: the expression of Cx43 contributes to aldosterone-induced mesangial cell proliferation via the ERK1/2 and PKC pathways. Cell Physiol Biochem 36:1210–1222
Zhu C, Huang S, Yuan Y, Ding G, Chen R, Liu B et al (2011) Mitochondrial dysfunction mediates aldosterone-induced podocyte damage: a therapeutic target of PPARγ. Am J Pathol 178:2020–2031
Acknowledgement and Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Shrestha, A., Che, RC., Zhang, AH. (2019). Role of Aldosterone in Renal Fibrosis. In: Liu, BC., Lan, HY., Lv, LL. (eds) Renal Fibrosis: Mechanisms and Therapies. Advances in Experimental Medicine and Biology, vol 1165. Springer, Singapore. https://doi.org/10.1007/978-981-13-8871-2_15
Download citation
DOI: https://doi.org/10.1007/978-981-13-8871-2_15
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-8870-5
Online ISBN: 978-981-13-8871-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)