
Abstract
As the most abundant internal modification in eukaryotic messenger RNAs (mRNAs), N 6-methyladenosine (m6A) modification has been shown recently to posttranscriptionally regulate expression of thousands of messenger RNA (mRNA) transcripts in each mammalian cell type in a dynamic and reversible manner. This epigenetic mark is deposited by the m6A methyltransferase complex (i.e., the METTL3/METTL14/WTAP complex and other cofactor proteins) and erased by m6A demethylases such as FTO and ALKBH5. Specific recognition of these m6A-modified mRNAs by m6A-binding proteins (i.e., m6A readers) determines the fate of target mRNAs through affecting splicing, nuclear export, RNA stability, and/or translation. During the past few years, m6A modification has been demonstrated to play a critical role in many major normal bioprocesses including self-renewal and differentiation of embryonic stem cells and hematopoietic stem cells, tissue development, circadian rhythm, heat shock or DNA damage response, and sex determination. Thus, it is not surprising that dysregulation of the m6A machinery is also closely associated with pathogenesis and drug response of both solid tumors and hematologic malignancies. In this chapter, we summarize and discuss recent findings regarding the biological functions and underlying mechanisms of m6A modification and the associated machinery in normal hematopoiesis and the initiation, progression, and drug response of acute myeloid leukemia (AML), a major subtype of leukemia usually associated with unfavorable prognosis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
4.1 Introduction
Since the 1960s, over 150 modified RNA nucleotide variants have been identified in both protein-coding and noncoding RNAs, such as N 6-methyladenosine (m6A) in messenger RNA (mRNA) and primary microRNA (pri-miRNA) [1,2,3,4], N 1-methyladenosine (m1A) in mRNA and transfer RNA (tRNA) [5,6,7], 5-methylcytosine (m5C) and 5-hydroxymethylcytosine (hm5C) in mRNA and long noncoding RNA (lncRNA) [8,9,10,11], and pseudouridine (ψ) in tRNA, ribosomal RNA (rRNA), small nuclear RNA (snRNA), and small nucleolar RNA (snoRNA) [12,13,14,15,16] (Figs. 4.1 and 4.2). Of them, m6A is the most prevalent and abundant internal modification on eukaryotic mRNAs. m6A RNA modification was first identified in the 1970s [1,2,3]. However, due to the lack of knowledge on its dynamic regulation and no high-throughput technology available to map m6A modification to the RNA transcriptome, little attention had been paid to this RNA mark until 2011, when the fat mass and obesity-associated protein (FTO) was identified as a genuine demethylase of m6A modification [17], which implies that m6A modification is a reversible and dynamic process analogous to the well-studied modifications on DNA and histone [18]. Subsequent development of high-throughput m6A sequencing technologies further facilitates the understanding of m6A modification in a transcriptome-wide view, revealing that m6A modification may affect more than 7000 mRNAs in individual transcriptomes of mammalian cells, with a special enrichment in the 3′ untranslated regions (UTRs) near the stop codons of mRNAs and with a consensus sequence of RRACH (R = G or A; H = A, C, or U) [19, 20]. Such findings strongly suggest that m6A modification may have important biological functions. Indeed, emerging data demonstrate that m6A modifications in mRNAs or noncoding RNAs influence RNA fate and functions and are critical for many normal and pathological bioprocesses including self-renewal and differentiation of embryonic stem cells, tissue development, circadian rhythm, heat shock or DNA damage response, sex determination, and tumorigenesis, as reviewed elsewhere [4, 16, 21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37]. Evidence is emerging that m6A modification and the associated machinery also play essential roles in tumorigenesis and drug response (see reviews [16, 38, 39]). Here we summarize recent advances on our understanding of the functions and underlying molecular mechanisms of the m6A machinery in normal hematopoiesis and AML pathogenesis and drug response.

Chemical structures of representative modified RNA nucleotide variants. Modifications are shown in blue. The known writer (in orange) and eraser (in green) proteins are also indicated

Chemical modifications in eukaryotic mRNA. A schematic representation of common chemical modifications across eukaryotic mRNA transcript including 5′ untranslated region (5′UTR), coding region (CDS), and 3′UTR. Reported roles of these modifications are summarized on top of the corresponding modifications. Note that the same modification in different mRNA regions may have different functions in regulating mRNA fate
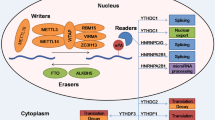
4.2 Regulators of m6A Modifications
Similar to other epigenetic modifications, m6A modification is regulated by its formation and removal catalyzed by the methyltransferases and demethylases, known as “writers” and “erasers,” respectively [16, 40, 41]. m6A marks are installed by a multicomponent methyltransferase complex (MTC) consisting of the core methyltransferase-like 3 and 14 (METTL3 and METTL14) heterodimer and their cofactors including Wilms’ tumor 1-associating protein (WTAP), vir like m6A methyltransferase associated (VIRMA, also known as KIAA1429), RNA-binding motif protein 15 (RBM15), and zinc finger CCCH domain-containing protein 13 (ZC3H13) [22, 42,43,44,45,46,47,48,49] (Fig. 4.3). Structural studies demonstrated that METTL3 is the sole catalytic subunit, while METTL14 offers an RNA-binding scaffold to allosterically activate and enhance the catalytic activity of METTL3 [50,51,52]. WTAP, VIRMA, RBM15, and ZC3H13 are the regulatory subunits of the MTC to facilitate m6A installation in cellulo. In addition, the METTL3 homolog METTL16 (methyltransferase-like 16) was recently shown to control cellular S-adenosyl methionine (SAM) level and install m6A marks onto the U6 small nuclear RNA [53, 54]. FTO, previously known to function as a demethylase for N 3-methylthymidine in single-stranded DNA and N 3-methyluridine in single-stranded RNA in vitro [55, 56], was identified as the first m6A demethylase that could demethylate m6A in both DNA and RNA in vivo [17]. A recent study reported that FTO also demethylates m6Am, a modification exclusively found at the first encoded nucleotide after the 7-methylguanosine cap structure of mRNAs [57]. AlkB homolog 5 (ALKBH5) is the second identified m6A demethylase that was found to be highly expressed in the testes [23]. Both FTO and ALKBH5 belong to the AlkB subfamily of the Fe(II)/2-oxoglutarate (2OG) dioxygenase superfamily that requires 2OG and molecular oxygen as co-substrates and ferrous iron Fe(II) as a cofactor to catalyze the oxidation of a substrate [56, 58].

