
Abstract
Rhythmical contractility of blood vessels was first observed in bat wing veins by Jones (Philos Trans R Soc Lond 1852:142, 131–136), and subsequently described in arteries and arterioles of multiple vascular beds in several species. Despite an abundance of descriptive literature regarding the presence of vasomotion, to date we do not have an accurate picture of the cellular and ionic basis of these oscillations in tone, or the physiological relevance of the changes in pulsatile blood flow arising from vasomotion. This chapter reviews our current understanding of the cellular and ionic mechanisms underlying vasomotion in resistance arteries and arterioles. Focus is directed to the ion channels, changes in cytosolic Ca2+ concentration, and involvement of intercellular gap junctions in the development and synchronization of rhythmic changes in membrane potential and cytosolic Ca2+ concentration within the vessel wall that contribute to vasomotion. The physiological consequences of vasomotion are discussed with a focus on the cerebral vasculature, as recent advances show that rhythmic oscillations in cerebral arteriolar diameter appear to be entrained by cortical neural activity to increase the local supply of blood flow to active regions of the brain.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Rhythmic oscillations in the diameter of blood vessels have been observed for more than 150 years beginning with the initial description by Jones [1]. In his analysis of veins in the bat wing, Jones [1] stated that there was “something peculiar in the flow of blood in the veins; that they contracted and dilated rhythmically,” with an average of 7–13 contractions per minute. Similar spontaneous and evoked oscillations in diameter, now generally referred to as vasomotion, are accepted to be a common feature of arteries and veins ranging in size from conduit vessels (e.g., carotid arteries) to microvasculature of animals and humans studied in vitro and in vivo under various experimental and physiological conditions (reviewed in [2,3,4,5,6,7]). Vasomotion is dependent on mechanisms within the vascular wall, as it is observed in isolated segments of arterioles maintained in vitro in the absence of neural activity and blood flow, but it can be modulated by neurohumoral factors in a vessel- and species-specific manner [4, 5, 7]. An example of spontaneous vasomotion in a segment of rat middle cerebral artery studied in vitro by pressure myography is presented in Fig. 12.1. Varied patterns of modulation and different sensitivities to pharmacological manipulation and endothelium removal have been described for vessels of different vascular beds and varied caliber, suggesting that multiple mechanisms may contribute to the appearance and maintenance of vasomotion in a vessel-specific manner [4,5,6,7,8]. Despite these complexities, considerable progress has been made in understanding the factors and mechanisms responsible for vasomotion. This chapter presents a current understanding of vasomotion in arteries and arterioles. Emphasis has been placed on the cellular and ionic mechanisms that have been postulated to contribute to the rhythmic oscillations in membrane potential, cytosolic Ca2+ concentration, and cross-bridge cycling in vascular smooth muscle cells during vasomotion. Furthermore, we explore recent advancements in our understanding of the influence of neural activity in the brain on vasomotion in the cerebral vasculature, and the link between the presence of vasomotion and metabolic demand associated with information processing in the brain cortex.

Vasomotion in pressurized middle cerebral arteries in vitro. Panel a: Continuous recording of arterial diameter over a range of intraluminal pressures from 20 to 100 mmHg. The “noisy” appearance of the recording results from oscillations in diameter of decreasing amplitude with increased intraluminal pressure. Panel b: Representative expanded segments of the trace in panel a at 20, 40, and 100 mmHg illustrating the change in amplitude and frequency of vasomotion associated with increased intraluminal pressure
2 Vasomotion is a Common Feature of the Macrovasculature
Vasomotion has been observed in segments of conduit arteries, as well as small-resistance arteries and arterioles studied in vitro by wire (isometric) and pressure (isobaric) myography (Fig. 12.1), and in vessels in vivo through the application of multiple techniques that assess blood flow (e.g., laser Doppler flow, blood cell velocity, capillary pressure). For a more comprehensive presentation and historical perspective the interested reader should consult the following review articles [2,3,4, 6, 8]. In the case of blood flow measurements, the rhythmic oscillations in arterial diameter during vasomotion evoke corresponding variations in flow velocity, or “flowmotion” [9, 10]. Blood flow measurements include variations in flow due to multiple causes; these include a “myogenic” component of temporal variability resulting from vasomotion, as well as fluctuations in flow due to cardiac and respiratory activity, and rhythmic endothelium-dependent and neurogenic mechanisms [11, 12].
Rhythmic, synchronous contraction over several millimeters of individual vessel segments is a ubiquitous feature of the arterial vasculature [4, 8]; examples include cerebral [13, 14], mesenteric [15,16,17,18], irideal [19], skeletal muscle [20], and cutaneous arteries and arterioles [21, 22]. In some instances, vasomotion can exhibit a more irregular character owing to the superimposition of multiple rhythms (Fig. 12.1), generated by multiple intrinsic oscillators at different frequencies [23, 24], or superimposed activity originating from multiple initiating sites along the vessel wall. Reported variations in the amplitude and frequency (0.01–0.3 Hz) of spontaneous vasomotion, or vasomotion observed in the presence of intraluminal pressure, stretch, vasoconstrictors, or vasodilators, arise from varied physiological or experimental conditions, species and vascular bed, and vessel caliber (Fig. 12.1). Different stable patterns of vasomotion at varied frequencies can also emerge in the presence of various ion channel and transport-blocking drugs, consistent with the presence of multiple oscillatory mechanisms in the vessel wall [7, 8]. In general, branch vessels of similar size behave independently and differences in frequency are detected at the branch points, with smaller downstream vessels oscillating at a higher frequency [5,6,7, 21, 25, 26]. Vasomotion is generally observed at intermediate levels of tone development, and may be reduced in amplitude or not detected towards extremes of full dilation or constriction [27]. The characteristics of noradrenaline- and arginine vasopressin-evoked vasomotion in rat mesenteric arteries assessed in vitro and in vivo were similar [16, 17, 28]. However, differences in amplitude and frequency of the oscillations were detected in vivo depending on the combination of anesthetic and vasoconstrictor employed [28]. Effects of anesthesia on vasomotion were previously reported in several studies, i.e., including an inhibition or a stimulation of rhythmic contractions [7, 9, 21, 29,30,31,32]. For this reason, analysis of vasomotion in vessels in vivo is best performed in the awake, unanesthetized condition [14, 22, 31, 32].
3 Cellular Mechanisms of Arterial Vasomotion
No clear pattern of pharmacological sensitivity, endothelium removal, or neuronal dependence has emerged for arterial vasomotion, prompting the view that multiple mechanisms may be involved in a vessel- and species-dependent manner [4, 6,7,8]. This section provides a summary of the major mechanisms postulated to underlie vasomotion that are largely based on an analysis of vasomotion in rat mesenteric arteries. Also presented are recent advances that (1) identify a role for bestrophin- and/or ANO1/TMEM16A-containing Ca2+-sensitive chloride channels as a cause of rhythmic depolarizations in vasomotion [33,34,35] and (2) present contrasting views concerning the role of asynchronous Ca2+ waves in the initiation of vasomotion in vitro and in vivo [7, 16, 22, 32, 36].
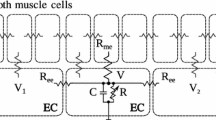
Vasomotion is dependent on simultaneous, rhythmic contraction and relaxation of smooth muscle cells over the length of individual vessel segments that may span several millimeters. By necessity this contractile behavior requires two key elements, a mechanism that synchronizes electrical and contractile activity in individual myocytes along the vessel wall, and an oscillatory mechanism that provides temporal control over Ca2+-dependent activation of cross-bridge cycling. Synchronized contractile behavior in individual vascular smooth muscle cells is not attributed to innervation, as neurotransmitters are released “en passant” from intermittent varicosities within a loose network of perivascular (mostly sympathetic) nerves in the outer adventitial connective tissue layer surrounding vessels [37]. Rather, there is general agreement that synchronization is dependent on the presence of gap junctions that permit intercellular electrical communication between individual smooth muscle cells (homocellular junctions), and between smooth muscle and endothelial cells (heterocellular junctions) at myoendothelial projections (i.e., myoendothelial gap junctions; Fig. 12.2) [5, 7, 8, 38,39,40]. Gap junctions are formed by the alignment of two connexon hemichannels in opposing cell membranes, with each connexon composed of six transmembrane connexin proteins (i.e., connexins 37, 40, 43, and/or 45) that surround an aqueous cell-to-cell pathway that permits electrical coupling, as well as the movement of ions and small molecules (<~1 kDa) between the cytosol of connected cells (reviewed in [39, 40]). Evidence that functional gap junctions are essential for vasomotion in rabbit mesenteric arteries was provided through the use of inhibitor peptides that disrupt the interaction between gap junction hemichannels [41]. Vasomotion was suppressed by these peptides without an effect on basal tone suggesting that a specific block of gap junction communication was achieved [41]. Vasomotion was also assessed following genetic knockout of connexin 40 in mice, and only irregular contractile activity was observed in cremaster arterioles [42]. Finally, inhibition of gap junctions with heptanol or glycyrrhetinic acid derivatives was also reported to suppress vasomotion, with the caveat that these agents are known to have off-target effects [41, 43, 44].

