
Abstract
Gabapentinoids are effective in a wide range of animal pain models and in patients with neuropathic pain and has become one of first-line treatments for neuropathic pain. Because spinal plasticity and sensitization have been intensely studied in neuropathic pain, most laboratory studies have focused on actions of gabapentinoids in the spinal cord, where they reduce primary afferent traffic and excitation of spinal nociceptive neurons, via interaction with α2δ subunits of voltage-gated Ca2+ channels. However, a recent clinical study questioned the relevance of this in vitro and in vivo rodent studies by demonstrating a complete lack of clinical efficacy of intrathecal gabapentin in patients with chronic pain. Curiously, preclinical studies continue to focus on spinal cord actions of gabapentinoids despite this lack of translation to humans.
We and others demonstrated that gabapentin inhibits presynaptic GABA release and induces glutamate release from astrocytes in the locus coeruleus (LC), thereby increasing LC neuron activity and spinal noradrenaline release, and that gabapentin relies on this action in the LC for its analgesia. We also recently discovered that, with prolonged time after neuropathic injury, noradrenergic neurons in the LC become less responsive to gabapentin, leading to impaired gabapentin analgesia, and that astroglial glutamate dysregulation is critical to this impaired LC response. The clinically available drug valproate increases glutamate transporter-1 (GLT-1) expression in the LC to restore this impaired gabapentin analgesia.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
8.1 Introduction
Peripheral nerve injury can result in neuropathic pain, hyperalgesia, and allodynia which respond poorly to nonsteroidal anti-inflammatory drugs. Although opioids are effective acutely [15], their chronic use is complicated by tolerance and therapy-limiting side effects, and hence alternatives to opioids have been sought for decades. Only a few of these have shown efficacy in the clinic.
Gabapentin was licensed as an antiepileptic drug in 1993 and was rapidly recognized to be effective to reduce pain in patients with neuropathic pain. This clinical experience is paralleled in the laboratory, where gabapentin has been shown to possess analgesic properties in a wide range of neuropathic pain models [7, 9, 13, 19]. Gabapentin interacts with the α2δ subunit of voltage-gated calcium channels, which modulate the release of excitatory amino acids at the level of the spinal dorsal horn [3]. α2δ subunits are upregulated in the spinal cord and primary sensory afferents after nerve injury in rodents [11], and gabapentin blocks voltage-gated calcium channels in transgenic mice with upregulated α2δ-1 subunits but not in normal mice [10], suggesting efficacy of gabapentin depends on this subunit. For this reason, most laboratory studies have focused on the theory that gabapentin primarily acts on spinal pain mechanisms. However, a recent clinical study questioned this theory by demonstrating a complete lack of clinical efficacy of intrathecal gabapentin in patients with noncancer pain [16], despite the known efficacy of oral gabapentin in this patient population.
Tanabe et al. first reported the involvement of the locus coeruleus (LC)-spinal descending noradrenergic pathway in gabapentin analgesia by demonstrating that depletion or blockade of spinal noradrenergic signaling abolished the anti-hypersensitivity effect of systemically administered gabapentin in mice after peripheral nerve injury [19]. Similar behavioral results have also been reported in various rodent models of neuropathic and postoperative pain after systemic, intra-cerebroventricular, or intra-LC administration of gabapentin [6, 7, 9, 13]. Consistent with these behavioral observations, we demonstrated that systemically administered gabapentin increases phosphorylated cAMP response element-binding protein expression, as a measure of neuronal activation, in LC neurons and subsequently induces spinal noradrenaline release in rats 2–3 weeks after spinal nerve ligation (SNL) [7]. Gabapentin likely acts similarly in humans, since its oral administration, in a dose that produces postoperative analgesia, increases noradrenaline concentration in cerebrospinal fluid from patients scheduled for orthopedic surgery for painful degenerative joint disease (Fig. 8.1) [6]. Together, these laboratory and clinical observations suggest that gabapentin recruits descending inhibition and that this inhibition plays a key role in gabapentin-induced analgesia.

On the day of orthopedic surgery, patients with chronic, painful degenerative joint disease were randomly assigned to receive oral 1200 mg gabapentin (GBP) or placebo approximately 90 min before collecting cerebrospinal fluid (CSF) samples for noradrenaline (NA) measurement (left panel) and the amounts of opioid (right panel) administered in the first 24 h. (Adapted from Hayashida et al. [6])
This mini-review discusses the mechanisms by which gabapentin activates the LC for analgesia, how gabapentin loses its efficacy during chronification of neuropathic pain, and pharmacologic approaches to restore or enhance gabapentin analgesia in chronic neuropathic pain.