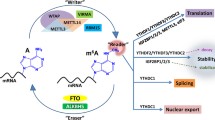
Roles of m6A RNA modification in determining mRNA fate. In nuclear, the methyltransferase complex composed of METTL3, METTL14, WTAP, VIRMA, RBM15, and ZC3H13 deposits m6A marks co-transcriptionally onto newly transcribed RNAs, while METTL16 is responsible for m6A deposition on the U6 snRNA. FTO and ALKBH5 function as m6A demethylases to remove m6A marks in selected sites of RNA. M6A-mediated structural switch of mRNA recruits hnRNP family proteins including hnRNPC, hnRNPG, and hnRNPA2B1. Nuclear reader protein YTHDC1 recognizes m6A to mediate alternative splicing. IGF2BP reader proteins bind to m6A mRNAs to stabilize these nascent transcripts. After exporting to cytoplasm, m6A-modified mRNAs could be subjected to degradation by YTHDF2 or YTHDC2 or protected by IGF2BP proteins and loaded to translation machinery. YTHDF1, YTHDF3, as well as METTL3 and eIF3a could also promote translation of m6A mRNAs
While the prevalence and distribution of m6A are determined by writers and erasers, the m6A-dependent functions are mediated by m6A-binding proteins, the so-called readers, which through specific recognition and binding to m6A-modified mRNAs determine the fate of these transcripts [16, 21, 59] (Fig. 4.3). The YT521-B homology (YTH) domain family of proteins, including YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2, are among the first identified m6A readers that possess a conserved m6A-binding pocket [19, 60,61,62,63,64,65,66]. Of them, YTHDC1 was found to be located in the nucleus, playing a role in splicing regulation, XIST-mediated X-chromosome silencing, and nuclear export of m6A-modified mRNAs [46, 63, 67]. The other YTH family proteins are all cytoplasmic m6A readers regulating mRNA fate through different mechanisms: YTHDF2 promotes degradation of target mRNAs [60, 68], and YTHDF1 promotes translation of target mRNAs [61], while YTHDF3 and YTHDC2 can both mediate mRNA decay and enhance translation [66, 69,70,71,72,73]. We recently identified insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs; including IGF2BP1/2/3) as a new family of m6A readers that could promote stability and translation of their target mRNAs [32], distinct from the functional manners of the YTH family proteins. The K homology (KH) domains of IGF2BPs are required for their recognition of m6A and are critical for their oncogenic functions. Interestingly, ELAV-like RNA-binding protein 1 (ELAVL1, also known as HuR), an mRNA stabilizer that was previously reported as an indirect m6A-binding protein [22], was found to be a cofactor of IGF2BPs and may mediate the mRNA stabilizing effect of IGF2BPs [32]. The heterogeneous nuclear ribonucleoprotein (HNRNP) HNRNPA2B1 was previously reported to regulate alternative splicing and primary microRNA processing as an m6A reader [74]; however, recent structural study suggested an “m6A switch” mechanism rather than direct m6A binding for the protein [75]. Two other members of the HNRNP family proteins, namely HNRNPC and HNRNPG, were shown to recognize m6A-induced changes in mRNA secondary structures [76]. Other proteins were also recently reported to be m6A interactors, including FMR1 and LRPPRC [77, 78], although the exact mode of binding still needs to be clarified.
4.3 Roles of m6A in Normal Hematopoiesis and HSC Self-Renewal
Hematopoiesis is a tightly regulated dynamic process where mature blood cells are generated from a small pool of multipotent hematopoietic stem cells (HSCs) [79, 80]. During the past few decades, it has been well acknowledged that transcriptional regulation by a variety of hematopoietic transcription factors (TFs) plays a big role in regulating the multistep normal hematopoiesis [81,82,83]. In particular, during myelopoiesis where HSCs are differentiated into myeloid progenitors and eventually mature myeloid cells, the sequential actions of master TFs are required to specify and re-enforce each cell fate decision. For instance, PU.1 (also known as SPI1, the product of the oncogene SPI1) is a transcriptional master regulator of myeloid cells which plays an essential role in generating early myeloid progenitors (i.e., common myeloid progenitors, CMPs), while the basic region leucine zipper transcription factor C/EBPα is required for the production of granulocyte/macrophage progenitors (GMPs) from CMPs [80, 84].