Cartoon representation of the key elements postulated to contribute to arterial vasomotion. The upper cartoon depicts a branching resistance arteriole showing the key structural elements, endothelial cells (ECs) lining the vessel lumen and arranged parallel to the direction of blood flow, surrounded by vascular smooth muscle cells (VSMCs) oriented in a perpendicular manner around the vessel wall, and the loose meshwork of sympathetic nerves and intermittent varicosities within the adventitial layer of connective tissue around the vessel. The lower cartoon is an expanded version showing that the ECs are electrically coupled to the surrounding VSMCs by heterocellular gap junctions (GJs) within myoendothelial projections that extend between the cell types. The postulated mechanisms of vasomotion within ECs and VSMCs include (1) Gq/11 G protein-coupled receptors, such as the α1-adrenoceptor (α1-AR), that evoke synthesis of inositol 1,4,5-trisphosphate (IP3) via phospholipase C (PLC) activation and PIP2 hydrolysis that is required to elicit the release of internal Ca2+ stores in the sarcoplasmic reticulum (SR) via IP3 receptors (IP3Rs) near the plasma membrane in VSMCs and within myoendothelial projections of ECs. The localized release of Ca2+ at these sites is denoted by the red shading in the cytosol between the IP3Rs and plasma membrane. (2) A cytosolic oscillator mechanism that involves cycles of Ca2+ release by IP3Rs to evoke depolarization by activating cGMP-dependent Ca2+-activated chloride channels (cGMP-ClCa), or nonselective cation channels consisting of TRPC3 (activation by a direct protein-protein interaction with IP3Rs is postulated for this conductance) or TRPM4 proteins, in the plasma membrane. The depolarization (+ΔVm) activates nearby voltage-gated Ca2+ channels resulting in Ca2+ influx and a global change in cytosolic Ca2+ concentration that evokes cross-bridge cycling in the myofilaments, and propagates electrotonically into adjacent VSMCs via homocellular GJs (ΔVm). Cytosolic Ca2+ concentration is subsequently reduced by transport into the SR via sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) for subsequent release during the next cycle of vasomotion as well as extrusion out of the cell (not shown). (3) IP3 from VSMCs may transit myoendothelial GJs to evoke Ca2+ release and activation of endothelial nitric oxide synthase (eNOS) and intermediate-conductance, Ca2+-activated K+ channels (IKCa) localized within and in close proximity to the myoendothelial projection (slightly displaced out of the projection for clarity in this cartoon), with the resultant synthesis and release of nitric oxide (NO) activating soluble and particulate guanylyl cyclase (GC) to produce the second messenger, cGMP, that is required for cGMP-ClCa activity
There is also consensus that the signal that promotes synchronization along the vessel wall must be electrical in nature, i.e., a change in membrane potential (ΔVm) [5,6,7,8]. Only an electrical signal appears sufficient to account for synchronization over millimeter lengths of arteriolar segments that are considerably greater than the width of individual myocytes (~3–5 μm) positioned perpendicularly around the vessel wall [5, 16]. In this scenario, independent oscillations in membrane potential within individual myocytes are thought to be entrained by rapid cell-to-cell spread of electrotonic current via gap junctions. This results in synchronized phases of depolarization, voltage-dependent Ca2+ channel activation, and Ca2+ influx that evoke a simultaneous, global rise in cytosolic Ca2+ concentration and cross-bridge cycling in all myocytes along the vessel wall (i.e., a coupled oscillator model [5, 16, 45]).
Three generalized mechanisms are thought to be responsible for rhythmic contractile behavior of smooth muscle (reviewed in [46]), and examples of each mechanism are presented in this book. The postulated mechanisms involve (1) oscillations in membrane potential in individual smooth muscle cells that are evoked by specialized pacemaker cells, such as interstitial cells of Cajal associated with gastrointestinal smooth muscle cells, and atypical smooth muscle cells within urogenital smooth muscle tissues; (2) a membrane oscillator mechanism within the plasma membrane that periodically depolarizes and hyperpolarizes membrane potential, leading to intermittent voltage-dependent Ca2+ entry and rhythmic activation of cross-bridge cycling; and (3) a cytosolic oscillator involving intermittent release of Ca2+ from internal Ca2+ stores via inositol trisphosphate receptors (IP3R) and/or ryanodine receptors (RyR) in the sarcoplasmic reticulum [46]. It is this third scenario involving oscillatory Ca2+ release and refilling of internal Ca2+ stores that is postulated to be involved in arterial vasomotion (Fig. 12.2) [5, 7, 16]. However, Ca2+ released from the internal stores does not activate contraction directly, as global elevations in cytosolic Ca2+ concentration are only achieved with depolarization and voltage-dependent Ca2+ channel activity in vascular smooth muscle cells [47]. Rather, localized Ca2+ release from IP3Rs in close proximity to the plasma membrane is postulated to cause the activation of a Ca2+-sensitive depolarizing current, and subsequent Ca2+ influx through voltage-dependent Ca2+ channels leading to contraction (Fig. 12.2) [7, 16]. Uptake of Ca2+ from the cytosol into the sarcoplasmic reticulum via sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) activity refills the internal Ca2+ store in preparation for the next release event. The delay required for store refilling is postulated to provide the necessary time lag needed for rhythmicity; that is, in this model, IP3R-dependent store release and refilling is the primary cytosolic oscillator [7, 8, 16], with release from the store dependent on the combination of IP3 concentration and luminal Ca2+ concentration [48]. Factors that influence the refilling process, including changes in the level of Ca2+ influx across the plasma membrane, cytosolic concentration of IP3 , and level of luminal Ca2+ in the store, may alter the frequency of vasomotion by affecting this cytosolic oscillator mechanism.
The current view that arterial vasomotion is dependent on a cytosolic oscillator involving the periodic release of internal Ca2+ stores via IP3Rs, coupled to rhythmic oscillations in membrane depolarization and elevations of global cytosol Ca2+ concentration due to the activation of a Ca2+-sensitive inward current, is supported by several observations:
-
1.
Oscillations in membrane potential are detected in a variety of vessels exhibiting vasomotion, with depolarization observed to precede the onset of constriction [5, 16, 17, 27, 49,50,51,52,53], with the apparent exception of irideal arterioles [19, 54]. That oscillations in membrane potential are required for vasomotion is indicated by experiments showing an inhibition of vasomotion in the presence of ATP-sensitive K+ channel-activator drugs that cause sustained smooth muscle hyperpolarization [16, 55].
-
2.
Rhythmic, synchronized oscillations in global cytosolic Ca2+ concentration in smooth muscle cells are detected during vasomotion in arterioles studied in vitro [15, 16] and in vivo [22, 32, 36], with cytosolic Ca2+ rapidly increasing prior to the onset of contraction in both experimental conditions. An ~0.3 s delay was detected between the peak elevation in cytosolic Ca2+ and maximal rate of constriction in murine ear arterioles in vivo [22], and a delay of ~0.9 s was detected in basilar arterial segments in vitro [53]. These values are consistent with the time lag between the rise in global Ca2+ concentration, and subsequent phosphorylation of myosin regulatory light chain (i.e., LC20) by Ca2+-calmodulin-dependent myosin light-chain kinase and contraction during depolarization-evoked contraction of urinary bladder smooth muscle following neural activation [56, 57].
-
3.
The oscillations in cytosolic Ca2+ concentration associated with vasomotion are suppressed by inhibiting L-type, voltage-gated Ca2+ channels, or removal of extracellular Ca2+ [16, 17, 56, 58, 59], whereas vasomotion is enhanced by treatment with the Ca2+ channel activator BayK8644 [59]. Ca2+ influx via voltage-dependent Ca2+ channels is required for cross-bridge cycling, and to refill the internal Ca2+ stores following Ca2+ release by IP3Rs (Fig. 12.2) [5, 7]. Global elevations in cytosolic Ca2+ in smooth muscle cells are widely recognized to be dependent on membrane electrical activity, in which depolarization evokes the activation of voltage-gated Ca2+ channels, and oscillations in Ca2+ entry lead to rhythmic variations in membrane potential and vasomotion through intermittent filling and release of internal Ca2+ stores [47].