8.2 Mechanisms of LC Activation by Gabapentin
Despite its name and structural similarity to GABA, gabapentin does not have direct effects on GABA receptors. However, analgesic effects of gabapentin may reflect modulation of GABA release. Gabapentin’s actions on GABA release are controversial and likely depend on the site in the central nervous system. For example, gabapentin does not affect GABA release from spinal dorsal horn synaptosomes in rats [23]. Some studies show an increase in GABA release in rat and human brain from gabapentin [4, 14, 21], whereas other studies show a direct reduction in GABA release when rat cortical synaptosomes are exposed to gabapentin [1]. In the LC, gabapentin and other α2δ ligands reduce presynaptic GABA release and thereby disinhibit noradrenergic neurons in rodents [18, 23], suggesting that gabapentin reduces the influence of GABA in the LC as one of the mechanisms by which it activates descending inhibition.
In rats 2–3 weeks after SNL surgery, gabapentin-induced analgesia and activation of LC neurons were blocked by local application of a AMPA glutamate receptor antagonist [7]. This suggests that, other than reducing the influence of GABA, gabapentin also relies on glutamate-dependent mechanisms in the LC to activate descending inhibition. In vivo microdialysis, systemic or local perfusion of gabapentin in rats consistently increases extracellular glutamate concentrations in the LC but not in the spinal cord [17]. In addition to its direct inhibition on LC neurons via postsynaptic GABAA receptors, GABA is known to inhibit glutamate release via presynaptic GABAB receptors [5, 20]. However, blockade of GABAB receptors in the LC affects neither basal glutamate levels nor the gabapentin-induced glutamate increase in rats [17], suggesting that tonic influence of GABA on glutamatergic terminals via presynaptic GABAB receptors is either minor or absent in the LC and that gabapentin’s effect on the glutamate levels is independent of GABA-mediated mechanisms.
We proposed that gabapentin increases extracellular glutamate in the LC via actions on the glutamate transporter-1 (GLT-1) in astroglia based on our observations that gabapentin and its related α2δ ligand pregabalin increase co-transport of Na+ ions and glutamate via glutamate transporters and enhance glutamate-induced intracellular Ca2+ response via the reverse mode of Na+-Ca2+ exchange and by these mechanisms facilitate glutamate release in cultured astrocytes [22]. We also showed the in vivo relevance of gabapentin’s action in astrocytes, by demonstrating in rats 2–3 weeks after SNL that selective blockade or knockdown of GLT-1 abolished gabapentin’s effects on glutamate increase in the LC and on gabapentin-induced anti-hypersensitivity [8, 17]. Since there is no evidence for the presence of α2δ subunits in astrocytes, α2δ interactions probably are not involved in the effect of gabapentin on astrocytes. This is further supported by our observation that pregabalin increases extracellular glutamate concentrations in the LC, whereas 3-exo-aminobicyclo[2.2.1]heptane-2-exo-carboxylic acid does not, despite its high binding affinity to α2δ subunits [12, 17].
Taken together, these results suggest that gabapentin inhibits presynaptic GABA release and induces glutamate release from astrocytes in the LC, thereby increasing LC neuronal activity and spinal noradrenaline release, and that gabapentin relies on this action in the LC for its analgesia, at least during the early phase (2–3 weeks after nerve injury) of neuropathic pain (Fig. 8.2).

Proposed mechanisms of gabapentin (GBP) action in the locus coeruleus (LC). GBP interacts with the α2δ subunit of voltage-gated calcium channels (VGCC) to reduce presynaptic GABA release and activates glutamate transferase-1 (GLT-1)-dependent mechanisms to induce glutamate (Glu) release from astrocytes in the LC, thereby increasing LC neuronal activity and spinal noradrenaline (NA) release to recruit descending inhibition
8.3 Impaired Gabapentin Analgesia in Chronic Neuropathic Pain
In various neuropathic pain models in rodents, gabapentin is remarkably and uniformly effective [7, 9, 11, 19]. However, gabapentin often fails to provide sufficient analgesia in patients with neuropathic pain [2]. In the past two decades, pain research has primarily focused on the mechanisms of clinical efficacy of gabapentin, but less attention has paid to understand why all rodents, but only some patients respond to gabapentin. From a public health perspective, understanding mechanisms of this heterogeneity in response and how efficacy might be improved is arguably as important as understanding therapeutic mechanisms.