In recent years, emerging studies reveal m6A modification at the RNA level as an additional layer of the posttranscriptional regulation in governing HSC activity and normal hematopoiesis (see Fig. 4.4). During the endothelial-to-hematopoietic transition (EHT) of zebrafish embryogenesis, a key developmental event leading to the formation of the earliest hematopoietic stem and progenitor cells (HSPCs), m6A modification was reported to play a role [30]. Deficiency of mettl3 in zebrafish embryos leads to decreased levels of m6A and blockage of HSPC emergence, likely due to the reduced m6A modification on the arterial endothelial genes notch1a and rhoca that delayed YTHDF2-mediated mRNA decay of these transcripts [30]. Vu and colleagues used short hairpin RNAs (shRNAs) to knock down METTL3 expression in human HSPCs and observed cell growth inhibition and increase of myeloid differentiation [34]. Conversely, overexpression of wild-type, but not catalytically dead mutant of METTL3, promotes proliferation and colony formation and inhibits myeloid differentiation. We recently showed that METTL14 is highly expressed in murine HSCs and Lin− Sca-1+c-kit+ (LSK) cells and was downregulated during myelopoiesis, showing reduced expression in CMP and GMP progenitors and especially in mature myeloid cells [33]. Consistent with the expression pattern, depletion of METTL14 expression in human HSPCs by shRNAs promotes myeloid differentiation in vitro. Moreover, by utilizing a Mettl14 conditional knockout mouse model, we demonstrated that induced deletion of Mettl14 impairs HSC self-renewal ability in vivo. Such effects were mediated by decreased MYB and MYC expression owing to the reduction of m6A modification on these transcripts upon METTL14 knockdown/knockout. Considering the role of MYB [85, 86] and MYC [87, 88] transcription factors in regulating HSC self-renewal and differentiation, METTL14-mediated m6A regulation on the mRNA transcripts of these TFs adds a new layer of complexity to the regulatory networks in normal hematopoiesis.

Modifiers of m6A RNA modification in normal hematopoiesis. METTL3 and METTL14 promote self-renewal of HSC and inhibit myeloid differentiation. In contrast, YTHDF2 inhibits HSC self-renewal. RBM15 inhibits myeloid and megakaryocytic differentiation while promotes B cell expansion, although it is unclear whether these effects are m6A-related
A very recent study took advantage of Mettl3 and Mettl14 conditional knockout mice to investigate the roles of these m6A writer proteins on regulation of HSC self-renewal in adult mouse bone marrow [89]. They found that deletion of Mettl3 alone or together with Mettl14 in the hematopoietic system substantially increases HSC frequency in the bone marrow; in contrast, deletion of Mettl14 alone has little effect [89]. Conditional deletion of Mettl14 and especially that of Mettl3 suppresses HSC self-renewal activity in recipient mice. Notably, although deletion of either Mettl3 or Mettl14 deletion leads to significant reduction of donor-derived myeloid cells in the peripheral blood, only deletion of Mettl3 results in a significant reduction of B- and T-cell lineage [89].
RBM15, recently identified as a component of the m6A methyltransferase complex, was also reported to play a role in normal hematopoiesis [90,91,92]. Conditional knockout of Rbm15 in adult mice blocks B cell differentiation and results in an increase of the Lin−Sca-1+c-Kit+ (LSK) HSPCs and an expansion of myeloid and megakaryocytic cells in spleen and bone marrow, demonstrating a role of RBM15 in hematopoietic development [90]. Rbm15 is expressed at highest levels in HSCs and inhibits myeloid differentiation and megakaryocytic expansion through stimulation of the Notch signaling and regulation of MYC expression, respectively [91, 92].
YTHDF2 is the first well-characterized m6A reader that promotes mRNA decay of target transcript with m6A modification [60]. Li and colleagues studied the role of YTHDF2 in adult stem cell maintenance and reported an increase of HSCs in Ythdf2 conditional knockout mice as well as in human umbilical cord blood upon YTHDF2 knockdown [93]. Such effects are partially mediated by the stabilization of mRNA transcripts encoding TFs critical for stem cell self-renewal. Overall, the studies of m6A in normal hematopoiesis are just in the beginning. It is of great interest to explore the functions and underlying mechanisms of other m6A modulators, including other writers, erasers, and readers, in stem cell biology and normal hematopoiesis.
4.4 Dysregulation of m6A Regulators in Malignant Hematopoiesis
Dysregulation of the regulatory networks of normal hematopoiesis, such as incorrect activity of the hematopoietic TFs attributed to either aberrant expression or mutation, breaks the balance between HSC self-renewal and differentiation and places the progenitor cells at a higher risk of developing leukemia [80, 94]. Acute myeloid leukemia (AML) is a clonal hematopoietic disorder where a stem cell-like self-renewal capacity is gained and the differentiation capacity is blocked [95, 96]. According to the Cancer Statistics 2017, the 5-year survival of AML between 2007 and 2013 is 26.9%, much lower than other types of leukemia and many other common cancer types [97]. For instance, during the same period, the 5-year survival rate is 89.7% for breast cancer and 66.9% for chronic myeloid leukemia (CML). Therefore, there is an urgent need to better understand the mechanisms underlying AML leukemogenesis and, based on the gained knowledge, to develop effective targeted therapies.