-
4.
Inhibition of the release of internal Ca2+ stores in vascular myocytes by blocking phospholipase C-dependent inositol 1,4,5-trisphosphate (IP3) synthesis from phosphoinositide 4,5-bisphosphate (PIP2) with U73122, or IP3Rs with the nonselective inhibitor 2-aminoethyl diphenylborinate (2-APB), was shown to prevent rhythmic oscillations in cytosolic Ca2+ concentration and vasomotion, as does blocking Ca2+ uptake into the sarcoplasmic reticulum with cyclopiazonic acid (CPA) or thapsigargin [16, 24, 27, 56, 58,59,60,61]. The key role played by IP3R is also indicated by the stimulation of vasomotion by different vasoconstrictor agonists, noradrenaline, serotonin, and vasopressin, that all act on Gq/11-coupled receptors to evoke phospholipase C-mediated generation of IP3 (Fig. 12.2) [7].
Treating arteries and arterioles with ryanodine to alter RyR-mediated release of internal Ca2+ stores has varied effects on vasomotion, including a block of cytosolic Ca2+ oscillations and rhythmic contraction [16, 56, 59], alteration in the frequency and amplitude of Ca2+ oscillations and vasomotion (as was also noted for CPA in rabbit mesenteric arteries), and a change in endothelium dependency of vasomotion from necessary to not required [60, 62]. These contrasting observations may reflect a varied contribution of RyR to the cytosolic oscillator in different vessels and experimental conditions, and/or that depleting internal Ca2+ stores with ryanodine or CPA may reveal the presence of additional oscillator mechanisms in the vascular wall that may or may not involve endothelium-dependent mechanisms [7]. Based on our current understanding, there is general agreement that periodic release of Ca2+ from the sarcoplasmic reticulum via IP3Rs in vascular myocytes represents the key component of the cytosolic oscillator necessary for vasomotion, but RyRs may be involved in vessel-specific manner, a view consistent with the evidence of a differential expression and functional contribution of RyRs, but not IP3Rs, to the regulation of cytosolic Ca2+ in smooth muscle cells within arteries and arterioles of varied size and location [63, 64].
Identification of the ionic conductance responsible for rhythmic depolarization of membrane potential during vasomotion has been hampered by a lack of selective pharmacological tools to inhibit or activate the suspected channel candidates, but the recent application of molecular approaches has provided novel insight. Several lines of evidence suggest that cGMP-dependent, Ca2+-activated chloride channels are stimulated by IP3R-dependent release of Ca2+ from the sarcoplasmic reticulum to evoke membrane potential depolarization during vasomotion in rat mesenteric arteries (i.e., ECl is ~−25 mV, so activation of the channels elicits outward Cl− flux and depolarization; Fig. 12.2) (reviewed in [7, 8]). Ca2+ released from the sarcoplasmic reticulum via IP3R may be localized within microdomains between the sarcoplasmic reticulum and plasma membrane such that chloride channel gating is stimulated without activation of cross-bridge cycling (Fig. 12.2). This mechanism parallels the regulation of membrane potential by localized Ca2+ sparks created by the focal release of Ca2+ from RyRs that stimulate juxtaposed large-conductance Ca2+-activated potassium channels in the plasma membrane to evoke hyperpolarization and relaxation [47].
The rhythmic depolarizations, oscillations in cytosolic Ca2+ concentration, and vasomotion of rat mesenteric arteries evoked by α1-adrenoceptor activation with phenylephrine were shown to be dependent on the presence of cytosolic cGMP [16]. Moreover, a cGMP-dependent, Ca2+-activated chloride current was observed in myocytes from these arteries [65]. This current was found to possess biophysical and pharmacological properties distinct from those of “classical” Ca2+-activated chloride channels in vascular smooth muscle [65] that are thought to be due to expression of ANO1/TMEM16A chloride channel proteins [66, 67]. Expression of another putative Ca2+-activated Cl− channel protein, bestrophin, was demonstrated to be essential for the cGMP dependence of the chloride current [33], and the amplitude of vasomotion in rat mesenteric arteries was reduced following siRNA knockdown of bestrophin-3 [34]. Although ANO1/TMEM16A silencing produced a similar suppression of vasomotion as bestrophin-3 knockdown, this strategy also reduced bestrophin expression, so it remains unclear if the two proteins form distinct channels, or that bestrophin confers cGMP sensitivity and unique properties to ANO1/TMEM16A channels by acting as an auxiliary subunit [35].
Alternatively, it is possible that rhythmic activation of a depolarizing, nonselective cation conductance is responsible for the IP3R-dependent depolarization of membrane potential in vasomotion (Fig. 12.2). Recent electrophysiological analysis has identified two mechanisms by which IP3Rs could potentially evoke rhythmic depolarization by activating nonselective cation channels in vascular smooth muscle cells. Specifically, a direct interaction of IP3Rs with transient receptor potential cation channels composed of TRPC3 proteins was identified [68]. In this case, the conformational change associated with IP3R activation is coupled to the gating of TRPC3 channels via a direct, protein-protein interaction [68]. Alternatively, localized Ca2+ release by IP3R near the plasma membrane could evoke the activation of cation channels containing Ca2+-sensitive TRPM4 subunits [69, 70]. Consistent with a role for nonselective cation channels, an increased level of vasomotion was observed in mesenteric arteries of spontaneously hypertensive rats (SHR) compared to normotensive controls (WKY), and found to be associated with a greater expression of TRPC1, TRPC3, and TRPC5 cation channel proteins [71]. The extent of vasomotion was reduced by exposing the arteries to putative nonselective cation channel inhibitors, gadolinium, SKF-96365, and 2-APB, or antibodies against TRPC1 and TRPC3 proteins [71]. Furthermore, chronic treatment of the SHRs with angiotensin AT1 receptor antagonist, but not the L-type Ca2+ channel blocker amlodipine, was demonstrated to suppress TRPC protein expression and vasomotion in the mesenteric vessels [71].
An intact, functional endothelium is essential for vasomotion in many, but not all, arteries [7]. For example, endothelial removal blocks vasomotion in rat mesenteric arteries [16, 59, 72,73,74], but it is potentiated by endothelial denudation or inhibition of NO synthesis in other vessels [23, 75, 76]. The endothelium appears to be required for nitric oxide synthesis and release (Fig. 12.2). Nitric oxide is thought to be necessary because it evokes cGMP synthesis by guanylyl cyclase in the myocytes, and cGMP is required to facilitate cGMP-dependent Ca2+-activated Cl− channel activity and depolarization during vasomotion in rat mesenteric arteries (Fig. 12.2) [7, 8]. Consistent with this view, inhibition of nitric oxide synthesis (e.g., with l-NG-nitroarginine (l-NNA)) also blocked vasomotion in these vessels, and it was partially restored with sodium nitroprusside or membrane-permeant analogs of cGMP [16, 59, 74, 77].
Alternatively, the endothelium may be required to facilitate electrical conduction and/or synchronization along the vessel wall. The longitudinal orientation of electrically coupled endothelial cells and presence of myoendothelial gap junctions would be expected to permit synchronization over greater distances along vessels than what is possible with electrotonic current spread within the smooth muscle layer alone [78, 79]. It is also possible that vasomotion is affected by endothelium-dependent hyperpolarization [73]. In this case, IP3 generated in smooth muscle cells by α1-adrenoceptor activation may diffuse into endothelial cells via myoendothelial gap junctions to cause the release of Ca2+ stores within the myoendothelial projections. Subsequent activation of intermediate-conductance Ca2+-activated potassium (IKCa) channels in the endothelial cells may evoke hyperpolarization that spreads back through the junctions to regulate smooth muscle membrane potential and contractility [80, 81]. This mechanism involving IP3- and endothelial IKCa channel-dependent feedback dilation is thought to account for endothelium-mediated inhibition of constriction evoked by α1-adrenoceptors (see [81]), but it may also modulate vasomotion [73] (Fig. 12.2).