We demonstrated that the early uniform anti-hypersensitivity effect of gabapentin in rats after SNL is lost as time progresses after surgery (Fig. 8.3) [8]. For a large effect size to reduce hypersensitivity (>30 g), this is uniformly lost around 5 weeks after injury, and for smaller effect sizes (>10 g), it is lost in nearly half of the animals 10 weeks after injury. This questions the relevance of much of the previous laboratory studies that were performed within the first 2–4 weeks of surgical injury, to clinical use of gabapentin in long-standing pain, and suggests that future studies aimed to better understand individual lack of response to gabapentin should focus on times more remote than 2–4 weeks after injury.

Upper panel: withdrawal thresholds in the ipsilateral hindpaw to spinal nerve ligation (SNL) were measured 1 h after an intraperitoneal injection of saline (SAL) or gabapentin (GBP, 100 mg/kg) in rats 0–10 weeks after SNL(upper right graph). Data (mean ± SD) are presented as change from baseline.*P < 0.05 vs. 2 weeks after SNL. #P < 0.05 vs. SAL. Lower panel: the likelihood, assessed by logistic regression, that gabapentin (GBP) will produce an increase in mechanical withdrawal threshold of three levels from baseline hypersensitive level (30 g = strong effect, 20 g = moderate effect, or 10 g = weak effect) as a function of time after spinal nerve ligation (SNL) injury. (Adapted from Kimura et al. [8])
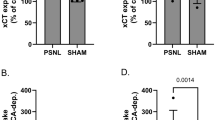
Given that expression of GLT-1 in the LC is essential to gabapentin’s analgesia and that GLT-1 is downregulated in the LC 6 weeks after SNL [8, 17], we recently hypothesized that downregulation of GLT-1 results in reduced gabapentin efficacy over time after nerve injury. Consistent with this hypothesis, inhibition of histone deacetylase (HDAC) by the clinically available drug valproate restores downregulated GLT-1 expression in the LC 8 weeks after SNL (Fig. 8.4a) and also restores the anti-hypersensitivity effect of gabapentin (Fig. 8.4b). This effect of valproate is reversed by the knockdown of GLT-1 in the LC. These results strongly suggest not only a causal relationship between GLT-1 expression in the LC and the anti-hypersensitivity effect of gabapentin but also a possible treatment strategy to restore impaired gabapentin analgesia using HDAC inhibitors in chronic neuropathic pain.

(a) Representative Western blotting images and quantification of GLT-1 in the LC from normal and SNL rats treated with oral vehicle or valproate (200 mg/5 mL/kg/day) for 14 days starting from 6 weeks after SNL surgery. Data (mean + SD) are presented as ratio to α-tubulin. (b) Vehicle or valproate-treated animals with or without repeated intra-LC injections of the control (8.3 pmol/0.5 μl) or GLT-1 (8.3 pmol/ 0.5 μl) siRNA for the last 5 days received an intraperitoneal gabapentin (100 mg/kg). Withdrawal thresholds in the hindpaw ipsilateral to SNL were measured 1 h after gabapentin administration. Data (mean + SD) are presented as change from baseline. (Adapted from Kimura et al. [8])
8.4 Conclusion
Although interactions with α2δ subunits of voltage-gated calcium channels are shown to be essential for gabapentin’s effects in rodents [10, 11], the lack of efficacy of intrathecal gabapentin in patients [16] questions whether gabapentin relies exclusively on spinal actions for its analgesic efficacy. We demonstrated that gabapentin inhibits presynaptic GABA release and induces glutamate release from astrocytes in the LC, thereby increasing LC neuron activity and spinal noradrenaline release, and that gabapentin relies on this action in the LC for its analgesia, at least during the early period after neuropathic injury. However, when neuropathic pain becomes chronic, noradrenergic neurons in the LC become less responsive to gabapentin, leading to impaired gabapentin analgesia. Downregulation of astroglial GLT-1 in the LC is critical to this impaired response of the LC to gabapentin. Oral valproate increases GLT-1 expression in the LC, possibly via HDAC inhibition, to restore the impaired anti-hypersensitivity effect of gabapentin. Given clinical availability and established safety profiles, valproate should be tested to rescue gabapentin efficacy in those who initially fail to respond to gabapentin.