The first evidence of a role of m6A modification on leukemia, specifically on AML, came from the study of FTO, a major m6A demethylase [31]. FTO was previously known as a gene associated with fat mass, adipogenesis, and body weight [98,99,100], and single-nucleotide polymorphisms (SNPs) in FTO were linked to higher risk of developing cancers including leukemia and lymphoma by large-scale epidemiology studies [101,102,103]. We found that FTO is highly expressed in certain subtypes of AML, including those carrying t(11q23)/MLL rearrangements, t(15;17)/PML-RARA, FLT3-ITD, and/or NPM1 mutations. Modulation of FTO expression by depletion of FTO or forced expression of wild-type FTO (but not catalytically inactive mutant) could significantly influence AML cell survival and leukemogenesis and affect the response of AML cells to all-trans retinoic acid (ATRA) [31]. Importantly, the oncogenic function of FTO in AML relies on its m6A demethylase activity. By reducing m6A abundance on the transcripts of ASB2 and RARA, two genes with reported roles in cell proliferation and drug response of leukemia cells, FTO posttranscriptionally regulates expression of ASB2 and RARA through reducing m6A abundance, thereby decreasing mRNA stability of these transcripts. These data provide compelling evidence on the role of FTO in leukemogenesis and establish a first link between m6A modification and leukemia pathogenesis (Fig. 4.5) [31].

Role of FTO in leukemogenesis. FTO removes m6A modification on its target mRNAs (e.g., ASB2, RARA, MYC, and CEBPA), resulting in the decreased or increased stability of these transcripts, and promotes leukemogenesis. R-2HG inhibits FTO and exhibits antileukemia effects
More recently, we showed that, by targeting FTO directly, R-2-hydroxyglutarate (R-2HG), a previously reported oncometabolite produced by mutant isocitrate dehydrogenase 1/2 (IDH1/2) enzymes [104,105,106], exhibits a broad and intrinsic antitumor activity in leukemia. By inhibiting the m6A demethylase activity of FTO, R-2HG results in an increase of m6A abundance on FTO target genes, such as MYC and CEBPA, leading to decay of these transcripts in R-2HG sensitive cells (see Fig. 4.5). Our data indicate that FTO/MYC homeostasis controls the sensitivity of leukemic cells to 2HG: high abundance of FTO confers R-2HG sensitivity, whereas hyper-activation of MYC signaling renders leukemic cells resistant to R-2HG [37]. Consistent with this notion, pharmaceutical or genetic inhibition of MYC signaling by JQ1 or MYC shRNAs can resensitize R-2HG-resistant leukemic cells to R-2HG. In addition, we showed that R-2HG treatment or FTO inhibition also sensitized AML cells to first-line chemotherapy drugs such as ATRA, Azacitidine (AZA), Decitabine, and Daunorubicin ([37] and unpublished data). Collectively, our studies demonstrate the critical role of FTO in leukemia pathogenesis and drug response and also highlight the therapeutic potential of targeting FTO signaling for AML treatment.
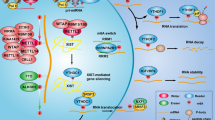
Subsequently, the dysregulation of the m6A installing machinery was also reported to be involved in AML pathogenesis [107] (see Fig. 4.6). METTL3 and METTL14 were both expressed at a higher level in AML than in the vast majority of other cancer types according to The Cancer Genome Atlas (TCGA) genome-wide gene expression datasets [33,34,35]. We found that METTL14 is aberrantly overexpressed in certain subtypes of AMLs, such as those carrying t(11q23)/MLL rearrangements, t(15;17)/PML-RARA, and t(8;21)/AML1-ETO [33]. We next conducted in vitro and in vivo gain- and loss-of-function studies and demonstrated that METTL14 plays a critical oncogenic role in AML pathogenesis [33]. Depletion of METTL14 inhibited survival and proliferation of AML cells, promoted myeloid differentiation, and suppressed leukemic oncofusion protein (e.g., MLL-AF9, MLL-AF10, and AML1-ETO9a)-mediated immortalization of normal HSPCs. The opposite is true when wild type, but not mutant METTL14 (i.e., R298P), was forced expressed. Moreover, knockdown of METTL14 significantly inhibited progression of human AML cells in xenotransplantation recipient mice, while inducible knockout of Mettl14 greatly inhibited AML development and maintenance in bone marrow transplantation (BMT) recipient mice [33]. High-throughput RNA-seq and transcriptome-wide m6A-seq, coupled with gene-specific m6A-qPCR assays, cross-linking and immunoprecipitation (CLIP) assays, luciferase reporter and mutagenesis assays, mRNA stability assays, and polysome profiling assays, demonstrated that METTL14 posttranscriptionally regulates the expression of its critical target mRNA transcripts, such as MYB and MYC, two well-known TF genes involved in leukemogenesis, in an m6A-dependent manner [33]. Silencing of METTL14 reduces m6A abundance of MYB and MYC transcripts, especially near the 3′ end of the mRNAs, resulting in decreased mRNA stability and translation of these transcripts. Such effects were not mediated by the YTH family proteins, and therefore other readers (such as IGF2BPs [32]) may mediate the effect of METTL14 on MYB and MYC. Furthermore, we also identified SPI1 (also called PU.1), a transcriptional master regulator of myelopoiesis, as a negative regulator of METTL14 expression in AML [33]. Taken together, our work reveals a previously unappreciated SPI1-METTL14-MYB/MYC signaling axis in leukemogenesis and highlights the critical roles of METTL14 and m6A modification in malignant hematopoiesis [33].