The cellular mechanism by which vasomotion is initiated is controversial; recent studies (e.g., [22, 32]) using in vivo experimental approaches have cast doubt on the established model involving asynchronous Ca2+ waves arising from analyses of vasoconstrictor-evoked vasomotion in rat mesenteric arterioles under isometric recording conditions in vitro [7, 8, 16]. An abundant literature on Ca2+ signaling in arteries in vitro indicates that activation of Gq/11-coupled receptors by vasoconstrictor agonists is associated with the presence of asynchronous, IP3R-dependent (and/or RyR-dependent) oscillations in cytosolic Ca2+ concentration within individual vascular myocytes [47, 82]. These oscillations in cytosolic Ca2+ occur as waves of Ca2+ elevation that spread through the cytosol due to regenerative Ca2+-induced Ca2+ release from nearby IP3Rs [47, 82]. Significantly, asynchronous Ca2+ waves are consistently detected in individual myocytes within the vessel wall prior to the appearance of synchronous, global elevations in cytosolic Ca2+ concentration and the onset of vasomotion in arteries studied in vitro [7, 8, 15, 16]. Peng et al. [16] postulated that the initiation of vasomotion occurs when these asynchronous Ca2+ wave events in individual cells are entrained to produce synchronized, global Ca2+ elevations in myocytes along the length of the vessel wall. Entrainment was postulated to involve Ca2+-dependent activation of depolarizing cGMP-dependent, Ca2+-activated chloride current and cell-to-cell electrical communication via gap junctions [16]. In this case, the oscillations in membrane potential in individual myocytes influence the electrical behavior of adjacent cells via electrotonic current flow, leading to simultaneous depolarization, global Ca2+ elevation, and contraction of increasingly greater numbers of myocytes along the vessel in the absence of a defined pacemaker. This mechanism of self-organizing behavior is comparable to that of crowds at sports events, such as soccer matches, in which songs initiated by small groups of supporters spread through the crowd, and unison is achieved in the absence of a conductor [83].
The Peng et al. [16] model for the initiation of vasomotion is compelling, but it remains to be established that this mechanism is applicable to arteries and arterioles in vivo. Indeed, although synchronous elevations in global cytosolic Ca2+ concentration and vasomotion are readily observed in small arteries and arterioles in vivo (or surgically exposed vessels in situ), asynchronous propagating Ca2+ waves were only rarely detected in cremaster skeletal muscle arterioles [36, 84], femoral arteries [85, 86], and intact ear arterioles [22, 32]. The genetically encoded GCaMP2 and exMLCK fluorescent probes used to monitor cytosolic Ca2+ concentration by confocal microscopy in these studies were shown to be appropriate for detecting asynchronous Ca2+ waves in arterial segments exposed to α1-adrenoceptor agonist in vitro [36]. These observations suggest that technical limitations are likely not involved. Alternatively, Mauban et al. [36] indicated that a Ca2+-dependent desensitization of IP3Rs may underlie the absence of asynchronous Ca2+ waves in vivo. An increased cytosolic Ca2+ concentration is expected in arterial myocytes in vivo due to myogenic constriction evoked by intraluminal pressure, and the influence of neurohumoral vasoconstrictors that are not present under in vitro isometric recording conditions [36]. This elevated cytosolic Ca2+ may cause desensitization of the IP3Rs and preclude propagating Ca2+ waves, but have a limited impact on Ca2+ release due to receptor- and phospholipase C-dependent IP3 synthesis [36]. This hypothesis needs to be tested experimentally. Alternatively, it is possible that there are multiple routes to vasomotion initiated by different mechanisms under varied physiological conditions [7, 8]. Further study is required to resolve this key issue.
4 Cerebral Arterial Vasomotion and Neural Activity in the Brain
A long-held, but widely debated, view holds that the physiological function of vasomotion is to increase blood flow to supply the metabolic needs of downstream parenchyma [2, 21, 87,88,89,90,91,92,93,94]. However, whether vasomotion is a physiological or pathophysiological characteristic of the vasculature remains unresolved [7, 8]. Historically, attention has focused on the peripheral vasculature due to ease of access for experimentation. Multiple studies have shown that flowmotion in cutaneous microcirculation is associated with increased tissue perfusion, greater O2 content, and increased O2 extraction, consistent with a physiological role [5, 7, 8, 94]. Recent analysis of the cerebral vasculature reinforces this view, indicating that vasomotion is associated with increased oxygen delivery to active regions and a direct consequence of neural activity in the brain [14]. These new findings coupled with evidence that the regulation of the cerebral vasculature and vasomotion may be impaired in situations of cognitive dysfunction such as Alzheimer’s disease [95, 96] highlight the importance of understanding the physiological role(s) and regulation of vasomotion, and the differences between the peripheral and cerebral vasculature.
The high metabolic cost of neural electrical activity and lack of substantive energy reserves in the brain require that a mechanism known as neurovascular coupling mediates dynamic regulation of blood flow in response to changes in neural activity. This regulation permits precise, spatiotemporal matching of neuronal metabolic demand with the supply of O2 and glucose, and removal of metabolic by-products [97,98,99]. Blood oxygen level-dependent (BOLD) functional magnetic resonance imaging (BOLD fMRI) [100, 101] and intrinsic optical signal (IOS) imaging techniques have been widely employed as a proxy for neural activity related to sensory stimulation, movement, and decision-making. A local positive BOLD signal (i.e., decreased deoxyhemoglobin) is indicative of augmented blood flow in response to an increase in neural activity. For example, sensory stimulation evokes a rapid increase in penetrating and pial arterial diameter and blood flow that is accompanied by a local increase in BOLD signal within the cortical region responsible for processing the sensory signal [31, 102].
Growing attention has recently focused on localized, spontaneous fluctuations in BOLD signals that are detected in the absence of sensory stimulation, but synchronized in functionally related yet distant regions of the brain that are connected by long range and commissural neural pathways, for example in symmetric regions across the brain midline [103,104,105,106]. Significantly, these so-called resting-state BOLD signals occur at frequencies focused around ~0.1 Hz, matching the frequency of the oscillations in arterial diameter associated with cerebral vasomotion and flowmotion [9, 14, 31, 107, 108].
The precise neural correlate of resting-state BOLD signal fluctuations has been widely debated [104, 109,110,111,112,113], and the interested reader is directed to a 2016 themed issue in Philosophical Transactions of the Royal Society B entitled “Interpreting BOLD: a dialogue between cognitive and cellular neuroscience” [114]. However, consensus has developed around the idea that they are the result of rhythmic fluctuations in the activity of intracortical neural networks which can be detected via extracellular electrophysiological recordings of local field potentials [111, 112, 115,116,117,118]. Specifically, the resting-state BOLD signal correlates with γ-band (30–80 Hz) rhythms in the local field potential that are generated by tightly synchronized electrical activity within local networks of fast-spiking cortical interneurons [111, 112, 115,116,117,118]. For example, such γ-band activity is observed in response to increased sensory drive in the somatosensory cortex [119], and increased neural activity associated with working memory and learning [120, 121].
Direct evidence that cerebral vasomotion is entrained by oscillations in γ-band power and is responsible for resting-state BOLD signal was recently provided by Mateo and co-workers [14]. In their study, Mateo et al. [14] assessed the local field potentials generated by superficial layers of neurons within the vibrissa area of the parietal cortex in head-restrained, conscious mice fitted with a transcranial window to permit two-photon fluorescence microscopy of vasomotion in pial arterioles. Vasomotion in the arterioles was found to be phase-locked to γ-band oscillations produced by cortical neuronal activity with a lag of ~2 s, and accompanied by a positive BOLD IOS signal due to increased blood flow that followed the arteriolar dilation by ~0.7 s [14]. Significantly, the vasomotion was transhemispheric in nature, with arterioles in mirrored regions across the midline showing identical behavior that was markedly reduced in mice lacking callosal connections [14]. An optogenetic approach was further used to establish causality between the γ-band oscillations and vasomotion. Specifically, transgenic mice expressing the channelrhodopsin protein in layer 5b pyramidal neurons were stimulated with a 40 Hz γ-like train of laser light pulses with a sinusoidal variation in intensity around 0.1 Hz [14] (i.e., 0.05–0.3 Hz consistent with the frequency range of vasomotion in the preparation; [31]). Driving the cortical circuitry in this manner caused phase-locked vasomotion in pial arterioles identical to that observed during spontaneous activity [14]. Identical illumination in wild-type mice did not elicit a vasomotor response, ruling out a direct effect of the light stimulation protocol on blood flow. In contrast, mice with smooth muscle-specific expression of the light-sensitive chloride pump protein, halorhodopsin, exhibited light-driven vasomotion consisting of ~20% changes in resting arterial diameter, but with no coherent neural activity [14]. This data set demonstrates that the functional interaction is unidirectional, with the neural activity entraining vasomotion in the arterioles, but not the reverse. Significantly, the amplitude of vasodilations observed during spontaneous vasomotion and functional hyperemia in response to whisker stimulation were of similar magnitude and both were attenuated by urethane anesthesia [31], reminiscent of the sensitivity of vasomotion in other vascular beds to anesthetics [7].