References
Brawek B et al (2009) Effects of gabapentin and pregabalin on K+−evoked 3H-GABA and 3H-glutamate release from human neocortical synaptosomes. Naunyn Schmiedeberg’s Arch Pharmacol 379:361–369
Finnerup NB et al (2015) Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 14:162–173
Gee NS et al (1996) The novel anticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J Biol Chem 271:5768–5776
Gotz E et al (1993) Effects of gabapentin on release of gamma-aminobutyric acid from slices of rat neostriatum. Arzneimittelforschung 43:636–638
Harte M, O’Connor WT (2005) Evidence for a selective prefrontal cortical GABA(B) receptor-mediated inhibition of glutamate release in the ventral tegmental area: a dual probe microdialysis study in the awake rat. Neuroscience 130:215–222
Hayashida K et al (2007) Gabapentin activates spinal noradrenergic activity in rats and humans and reduces hypersensitivity after surgery. Anesthesiology 106:557–562
Hayashida K et al (2008) Gabapentin acts within the locus coeruleus to alleviate neuropathic pain. Anesthesiology 109:1077–1084
Kimura M, Eisenach JC, Hayashida K (2016) Gabapentin loses efficacy over time after nerve injury in rats: role of glutamate transporter-1 in the locus coeruleus. Pain 157:2024–2032
Kitamura R et al (2014) Gabapentin inhibits bortezomib-induced mechanical allodynia through supraspinal action in mice. J Pharmacol Sci 124:502–510
Li CY et al (2006) Calcium channel alpha2delta1 subunit mediates spinal hyperexcitability in pain modulation. Pain 125:20–34
Luo ZD et al (2002) Injury type-specific calcium channel alpha 2 delta-1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. J Pharmacol Exp Ther 303:1199–1205
Lynch JJ 3rd et al (2006) (L)-Phenylglycine, but not necessarily other alpha2delta subunit voltage-gated calcium channel ligands, attenuates neuropathic pain in rats. Pain 125:136–142
Miyazaki R, Yamamoto T (2012) The efficacy of morphine, pregabalin, gabapentin, and duloxetine on mechanical allodynia is different from that on neuroma pain in the rat neuropathic pain model. Anesth Analg 115:182–188
Petroff OA et al (1996) The effect of gabapentin on brain gamma-aminobutyric acid in patients with epilepsy. Ann Neurol 39:95–99
Raja SN et al (2002) Opioids versus antidepressants in postherpetic neuralgia: a randomized, placebo-controlled trial. Neurology 59:1015–1021
Rauck R et al (2013) Intrathecal gabapentin to treat chronic intractable noncancer pain. Anesthesiology 119:675–686
Suto T et al (2014) Gabapentin increases extracellular glutamatergic level in the locus coeruleus via astroglial glutamate transporter-dependent mechanisms. Neuropharmacology 81:95–100
Takasu K, Ono H, Tanabe M (2008) Gabapentin produces PKA-dependent pre-synaptic inhibition of GABAergic synaptic transmission in LC neurons following partial nerve injury in mice. J Neurochem 105:933–942
Tanabe M et al (2005) Role of descending noradrenergic system and spinal alpha2-adrenergic receptors in the effects of gabapentin on thermal and mechanical nociception after partial nerve injury in the mouse. Br J Pharmacol 144:703–714
Tanaka S et al (2003) GABAergic modulation of hippocampal glutamatergic neurons: an in vivo microdialysis study. Eur J Pharmacol 465:61–67
Taylor CP et al (1998) A summary of mechanistic hypotheses of gabapentin pharmacology. Epilepsy Res 29:233–249
Yoshizumi M, Eisenach JC, Hayashida K (2012a) Riluzole and gabapentinoids activate glutamate transporters to facilitate glutamate-induced glutamate release from cultured astrocytes. Eur J Pharmacol 677:87–92
Yoshizumi M et al (2012b) Gabapentin inhibits gamma-amino butyric acid release in the locus coeruleus but not in the spinal dorsal horn after peripheral nerve injury in rats. Anesthesiology 116:1347–1353
Acknowledgments
This work was supported by grants DA27690 from the National Institutes of Health (Bethesda, Maryland, USA) and KAKENHI 16K08985 from the Japan Society for the Promotion of Science (Tokyo, Japan) to K. H.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Hayashida, Ki., Eisenach, J.C. (2018). Descending Noradrenergic Inhibition: An Important Mechanism of Gabapentin Analgesia in Neuropathic Pain. In: Shyu, BC., Tominaga, M. (eds) Advances in Pain Research: Mechanisms and Modulation of Chronic Pain. Advances in Experimental Medicine and Biology, vol 1099. Springer, Singapore. https://doi.org/10.1007/978-981-13-1756-9_8
Download citation
DOI: https://doi.org/10.1007/978-981-13-1756-9_8
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-1755-2
Online ISBN: 978-981-13-1756-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)