Roles of m6A writer genes in leukemogenesis. METTL3 and METTL14 function as oncogenes in AML through depositing m6A modification on their target transcripts (e.g., MYC, BCL2, and PTEN for METTL3 and MYB and MYC for METTL14) to enhance translation and/or increase mRNA stability of these transcripts. METTL3 can also be recruited to promoter of its target gene by CEBPZ and deposit m6A modification on the coding region of the target transcripts (e.g., SP1 and SP2) to promote their translation and eventually leads to MYC activation
Meanwhile, two other groups have demonstrated that METTL3 also plays an essential oncogenic role in AML, by showing that depletion of METTL3 expression results in cell growth inhibition, cell cycle arrest, and induction of differentiation and apoptosis, whereas overexpression of wild-type METTL3, but not a catalytically inactive mutant, promotes proliferation in AML cells and primary blasts [34, 35]. Similar to METTL14, METTL3 is also required for AML development as demonstrated by the xenotransplantation assay data that shRNA-mediated knockdown or CRISPR/Cas9-mediated knockout of METTL3 substantially inhibited AML progression and prolonged survival in recipient mice [34, 35]. Such findings support the oncogenic role of METTL3 as an m6A-catalyzing enzyme. Despite of the similar functions of METTL3 reported by the two groups, distinct underlying mechanisms of METTL3 in AML were reported [34, 35]. Vu et al. performed m6A individual-nucleotide-resolution cross-linking and immunoprecipitation (miCLIP), RNA-seq, and Ribo-Seq, and identified MYC, BCL2 and PTEN as direct RNA targets of METTL3, whose m6A abundances were substantially reduced when METTL3 was knocked down [34]. However, upon METTL3 depletion, while expression of MYC, BCL2, and PTEN transcripts showed a great increase, the corresponding protein levels decreased on day 3 after shRNA transduction and recovered 1 day later [34]. Although the authors proposed that an alternative internal ribosome entry site (IRES)-mediated translational mechanism may be involved to reactivate translation of MYC and BCL2 under METTL3 depletion-induced cell apoptosis, it remains unclear why the expression changes of these target genes at the mRNA level are inconsistent with that at the protein levels. A systematic understanding of the changes and functions of m6A readers under this circumstance may help to address this question. Different from Vu’s study [34], Barbieri et al. found that METTL3 and METTL14 could bind chromatin, mainly localizing to the transcriptional start sites (TSSs) of coding genes characterized by bimodal H3K4me3 peaks [35]. However, METTL3 and METTL14 did not bind the same TSSs, suggesting independent regulation on target genes. CEBPZ, a transcription factor critical for hematopoietic differentiation, recruits METTL3, but not METTL14, to chromatin [35]. Promoter-bound METTL3 induces m6A modification within the coding region of the associated mRNA transcript, such as SP1 and SP2, and enhances their translation by relieving ribosome stalling [35].
In addition to METTL3 and METTL14, other m6A methyltransferases or cofactors of the m6A methyltransferase complex have also been implicated to function in AML. A genome-wide CRISPR-Cas9 screening identified METTL16, as well as METTL3 and METTL14, as critical genes for AML survival [35], while detailed study is yet to be done. Before it was identified as a component of the m6A methyltransferase complex [43], WTAP was shown to be upregulated in AML and is a novel client protein of HSP90 [108]. Knockdown of WTAP in AML cells led to reduced proliferation and promotion of phorbol 12-myristate 13-acetate (PMA)-induced myeloid differentiation and inhibited growth of human leukemia cells in xenograft mice [108]. RBM15 was found to be a fusion partner of the MKL1 gene in t(1,22)(p13;q13) acute megakaryoblastic leukemia (AMKL), a subtype of pediatric AML [109]. It is therefore reasonable to speculate that RBM15 may play a role in leukemogenesis, possibly related to m6A modification, for which further studies are warranted.
4.5 m6A and Leukemia Stem Cells
Malignant stem cells are considered as the potential origin of and a key therapeutic target for AML, similar to what is believed for other cancer types [110,111,112,113,114,115]. Leukemia stem cells (LSCs) are defined as cells with two important properties: (1) capable of engrafting and initiating the disease when transplanted into immunodeficient animals and can self-renew by giving rise to leukemia in serial transplantations and (2) produce non-LSC bulk blasts that resemble the original disease but are unable to self-renew [116]. During the last few decades, investigators have been dedicated to the characterization of LSCs using different combinations of cell surface markers. It was found that LSCs immunophenotypically resemble certain normal hematopoietic progenitor populations and usually reside in the CD34+CD38− fraction, although approximately 25% of AML cases lack CD34 expression [116]. It is believed that therapeutic targeting and eliminating of LSC is the key to eradicate leukemia and achieve long-term remissions.