To appreciate how cerebral vasomotion may be phase-locked to γ-band neural activity, it is appropriate to consider the mechanisms of neurovascular coupling, as they may foster vasomotion. Our understanding of neurovascular coupling is still evolving, but the contemporary view presented by Iadecola [99] indicates that vasomotor responses are caused by stimulus- and brain region-dependent release of vasoactive mediators, such as nitric oxide, prostaglandins, vasoactive peptides, ATP, adenosine, and/or potassium ions (K+) from neurons and/or astrocytes, that affect membrane potential within capillary endothelial cells [122], pericytes [123], and/or nearby smooth muscle cells in the cerebral microvasculature to evoke dilation or constriction (a decline in local O2 or glucose content may also be involved; see [99]).
For example, evidence consistent with the potential involvement of capillary endothelial cells in sensing modest elevations in extracellular K+ concentration due to neural activity, and communicating this vasodilatory signal to upstream arterioles was recently provided by Longden et al. [122]. The cortical capillary endothelium of mice was demonstrated to possess the Ba2+-sensitive Kir2.1 subtype of inward-rectifier K+ channel that conducts hyperpolarizing K+ current of increased magnitude in the presence of elevated extracellular K+ concentration (from ~4 to 8–10 mM) [122], as previously shown for endothelial cells in other vascular beds [124]. Exposing cerebral capillary endothelial cells to 10 mM external K+ resulted in a rapid (~2 mm/s) propagating hyperpolarization and dilation in upstream arterioles in an ex vivo parenchymal arteriolar-capillary preparation, and increased red blood cell flux in the cortical vasculature of mice in vivo, that were similarly suppressed by either Ba2+ treatment or endothelium-specific knockout of Kir2.1 channels [122].
Hyperpolarization and vasodilation in the cerebral microvasculature in response to extracellular K+ accumulation, prostaglandin, or nitric oxide release during neurovascular coupling [99] evoke vasodilation in upstream penetrating and pial arterioles by a mechanism of ascending, retrograde vasodilation, identical to that described for the peripheral vasculature [81, 122, 125,126,127]. The functional hyperemic response in the cerebral vasculature consists of: (1) a propagating electrical signal of membrane potential hyperpolarization that rapidly spreads (~2–2.5 mm/s) into upstream pial and penetrating arterioles to evoke immediate vasodilation and (2) a second, slower mechanism of ascending vasodilation involving cytosolic Ca2+-dependent release of nitric oxide and prostacyclin from the endothelium in response to increased flow and shear stress due to dilation of distal terminal arterioles [102, 122, 125, 126]. Retrograde propagation of the hyperpolarization involves cell-to-cell electrical communication through the endothelium and then into smooth muscle cells via myoendothelial gap junctions to evoke vasodilation, being inhibited by focal disruption of the endothelium or suppression of endothelial Kir2.1 expression [102, 122, 126].
Retrograde, ascending propagation of hyperpolarization evoked by neurovascular coupling thus represents a likely mechanism to account for the entrainment of vasomotion by neural activity in the cerebral vasculature. Rhythmic ascending waves of hyperpolarization evoked by neurovascular coupling may serve to entrain the oscillations in membrane potential responsible for vasomotion at the level of the penetrating and pial arterioles. Periodic hyperpolarization may additionally reinforce the subsequent depolarization phase of each oscillation by favoring recovery from inactivation and increased availability of voltage-gated Ca2+ channels. Additionally, release of nitric oxide owing to increased shear stress could facilitate cGMP-dependent Ca2+-activated chloride current and oscillatory depolarization. A role for perivascular nerves in the modulation of vasomotion may also be postulated. Further research is required to delineate the precise mechanism(s) contributing to the entrainment of vasomotion by neural activity in the brain.
As a final point, vasomotion in the peripheral vasculature is similarly affected by nerve activity, and oscillations in separate vessel segments can be entrained by bursts of sympathetic nerve activity [128, 129]. This is achieved through a prolongation of cycle length when nerve activity occurs after peak relaxation until a critical point after which cycle length is reduced [129]. However, in contrast to the role of neurovascular coupling in entrainment in the cerebral vasculature, entrainment and vasomotion in peripheral vessels are facilitated by the release of noradrenaline from adventitial sympathetic nerve varicosities. Oscillations in forearm skin blood flow at 0.1 Hz were suppressed by sympathetic blockade following anesthesia by brachial plexus infiltration in humans [128]. Suppression of sympathetic nerve activity following application of the ganglionic blocker hexamethonium was shown to inhibit synchronous oscillations in cytosolic Ca2+ concentration and vasomotion in rabbit ear arterioles in vivo [22, 36]. Finally, 0.1 Hz fluctuations in cutaneous microcirculatory blood flow disappear in conditions of sympathetic dysfunction such as in diabetes [130, 131]. The contribution of rhythmic ascending vasodilation to vasomotion in peripheral vasculature is not known at present. The presence of these different mechanisms for entrainment of vasomotion in cerebral and peripheral vasculature illustrates the challenge in developing a comprehensive understanding of vasomotion and its regulation in varied vascular beds.
5 Summary
Despite more than 150 years of research, there are many questions concerning vasomotion that remain to be adequately addressed. Key issues for future study should include (1) identification of the inward current activated by the IP3R-dependent release of Ca2+ stores: Specifically, is the contribution of cGMP-dependent, Ca2+-activated Cl− channels ubiquitous or restricted to mesenteric arteries, and what is the role of nonselective cation channels? (2) Why does the contribution of the endothelium to vasomotion vary in a vessel-specific manner? (3) What is the mechanism by which vasomotion in cerebral pial and penetrating arterioles is entrained to cortical neural activity: specifically, is this process dependent on rhythmic hyperpolarization associated with ascending vasodilation and Kir2.1 channels in the cerebrovascular endothelium and capillaries? (4) If asynchronous Ca2+ waves are not involved in synchronization of contractile activity in individual myocytes in vivo, what is the mechanism responsible for the self-organizing behavior? (5) Does entrainment of vasomotion in peripheral and cerebral vasculature involve a differential contribution of neural- versus endothelium-dependent mechanisms of modulation, respectively? Novel understanding in these areas will undoubtedly be facilitated by technological advancements permitting in vivo imaging of arterial diameter and cytosolic Ca2+ concentration, as well as cell type-specific expression of functional and genetically compromised proteins to selectively augment and disrupt cellular processes postulated to contribute to vasomotion and its regulation in health and disease.
References
Jones TW. Discovery that veins of the bat’s wing (which are furnished with valves) are endowed with rhythmical contractility and that the onward flow of blood is accelerated by each contraction. Philos Trans R Soc Lond. 1852;142:131–6.
Funk W, Intaglietta M. Spontaneous arteriolar vasomotion. Prog Appl Microcirc. 1983;3:66–82.
Shimamura K, Sekiguchi F, Sunano S. Tension oscillation in arteries and its abnormality in hypertensive animals. Clin Exp Pharmacol Physiol. 1999;26:275–84.
Nilsson H, Aalkjaer C. Vasomotion: mechanisms and physiological importance. Mol Interv. 2003;3:79–89.
Aalkjaer C, Nilsson H. Vasomotion: cellular background for the oscillator and for the synchronization of smooth muscle cells. Br J Pharmacol. 2005;144:605–16.
Haddock RE, Hill CE. Rhythmicity in arterial smooth muscle. J Physiol. 2005;566:645–56.
Aalkjaer C, Boedtkjer D, Matchkov V. Vasomotion—what is currently thought? Acta Physiol (Oxford). 2011;202:253–69.
Matchkov VV. Mechanisms of cellular synchronization in the vascular wall. Mechanisms of vasomotion. Dan Med Bull. 2010;57:B4191.
Intaglietta M. Vasomotion and flowmotion: physiological mechanisms and clinical evidence. Vasc Med Rev. 1990;1:101–12.
Schmidt JA. Periodic hemodynamics in health and disease. Georgetown, TX: R.G. Landes Company; 1996.
Kvandal P, Landsverk SA, Bernjak A, Stefanovska A, Kvernmo HD, Kirkebøen KA. Low-frequency oscillations of the laser Doppler perfusion signal in human skin. Microvasc Res. 2006;72:120–7.
Stefanovska A. Coupled oscillators. Complex but not complicated cardiovascular and brain interactions. IEEE Eng Med Biol Mag. 2007;26:25–9.