Retroviral transduction of the MLL-AF9 oncofusion gene into mouse HSPCs followed by transplantation into recipient mice represents one of the best mouse models for AML LSC studies [116]. By using this model, we have shown that depletion of either Fto or Mettl14 in mouse HSPCs could inhibit leukemia initiation in primary BMT recipients and maintenance in secondary transplantations [31, 33], suggesting a role of these m6A modifying proteins on AML LSC function. Indeed, we conducted limiting dilution assays using bone marrow cells harvested from MLL-AF9 primary leukemia mice to directly evaluate the effect of Mettl14 depletion on the frequency of leukemia stem/initiating cells (LSCs/LICs) and found that the estimated LSC/LIC frequency was significantly reduced when Mettl14 was knocked out [33]. While our data provide the first link between m6A modification and LSC self-renewal [33], further systematic studies are warranted to better understand the role of m6A modification on LSC biology.
4.6 Conclusions and Perspectives
Evidence is emerging that m6A modification and the associated machinery play essential roles in both normal and malignant hematopoiesis, including the self-renewal and differentiation of normal and malignant HSCs. Systematical studies of the functions and underlying molecule mechanisms of individual m6A writer, eraser, and reader genes will further advance our understanding of the complex networks and molecular mechanisms underlying normal and malignant hematopoiesis. In addition, it is also important to understand how expression of individual m6A regulators is regulated during normal and malignant hematopoiesis.
The impact of m6A modification on the fate of its embedded RNA mediates the functions of such modification. Therefore, it is of great importance to identify key functionally important RNA targets (including mRNA, lncRNA, etc.) that when manipulated could phenocopy or reverse the effects of knockdown or overexpression of individual m6A regulators. For instance, recent studies show the oncogenic MYC transcript as an important target of many m6A regulators in cancer including leukemia [32,33,34, 37], highlighting the importance of precise regulation of MYC expression at the posttranscription level in normal development and the big impact of its dysregulation in tumorigenesis. Further systematic identification and functional studies of all critical target genes of m6A modification by combined use of high-throughput sequencing such as CLIP-seq, m6A-seq, and RNA-seq, followed by the validation assays and in vitro and in vivo functional studies, are warranted. Such knowledge will be not only important for the understanding of the effects of manipulation of individual m6A regulator but also may lead to the discovery of novel therapeutic targets for AML and other types of leukemia.
As demonstrated by the published data thus far, m6A regulators represent promising targets for treatment of cancer, including leukemia. Development of effective and selective inhibitors targeting oncogenic m6A regulators (e.g., FTO and METTL14) that play more essential roles in leukemogenesis, especially in self-renewal of LSCs/LICs, than in normal hematopoiesis may hold great therapeutic potential in treating leukemia, especially when in combination with other therapeutic agents.
Abbreviations
- AMKL:
-
Acute megakaryoblastic leukemia
- AML:
-
Acute myeloid leukemia
- BMT:
-
Bone marrow transplantation
- CLIP:
-
Cross-linking immunoprecipitation
- CMP:
-
Common myeloid progenitor
- EHT:
-
Endothelial-to-hematopoietic transition
- GMP:
-
Granulocyte/macrophage progenitor
- HSCs:
-
Hematopoietic stem cells
- HSPCs:
-
Hematopoietic stem and progenitor cells
- lncRNA:
-
Long noncoding RNA
- LSCs:
-
Leukemia stem cells
- LSCs/LICs:
-
Leukemia stem/initiating cells
- m6A:
-
N 6-methyladenosine
- miCLIP:
-
m6A individual-nucleotide-resolution cross-linking and immunoprecipitation
- mRNA:
-
Messenger RNA
- MTC:
-
Methyltransferase complex
- pri-miRNA:
-
Primary microRNA
- rRNA:
-
Ribosomal RNA
- snoRNA:
-
Small nucleolar RNA
- SNPs:
-
Single-nucleotide polymorphisms
- snRNA:
-
Small nuclear RNA
- TF:
-
Transcription factor
- tRNA:
-
Transfer RNA
- TSS:
-
Transcriptional start site
References
Wei CM, Moss B (1977) Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry 16:1672–1676
Adams JM, Cory S (1975) Modified nucleosides and bizarre 5′-termini in mouse myeloma mRNA. Nature 255:28–33
Krug RM, Morgan MA, Shatkin AJ (1976) Influenza viral mRNA contains internal N6-methyladenosine and 5′-terminal 7-methylguanosine in cap structures. J Virol 20:45–53
Alarcon CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF (2015) N6-methyladenosine marks primary microRNAs for processing. Nature 519:482–485
Dunn DB (1961) The occurrence of 1-methyladenine in ribonucleic acid. Biochim Biophys Acta 46:198–200
Hall RH (1963) Method for isolation of 2 -O-methylribonucleosides and N1-methyladenosine from ribonucleic acid. Biochim Biophys Acta 68:278–283
El Yacoubi B, Bailly M, de Crecy-Lagard V (2012) Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu Rev Genet 46:69–95
Squires JE et al (2012) Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res 40:5023–5033
Amort T et al (2013) Long non-coding RNAs as targets for cytosine methylation. RNA Biol 10:1003–1008
Fu L et al (2014) Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J Am Chem Soc 136:11582–11585
Huber SM et al (2015) Formation and abundance of 5-hydroxymethylcytosine in RNA. Chembiochem 16:752–755
Carlile TM et al (2014) Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515:143–146
Cohn WE, Volkin E (1951) Nucleoside-5′-phosphates from ribonucleic acid. Nature 167:483–484
Charette M, Gray MW (2000) Pseudouridine in RNA: what, where, how, and why. IUBMB Life 49:341–351
Roundtree IA, Evans ME, Pan T, He C (2017) Dynamic RNA modifications in gene expression regulation. Cell 169:1187–1200
Deng X et al (2018) RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res 28:507–517
Jia G et al (2011) N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7:885–887
Zhao BS, Roundtree IA, He C (2016) Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol 18:31–42
Dominissini D et al (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485:201–206
Meyer KD et al (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149:1635–1646
Zhao BS, Roundtree IA, He C (2017) Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol 18:31–42
Wang Y et al (2014) N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16:191–198
Zheng G et al (2013) ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49:18–29
Zhao X et al (2014) FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res 24:1403–1419
Geula S et al (2015) Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347:1002–1006
Chen T et al (2015) m(6)A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell 16:289–301
Zhou J et al (2015) Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature 526:591–594
Xiang Y et al (2017) RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 543:573–576
Zhao BS et al (2017) m(6)A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 542:475–478
Zhang C et al (2017) m(6)A modulates haematopoietic stem and progenitor cell specification. Nature 549:273–276
Li Z et al (2017) FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell 31:127–141
Huang H et al (2018) Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol 20:285–295. In Press
Weng H et al (2018) METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell 22:191–205 e199
Vu LP et al (2017) The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med 23:1369–1376
Barbieri I et al (2017) Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 552:126–131
Zhang S et al (2017) m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell 31:591–606 e596
Su R et al (2018) R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell 172:90–105 e123
Deng X, Su R, Stanford S, Chen J (2018) Critical enzymatic functions of FTO in obesity and cancer. Front Endocrinol (Lausanne) 9:396
Deng X, Su R, Feng X, Wei M, Chen J (2018) Role of N(6)-methyladenosine modification in cancer. Curr Opin Genet Dev 48:1–7
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21:381–395
Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM (1997) Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 3:1233–1247
Ping XL et al (2014) Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 24:177–189
Schwartz S et al (2014) Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep 8:284–296
Liu J et al (2014) A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 10:93–95
Patil DP et al (2016) m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537:369–373
Wen J et al (2018) Zc3h13 regulates nuclear RNA m(6)A methylation and mouse embryonic stem cell self-renewal. Mol Cell 69:1028–1038 e1026
Knuckles P et al (2018) Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev 32:415–429
Guo J, Tang HW, Li J, Perrimon N, Yan D (2018) Xio is a component of the Drosophila sex determination pathway and RNA N(6)-methyladenosine methyltransferase complex. Proc Natl Acad Sci U S A 115:3674–3679
Wang P, Doxtader KA, Nam Y (2016) Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell 63:306–317
Wang X et al (2016) Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 534:575–578
Sledz P, Jinek M (2016) Structural insights into the molecular mechanism of the m(6)A writer complex. Elife 5:e18434
Warda AS et al (2017) Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep 18:2004–2014
Pendleton KE et al (2017) The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell 169:824–835 e814
Jia G et al (2008) Oxidative demethylation of 3-methylthymine and 3-methyluracil in single-stranded DNA and RNA by mouse and human FTO. FEBS Lett 582:3313–3319
Gerken T et al (2007) The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 318:1469–1472
Mauer J et al (2017) Reversible methylation of m(6)Am in the 5′ cap controls mRNA stability. Nature 541:371–375
Kurowski MA, Bhagwat AS, Papaj G, Bujnicki JM (2003) Phylogenomic identification of five new human homologs of the DNA repair enzyme AlkB. BMC Genomics 4:48
Yang Y, Hsu PJ, Chen YS, Yang YG (2018) Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res 28:616–624
Wang X et al (2014) N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505:117–120
Wang X et al (2015) N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161:1388–1399
Xu C et al (2014) Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol 10:927–929
Xiao W et al (2016) Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell 61:507–519
Luo S, Tong L (2014) Molecular basis for the recognition of methylated adenines in RNA by the eukaryotic YTH domain. Proc Natl Acad Sci U S A 111:13834–13839
Zhu T et al (2014) Crystal structure of the YTH domain of YTHDF2 reveals mechanism for recognition of N6-methyladenosine. Cell Res 24:1493–1496
Hsu PJ et al (2017) Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res 27:1115–1127
Roundtree IA et al (2017) YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. eLife 6:e31311
Du H et al (2016) YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun 7:12626
Shi H et al (2017) YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res 27:315–328
Li A et al (2017) Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res 27:444–447
Bailey AS et al (2017) The conserved RNA helicase YTHDC2 regulates the transition from proliferation to differentiation in the germline. Elife 6:e26116
Wojtas MN et al (2017) Regulation of m(6)A transcripts by the 3′-->5′ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol Cell 68:374–387 e312