Gokina NI, Bevan RD, Walters CL, Bevan JA. Electrical activity underlying rhythmic contraction in human pial arteries. Circ Res. 1996;78:148–53.
Mateo C, Knutsen PM, Tsai PS, Shih AY, Kleinfeld D. Entrainment of arteriole vasomotor fluctuations by neural activity is a basis of blood-oxygenation-level-dependent “resting-state” connectivity. Neuron. 2017;96:936–48.
Mauban JR, Lamont C, Balke CW, Wier WG. Adrenergic stimulation of rat resistance arteries affects Ca2+ sparks, Ca2+ waves, and Ca2+ oscillations. Am J Physiol Heart Circ Physiol. 2001;280:H2399–405.
Peng H, Matchkov V, Ivarsen A, Aalkjaer C, Nilsson H. Hypothesis for the initiation of vasomotion. Circ Res. 2001;88:810–5.
Oishi H, Schuster A, Lamboley M, Stergiopulos N, Meister JJ, Bény JL. Role of membrane potential in vasomotion of isolated pressurized rat arteries. Life Sci. 2002;71:2239–48.
Zacharia J, Zhang J, Wier WG. Ca2+ signaling in mouse mesenteric small arteries: myogenic tone and adrenergic vasoconstriction. Am J Physiol Heart Circ Physiol. 2007;292(3):H1523–32.
Haddock RE, Hirst GD, Hill CE. Voltage independence of vasomotion in isolated irideal arterioles of the rat. J Physiol. 2002;540:219–29.
Bakker EN, Sorop O, Spaan JA, VanBavel E. Remodeling of resistance arteries in organoid culture is modulated by pressure and pressure pulsation and depends on vasomotion. Am J Physiol Heart Circ Physiol. 2004;286:H2052–6.
Colantuoni A, Bertuglia S, Intaglietta M. Quantitation of rhythmic diameter changes in arterial microcirculation. Am J Phys. 1984;246:H508–17.
Fairfax ST, Mauban JR, Hao S, Rizzo MA, Zhang J, Wier WG. Ca2+ signaling in arterioles and small arteries of conscious, restrained, optical biosensor mice. Front Physiol. 2014;5:387.
Griffith TM, Edwards DH. Mechanisms underlying chaotic vasomotion in isolated resistance arteries: roles of calcium and EDRF. Biorheology. 1993;30:333–47.
Griffith TM, Edwards DH. Ca2+ sequestration as a determinant of chaos and mixed-mode dynamics in agonist-induced vasomotion. Am J Phys. 1997;272:H1696–709.
Colantuoni A, Bertuglia S, Intaglietta M. Variations of rhythmic diameter changes at the arterial micro-vascular bifurcations. Pflugers Arch. 1985;403:289–95.
Oude Vrielink HHE, Slaaf DW, Tangelder GJ, Weijmer-Van Velzen S, Reneman RR. Analysis of vasomotion waveform changes during pressure reduction and adenosine application. Am J Phys. 1990;258:H29–37.
Gustafsson H. Vasomotion and underlying mechanisms in small arteries. An in vitro study of rat blood vessels. Acta Physiol Scand. 1993;149(Suppl. 614):1–44.
Nyvad J, Mazur A, Postnov DD, Straarup MS, Soendergaard AM, Staehr C, Brøndum E, Aalkjaer C, Matchkov VV. Intravital investigation of rat mesenteric small artery tone and blood flow. J Physiol. 2017;595:5037–53.
Burrows ME, Johnson PC. Diameter, wall tension, and flow in mesenteric arterioles during autoregulation. Am J Phys. 1981;241:H829–37.
Hundley WG, Renaldo GJ, Levasseur JE, Kontos HA. Vasomotion in cerebral microcirculation of awake rabbits. Am J Phys. 1988;254:H67–71.
Drew PJ, Shih AY, Kleinfeld D. Fluctuating and sensory-induced vasodynamics in rodent cortex extend arteriole capacity. Proc Natl Acad Sci U S A. 2011;108:8473–8.
Mauban JR, Fairfax ST, Rizzo MA, Zhang J, Wier WG. A method for noninvasive longitudinal measurements of [Ca2+] in arterioles of hypertensive optical biosensor mice. Am J Physiol Heart Circ Physiol. 2014;307:H173–81.
Matchkov VV, Larsen P, Bouzinova EV, Rojek A, Boedtkjer DM, Golubinskaya V, Pedersen FS, Aalkjaer C, Nilsson H. Bestrophin-3 (vitelliform macular dystrophy 2-like 3 protein) is essential for the cGMP-dependent calcium-activated chloride conductance in vascular smooth muscle cells. Circ Res. 2008;103:864–72.
Broegger T, Jacobsen JC, Secher Dam V, Boedtkjer DM, Kold-Petersen H, Pedersen FS, Aalkjaer C, Matchkov VV. Bestrophin is important for the rhythmic but not the tonic contraction in rat mesenteric small arteries. Cardiovasc Res. 2011;91:685–93.
Dam VS, Boedtkjer DM, Nyvad J, Aalkjaer C, Matchkov V. TMEM16A knockdown abrogates two different Ca2+-activated Cl− currents and contractility of smooth muscle in rat mesenteric small arteries. Pflugers Arch. 2014;466:1391–409.
Mauban JR, Zacharia J, Zhang J, Wier WG. Vascular tone and Ca2+ signaling in murine cremaster muscle arterioles in vivo. Microcirculation. 2013;220:269–77.
Westcott EB, Segal SS. Perivascular innervation: a multiplicity of roles in vasomotor control and myoendothelial signaling. Microcirculation. 2013;20:217–38.
Haddock RE, Grayson TH, Brackenbury TD, Meaney KR, Neylon CB, Sandow SL, Hill CE. Endothelial coordination of cerebral vasomotion via myoendothelial gap junctions containing connexins 37 and 40. Am J Physiol Heart Circ Physiol. 2006;291:H2047–56.
Sandow SL, Senadheera S, Bertrand PP, Murphy TV, Tare M. Myoendothelial contacts, gap junctions, and microdomains: anatomical links to function? Microcirculation. 2012;19:403–15.
de Wit C, Griffith TM. Connexins and gap junctions in the EDHF phenomenon and conducted vasomotor responses. Pflugers Arch. 2010;459:897–914.
Chaytor AT, Evans WH, Griffith TM. Peptides homologous to extracellular loop motifs of connexin 43 reversibly abolish rhythmic contractile activity in rabbit arteries. J Physiol. 1997;503:99–110.
de Wit C, Roos F, Bolz SS, Pohl U. Lack of vascular connexin 40 is associated with hypertension and irregular arteriolar vasomotion. Physiol Genomics. 2003;13:169–77.
Tsai ML, Watts SW, Loch-Caruso R, Webb RC. The role of gap junctional communication in contractile oscillations in arteries from normotensive and hypertensive rats. J Hypertens. 1995;13:1123–33.
Matchkov VV, Rahman A, Peng H, Nilsson H, Aalkjaer C. Junctional and nonjunctional effects of heptanol and glycyrrhetinic acid derivates in rat mesenteric small arteries. Br J Pharmacol. 2004;142:961–72.
Imtiaz MS, von der Weid PY, van Helden DF. Synchronization of Ca2+ oscillations: a coupled oscillator-based mechanism in smooth muscle. FEBS J. 2010;277:278–85.
Berridge MJ. Smooth muscle cell calcium activation mechanisms. J Physiol. 2008;586:5047–61.
Hill-Eubanks DC, Werner ME, Heppner TJ, Nelson MT. Calcium signaling in smooth muscle. Cold Spring Harb Perspect Biol. 2011;3:a004549.
Missiaen L, Taylor CW, Berridge MJ. Luminal Ca2+ promoting spontaneous Ca2+ release from inositol trisphosphate-sensitive stores in rat hepatocytes. J Physiol. 1992;455:623–40.
Mulvany MJ, Nilsson H, Flatman JA. Role of membrane potential in the response of rat small mesenteric arteries to exogenous noradrenaline stimulation. J Physiol. 1982;332:363–73.
Segal SS, Beny JL. Intracellular recording and dye transfer in arterioles during blood flow control. Am J Phys. 1992;263:H1–7.
von der Weid PY, Bény JL. Simultaneous oscillations in the membrane potential of pig coronary artery endothelial and smooth muscle cells. J Physiol. 1993;471:13–24.
Bartlett IS, Crane GJ, Neild TO, Segal SS. Electrophysiological basis of arteriolar vasomotion in vivo. J Vasc Res. 2000;37:568–75.
Haddock RE, Hill CE. Differential activation of ion channels by inositol 1,4,5-trisphosphate IP3- and ryanodine-sensitive calcium stores in rat basilar artery vasomotion. J Physiol. 2002;545:615–27.
Hill CE, Eade J, Sandow SL. Mechanisms underlying spontaneous rhythmical contractions in irideal arterioles of the rat. J Physiol. 1999;521:507–16.
Bouskela E, Grampp W. Spontaneous vasomotion in hamster cheek pouch arterioles in varying experimental conditions. Am J Phys. 1992;262:H478–85.
Isotani E, Zhi G, Lau KS, Huang J, Mizuno Y, Persechini A, Geguchadze R, Kamm KE, Stull JT. Real-time evaluation of myosin light chain kinase activation in smooth muscle tissues from a transgenic calmodulin-biosensor mouse. Proc Natl Acad Sci U S A. 2004;101:6279–84.
Ding HL, Ryder JW, Stull JT, Kamm KE. Signaling processes for initiating smooth muscle contraction upon neural stimulation. J Biol Chem. 2009;284:15541–8.
Gustafsson H, Nilsson H. 1993. Rhythmic contractions of isolated small arteries from rat: role of calcium. Acta Physiol Scand. 1993;149:283–91.
Gustafsson H, Mulvany MJ, Nilsson H. Rhythmic contractions of isolated small arteries from rat: influence of the endothelium. Acta Physiol Scand. 1993;143:153–63.
Lamont C, Wier WG. Different roles of ryanodine receptors and inositol (1,4,5)-trisphosphate receptors in adrenergically stimulated contractions of small arteries. Am J Physiol Heart Circ Physiol. 2004;287:H617–25.
Rahman A, Hughes A, Matchkov V, Nilsson H, Aalkjaer C. Antiphase oscillations of endothelium and smooth muscle [Ca2+]i in vasomotion of rat mesenteric small arteries. Cell Calcium. 2007;42:536–47.
Omote M, Mizusawa H. The role of sarcoplasmic reticulum in endothelium-dependent and endothelium-independent rhythmic contractions in the rabbit mesenteric artery. Acta Physiol Scand. 1993;149:15–21.
Westcott EB, Jackson WF. Heterogeneous function of ryanodine receptors, but not IP3 receptors, in hamster cremaster muscle feed arteries and arterioles. Am J Physiol Heart Circ Physiol. 2011;300:H1616–30.
Westcott EB, Goodwin EL, Segal SS, Jackson WF. Function and expression of ryanodine receptors and inositol 1,4,5-trisphosphate receptors in smooth muscle cells of murine feed arteries and arterioles. J Physiol. 2012;590:1849–69.
Matchkov VV, Aalkjaer C, Nilsson H. A cyclic GMP-dependent calcium-activated chloride current in smooth-muscle cells from rat mesenteric resistance arteries. J Gen Physiol. 2004;123:121–34.
Manoury B, Tamuleviciute A, Tammaro P. TMEM16A/anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J Physiol. 2010;588:2305–14.
Thomas-Gatewood C, Neeb ZP, Bulley S, Adebiyi A, Bannister JP, Leo MD, Jaggar JH. TMEM16A channels generate Ca2+-activated Cl− currents in cerebral artery smooth muscle cells. Am J Phys. 2011;301:H1819–27.
Adebiyi A, Zhao G, Narayanan D, Thomas-Gatewood CM, Bannister JP, Jaggar JH. Isoform-selective physical coupling of TRPC3 channels to IP3 receptors in smooth muscle cells regulates arterial contractility. Circ Res. 2010;106:1603–12.
Gonzales AL, Amberg GC, Earley S. Ca2+ release from the sarcoplasmic reticulum is required for sustained TRPM4 activity in cerebral artery smooth muscle cells. Am J Phys Cell Phys. 2010;299:C279–88.
Gonzales AL, Yang Y, Sullivan MN, Sanders L, Dabertrand F, Hill-Eubanks DC, Nelson MT, Earley S. A PLCγ1-dependent, force-sensitive signaling network in the myogenic constriction of cerebral arteries. Sci Signal. 2014;7:ra49.
Chen X, Yang D, Ma S, He H, Luo Z, Feng X, Cao T, Ma L, Yan Z, Liu D, Tepel M, Zhu Z. Increased rhythmicity in hypertensive arterial smooth muscle is linked to transient receptor potential canonical channels. J Cell Mol Med. 2010;14:2483–94.
Jackson WF. Oscillations in active tension in hamster aortas: role of the endothelium. Blood Vessels. 1988;25:144–56.
Mauban JR, Wier WG. Essential role of EDHF in the initiation and maintenance of adrenergic vasomotion in rat mesenteric arteries. Am J Physiol Heart Circ Physiol. 2004;287:H608–16.
Rahman A, Matchkov V, Nilsson H, Aalkjaer C. Effects of cGMP on coordination of vascular smooth muscle cells of rat mesenteric small arteries. J Vasc Res. 2005;42:301–11.
Dirnagl U, Lindauer U, Villringer A. Nitric oxide synthase blockade enhances vasomotion in the cerebral microcirculation of anesthetized rats. Microvasc Res. 1993;45:318–23.
Bertuglia S, Colantuoni A, Intaglietta M. Capillary reperfusion after L-arginine, L-NMMA, and L-NNA treatment in cheek pouch microvasculature. Microvasc Res. 1995;50:162–74.
Jackson WF, Mulsch A, Busse R. Rhythmic smooth muscle activity in hamster aortas is mediated by continuous release of NO from the endothelium. Am J Phys. 1991;260:H248–53.
Yamamoto Y, Klemm MF, Edwards FR, Suzuki H. Intercellular electrical communication among smooth muscle and endothelial cells in guinea-pig mesenteric arterioles. J Physiol. 2001;535:181–95.
Jacobsen JC, Aalkjaer C, Matchkov VV, Nilsson H, Freiberg JJ, Holstein-Rathlou NH. Heterogeneity and weak coupling may explain the synchronization characteristics of cells in the arterial wall. Philos Trans A Math Phys Eng Sci. 2008;366:3483–502.
Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, Tallini Y, Kotlikoff MI, Nelson MT. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci U S A. 2008;105:9627–32.
Segal SS. Integration and modulation of intercellular signaling underlying blood flow control. J Vasc Res. 2015;52:136–57.
Zang WJ, Balke CW, Wier WG. Graded α1-adrenoceptor activation of arteries involves recruitment of smooth muscle cells to produce ‘all or none’ Ca2+ signals. Cell Calcium. 2001;29:327–34.
Smith R, Imtiaz M, Banney D, Paul JW, Young RC. Why the heart is like an orchestra and the uterus is like a soccer crowd. Am J Obstet Gynecol. 2015;213:181–5.
Zhang J, Chen L, Raina H, Blaustein MP, Wier WG. In vivo assessment of artery smooth muscle [Ca2+]i and MLCK activation in FRET-based biosensor mice. Am J Physiol Heart Circ Physiol. 2010;299:H946–56.
Wang Y, Chen L, Wier WG, Zhang J. Intravital Förster resonance energy transfer imaging reveals elevated [Ca2+]i and enhanced sympathetic tone in femoral arteries of angiotensin II-infused hypertensive biosensor mice. J Physiol. 2013;591:5321–36.
Zacharia J, Mauban JR, Raina H, Fisher SA, Wier WG. High vascular tone of mouse femoral arteries in vivo is determined by sympathetic nerve activity via alpha1A- and alpha1D-adrenoceptor subtypes. PLoS One. 2013;8:e65969.
Funk W, Endrich B, Messmer K, Intaglietta M. Spontaneous arteriolar vasomotion as a determinant of peripheral vascular resistance. Int J Microcirc Clin Exp. 1983;2:11–25.
Bertuglia S, Colantuoni A, Coppini G, Intaglietta M. Hypoxia- or hyperoxia-induced changes in arteriolar vasomotion in skeletal muscle microcirculation. Am J Phys. 1991;260:H362–72.
Tsai AG, Intaglietta M. Evidence of flowmotion induced changes in local tissue oxygenation. Int J Microcirc Clin Exp. 1993;12:75–88.
Parthimos D, Edwards DH, Griffith TM. Comparison of chaotic and sinusoidal vasomotion in the regulation of microvascular flow. Cardiovasc Res. 1996;31:388–99.
Gratton RJ, Gandley RE, McCarthy JF, Michaluk WK, Slinker BK, McLaughlin MK. Contribution of vasomotion to vascular resistance: a comparison of arteries from virgin and pregnant rats. J Appl Physiol. 1998;85:2255–60.