Jain D et al (2018) ketu mutant mice uncover an essential meiotic function for the ancient RNA helicase YTHDC2. Elife 7:e30919
Alarcon CR et al (2015) HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell 162:1299–1308
Wu B et al (2018) Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat Commun 9:420
Liu N et al (2015) N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518:560–564
Edupuganti RR et al (2017) N(6)-methyladenosine (m(6)A) recruits and repels proteins to regulate mRNA homeostasis. Nat Struct Mol Biol 24:870–878
Arguello AE, DeLiberto AN, Kleiner RE (2017) RNA chemical proteomics reveals the N(6)-methyladenosine (m(6)A)-regulated protein-RNA interactome. J Am Chem Soc 139:17249–17252
Doulatov S, Notta F, Laurenti E, Dick JE (2012) Hematopoiesis: a human perspective. Cell Stem Cell 10:120–136
Rosenbauer F, Tenen DG (2007) Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol 7:105–117
Koschmieder S, Rosenbauer F, Steidl U, Owens BM, Tenen DG (2005) Role of transcription factors C/EBPalpha and PU.1 in normal hematopoiesis and leukemia. Int J Hematol 81:368–377
Goode DK et al (2016) Dynamic gene regulatory networks drive hematopoietic specification and differentiation. Dev Cell 36:572–587
Rosmarin AG, Yang Z, Resendes KK (2005) Transcriptional regulation in myelopoiesis: hematopoietic fate choice, myeloid differentiation, and leukemogenesis. Exp Hematol 33:131–143
Dakic A et al (2005) PU.1 regulates the commitment of adult hematopoietic progenitors and restricts granulopoiesis. J Exp Med 201:1487–1502
Sandberg ML et al (2005) c-Myb and p300 regulate hematopoietic stem cell proliferation and differentiation. Dev Cell 8:153–166
Mucenski ML et al (1991) A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell 65:677–689
Wilson A et al (2004) c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev 18:2747–2763
Satoh Y et al (2004) Roles for c-Myc in self-renewal of hematopoietic stem cells. J Biol Chem 279:24986–24993
Yao QJ et al (2018) Mettl3-Mettl14 methyltransferase complex regulates the quiescence of adult hematopoietic stem cells. Cell Res 28:952–954
Raffel GD et al (2007) Ott1(Rbm15) has pleiotropic roles in hematopoietic development. Proc Natl Acad Sci U S A 104:6001–6006
Ma X et al (2007) Rbm15 modulates Notch-induced transcriptional activation and affects myeloid differentiation. Mol Cell Biol 27:3056–3064
Niu C et al (2009) c-Myc is a target of RNA-binding motif protein 15 in the regulation of adult hematopoietic stem cell and megakaryocyte development. Blood 114:2087–2096
Li Z et al (2018) Suppression of m(6)A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res 28:904–917
Ye M et al (2015) Hematopoietic differentiation is required for initiation of acute myeloid leukemia. Cell Stem Cell 17:611–623
Testa U (2011) Leukemia stem cells. Ann Hematol 90:245–271
Dohner H, Weisdorf DJ, Bloomfield CD (2015) Acute myeloid leukemia. N Engl J Med 373:1136–1152
Siegel RL, Miller KD, Jemal A (2017) Cancer statistics, 2017. CA Cancer J Clin 67:7–30
Fischer J et al (2009) Inactivation of the Fto gene protects from obesity. Nature 458:894–898
Church C et al (2010) Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet 42:1086–1092
Merkestein M et al (2015) FTO influences adipogenesis by regulating mitotic clonal expansion. Nat Commun 6:6792
Soderberg KC et al (2009) Overweight, obesity and risk of haematological malignancies: a cohort study of Swedish and Finnish twins. Eur J Cancer 45:1232–1238
Castillo JJ, Mull N, Reagan JL, Nemr S, Mitri J (2012) Increased incidence of non-Hodgkin lymphoma, leukemia, and myeloma in patients with diabetes mellitus type 2: a meta-analysis of observational studies. Blood 119:4845–4850
Hernandez-Caballero ME, Sierra-Ramirez JA (2015) Single nucleotide polymorphisms of the FTO gene and cancer risk: an overview. Mol Biol Rep 42:699–704
Wang F et al (2013) Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340:622–626
Sasaki M et al (2012) IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 488:656–659
Losman JA et al (2013) (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339:1621–1625
Martin GH, Park CY (2018) Meddling with METTLs in normal and leukemia stem cells. Cell Stem Cell 22:139–141
Bansal H et al (2014) WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia 28:1171–1174
Ma Z et al (2001) Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat Genet 28:220–221
Pollyea DA, Jordan CT (2017) Therapeutic targeting of acute myeloid leukemia stem cells. Blood 129:1627–1635
Misaghian N et al (2009) Targeting the leukemic stem cell: the Holy Grail of leukemia therapy. Leukemia 23:25–42
Yang ZJ, Wechsler-Reya RJ (2007) Hit ‘em where they live: targeting the cancer stem cell niche. Cancer Cell 11:3–5
Borovski T, De Sousa EMF, Vermeulen L, Medema JP (2011) Cancer stem cell niche: the place to be. Cancer Res 71:634–639
Plaks V, Kong N, Werb Z (2015) The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 16:225–238
Beck B, Blanpain C (2013) Unravelling cancer stem cell potential. Nat Rev Cancer 13:727–738
Thomas D, Majeti R (2017) Biology and relevance of human acute myeloid leukemia stem cells. Blood 129:1577–1585
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Weng, H., Huang, H., Chen, J. (2019). RNA N 6-Methyladenosine Modification in Normal and Malignant Hematopoiesis. In: Zhang, H., Li, S. (eds) Leukemia Stem Cells in Hematologic Malignancies. Advances in Experimental Medicine and Biology, vol 1143. Springer, Singapore. https://doi.org/10.1007/978-981-13-7342-8_4
Download citation
DOI: https://doi.org/10.1007/978-981-13-7342-8_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-7341-1
Online ISBN: 978-981-13-7342-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)