Rücker M, Strobel O, Vollmar B, Roesken F, Menger MD. Vasomotion in critically perfused muscle protects adjacent tissues from capillary perfusion failure. J Physiol Heart Circ Physiol. 2000;279:H550–8.
Meyer C, de Vries G, Davidge ST, Mayes DC. Reassessing the mathematical modeling of the contribution of vasomotion to vascular resistance. J Appl Physiol. 2002;92:888–9.
Thorn CE, Kyte H, Slaff DW, Shore AC. An association between vasomotion and oxygen extraction. Am J Physiol Heart Circ Physiol. 2011;301:H442–9.
Di Marco LY, Farkas E, Martin C, Venneri A, Frangi AF. Is vasomotion in cerebral arteries impaired in Alzheimer’s disease? J Alzheimers Dis. 2015;46:35–53.
Tarantini S, Tran CHT, Gordon GR, Ungvari Z, Csiszar A. Impaired neurovascular coupling in aging and Alzheimer’s disease: contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp Gerontol. 2017;94:52–8.
Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–76.
Lecrux C, Hamel E. The neurovascular unit in brain function and disease. Acta Physiol (Oxford). 2011;203:47–59.
Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron. 2017;96:17–42.
Ogawa S, Lee TM, Nayak AS, Glynn P. Oxygenation-sensitive contrast in magnetic resonance image of rodent brain at high magnetic fields. Magn Reson Med. 1990;14:68–78.
Ogawa S, Lee TM, Kay AR, Tank DW. Brain magnetic resonance imaging with contrast dependent on blood oxygenation. Proc Natl Acad Sci U S A. 1990;87:9868–72.
Chen BR, Bouchard MB, McCaslin AF, Burgess SA, Hillman EM. High-speed vascular dynamics of the hemodynamic response. NeuroImage. 2011;54:1021–30.
Nir Y, Hasson U, Levy I, Yeshurun Y, Malach R. Widespread functional connectivity and fMRI fluctuations in human visual cortex in the absence of visual stimulation. NeuroImage. 2006;30:1313–24.
Fox MD, Raichle ME. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nat Rev Neurosci. 2007;8:700–11.
Bruyns-Haylett M, Harris S, Boorman L, Zheng Y, Berwick J, Jones M. The resting-state neurovascular coupling relationship: rapid changes in spontaneous neural activity in the somatosensory cortex are associated with haemodynamic fluctuations that resemble stimulus-evoked haemodynamics. Eur J Neurosci. 2013;38:2902–16.
Liu TT. Neurovascular factors in resting-state functional MRI. NeuroImage. 2013;80:339–48.
Mayhew JEW, Askew S, Zheng Y, Porrill J, Westby GWM, Redgrave P, Rector DM, Harper RM. Cerebral vasomotion: a 0.1-Hz oscillation in reflected light imaging of neural activity. NeuroImage. 1996;4:183–93.
Obrig H, Neufang M, Wenzel R, Kohl M, Steinbrink J, Einhäupl K, Villringer A. Spontaneous low frequency oscillations of cerebral hemodynamics and metabolism in human adults. NeuroImage. 2000;12:623–39.
Logothetis NK. The neural basis of the blood-oxygen-level-dependent functional magnetic resonance imaging signal. Philos Trans R Soc Lond B. 2002;357:1003–37.
Logothetis NK. The underpinnings of the BOLD functional magnetic resonance imaging signal. J Neurosci. 2003;23:3963–71.
Logothetis NK. What we can do and what we cannot do with fMRI. Nature. 2008;453:869–78.
Lauritzen M. Reading vascular changes in brain imaging: is dendritic calcium the key? Nat Rev Neurosci. 2005;6:77–85.
Devor A, Sakadžić S, Srinivasan VJ, Yaseen MA, Nizar K, Saisan PA, Tian P, Dale AM, Vinogradov SA, Franceschini MA, Boas DA. Frontiers in optical imaging of cerebral blood flow and metabolism. J Cereb Blood Flow Metab. 2012;32:1259–76.
Mishra A, Kurth-Nelson Z, Hall C, Howarth C. Interpreting BOLD: a dialogue between cognitive and cellular neuroscience. Theo Murphy Meeting Themed Issue. Philos Trans R Soc B. 2016;371(1705):20150348–61.
Logothetis NK, Pauls J, Augath M, Trinath T, Oeltermann A. Neurophysiological investigation of the basis of the fMRI signal. Nature. 2001;412:150–7.
Niessing J, Ebisch B, Schmidt KE, Niessing M, Singer W, Galuske RA. Hemodynamic signals correlate tightly with synchronized gamma oscillations. Science. 2005;309:948–51.
Nir Y, Mukamel R, Dinstein I, Privman E, Harel M, Fisch L, Gelbard-Sagiv H, Kipervasser S, Andelman F, Neufeld MY, Kramer U, Arieli A, Fried I, Malach R. Interhemispheric correlations of slow spontaneous neuronal fluctuations revealed in human sensory cortex. Nat Neurosci. 2008;11:1100–8.
He BJ, Raichle ME. The fMRI signal, slow cortical potential and consciousness. Trends Cogn Sci. 2009;13:302–9.
Henrie JA, Shapley R. LFP power spectra in V1 cortex: the graded effect of stimulus contrast. J Neurophysiol. 2005;94:479–90.
Pesaran B, Pezaris JS, Sahani M, Mitra PP, Andersen RA. Temporal structure in neuronal activity during working memory in macaque parietal cortex. Nat Neurosci. 2002;5:805–11.
Bauer EP, Paz R, Paré D. Gamma oscillations coordinate amygdalo-rhinal interactions during learning. J Neurosci. 2007;27:9369–79.
Longden T, Dabertrand F, Koide M, Gonzales AL, Tykocki NR, Brayden JE, Hill-Eubanks D, Nelson MT. Capillary K+-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat Neurosci. 2017;20:717–26.
Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O’Farrell FM, Buchan AM, Lauritzen M, Attwell D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508:55–60.
Jackson WF. Boosting the signal: Endothelial inward rectifier K+ channels. Microcirculation. 2017;24:e12319.
Tallini YN, Brekke JF, Shui B, Doran R, Hwang SM, Nakai J, Salama G, Segal SS, Kotlikoff MI. Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: measurements in Cx40BAC–GCaMP2 transgenic mice. Circ Res. 2007;101:1300–9.
Chen BR, Kozberg MG, Bouchard MB, Shaik MA, Hillman EMC. A critical role of the vascular endothelium in functional neurovascular coupling in the brain. J Am Heart Assoc. 2014;3:e000787.
Sinkler SY, Segal SS. Rapid versus slow ascending vasodilatation: intercellular conduction versus flow-mediated signalling with tetanic versus rhythmic muscle contractions. J Physiol. 2017;595:7149–65.
Bernardi L, Rossi M, Fratino P, Finardi G, Mevio E, Orlandi C. Relationship between phasic changes in human skin blood flow and autonomic tone. Microvasc Res. 1989;37:16–27.
Borovik A, Golubinskaya V, Tarasova O, Aalkjaer C, Nilsson H. Phase resetting of arterial vasomotion by burst stimulation of perivascular nerves. J Vasc Res. 2005;42:165–73.
Bernardi L, Rossi M, Leuzzi S, Mevio E, Fornasari G, Calciati A, Orlandi C, Fratino P. Reduction of 0.1 Hz microcirculatory fluctuations as evidence of sympathetic dysfunction in insulin-dependent diabetes. Cardiovasc Res. 1997;34:185–91.
Meyer MF, Rose CJ, Hülsmann JO, Schatz H, Pfohl M. Impaired 0.1-Hz vasomotion assessed by laser Doppler anemometry as an early index of peripheral sympathetic neuropathy in diabetes. Microvasc Res. 2003;65:88–95.
Acknowledgements
The authors thank Drs. HL Zhu and XZ Zhong for the original recordings of vasomotion presented in Fig. 12.1. The work is supported by a research operating grant from the Canadian Institutes of Health Research (PJT-155963).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Cole, W.C., Gordon, G.R., Braun, A.P. (2019). Cellular and Ionic Mechanisms of Arterial Vasomotion. In: Hashitani, H., Lang, R. (eds) Smooth Muscle Spontaneous Activity. Advances in Experimental Medicine and Biology, vol 1124. Springer, Singapore. https://doi.org/10.1007/978-981-13-5895-1_12
Download citation
DOI: https://doi.org/10.1007/978-981-13-5895-1_12
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-5894-4
Online ISBN: 978-981-13-5895-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)