
Abstract
Cumulative evidence indicates that cortical synapses not only play important roles in pain perception and related emotional functions but also undergo long-term potentiation (LTP) and contribute to chronic pain. LTP is found at two key cortical regions such as the anterior cingulate cortex (ACC) and insular cortex (IC), and inhibition of cortical LTP produces analgesic effects as well as anxiolytic effects. In this chapter, I will summarize our work on ACC and IC and provide evidence for calcium-stimulated AC1 as a key molecule for cortical LTP and chronic pain.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
13.1 Introduction
Chronic pain is a major health issue all over the world and is caused by tissue or nerve injury under different disease conditions. Due to poor understanding of the molecular mechanisms of chronic pain, especially at central regions, current pain medicines are ineffective. Consequently, patients mostly depend on the use of opioids to control pain. While opioids fail to erase chronic pain, they only produce analgesic effects by nonselectively inhibiting synaptic transmission. The long-term use of opioids has caused widespread drug abuse problems. In this chapter, I will discuss previous and recent discoveries related to the cortical mechanism of chronic pain and propose LTP as a key synaptic mechanism for chronic pain.
13.2 ACC and IC
Human and animal studies have consistently demonstrated that neurons in the anterior cingulate cortex (ACC) and insular cortex (IC) are two key cortical regions for pain perception and chronic pain [1, 2, 13, 14, 24, 26, 28, 29, 30]. Activation of the ACC and IC has been reported to be caused by various noxious heat, cold, and chemical stimuli. In animal models of chronic pain, activity-dependent immediate early genes have been reported to be activated in these two areas after peripheral inflammation or nerve injury. In genetic knockout mice in which chronic pain has been significantly reduced, these immediate early genes are also reduced in the ACC and IC [18, 20, 25]. Electrophysiological experiments have provided direct evidence that neurons in the ACC and IC are activated by noxious stimuli and most of these cells are likely excitatory pyramidal neurons [21, 25]. Moreover, activation of the ACC causes fearful memory, supporting the roles of ACC in the unpleasantness of pain [16]. In human brain imaging experiments, it has also been found that ACC and/or IC regions are triggered by psychological pain and social exclusion, providing further evidence for their importance in the process of pain [2]. Furthermore, biochemical and anatomic studies have indicated that plasticity-related signaling pathways are activated in the ACC and IC after peripheral injuries. It is believed that cortical synapses undergo long-term plastic changes after peripheral injuries.
Recently, activation of the ACC induced by peripheral nerve injury increases the turnover of specific synaptic proteins in a persistent manner. Ko et al. [5] demonstrate that neural cell adhesion molecule 1 (NCAM1) is one of the molecules involved and show that it mediates spine reorganization and contributes to the behavioral sensitization.
13.3 Long-Term Potentiation (LTP) in the ACC and IC
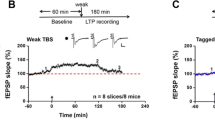
LTP can be readily induced using a variety of experimental methods, including field excitatory postsynaptic potential (EPSP) recordings, whole-cell patch-clamp recordings, and multielectrode array (MEA) recordings. Field recordings in slices from adult mice have shown that glutamatergic synapses in the ACC exhibit LTP lasting many hours in response to theta burst stimulation (TBS). The activation of NMDA receptors and L-type voltage-gated calcium channels (L-VGCCs) are required for ACC LTP [25]. ACC LTP can also be induced using other LTP inducing protocols such as stimulus-depolarization pairing and spike-EPSP pairing. In whole-cell patch-clamp recording experiments, LTP induced by different pairing protocols is typically NMDA receptor-dependent. Activation of L-VGCCs is not required.
NMDA receptors comprise a variety of subtypes that are composed of different combinations of subunits, typically two GluN1 (NR1) subunits and two GluN2 (NR2) subunits, of which there are four possible subtypes, GluN2A-D (NR2A-D). In the ACC, it has been shown that LTP, as detected by whole-cell patch-clamp recording, is sensitive to both GluN2A-preferring and GluN2B- preferring antagonists, indicating that tri-heteromers of the NMDA receptor may also be the dominant form of the receptor that contributes to LTP at ACC.
The activation of NMDA receptors leads to an increase in Ca2+ levels in dendritic spines, owing to Ca2+ entry through NMDA receptors and a consequent Ca2+-stimulated release of Ca2+ from intracellular stores [1]. The postsynaptic Ca2+ signal is an essential component for the induction of LTP. Electroporation studies indicate that activation of CaM-dependent signaling pathways by Ca2+ binding is essential for ACC LTP. The expression of ACC LTP requires AMPA receptor GluA1 subunit. Pharmacological experiments show that allocation of a peptide that mimics the PDZ domain at the C-terminal tail of the GluA1 subunit blocked the early expression of LTP. By contrast, peptides that interfere with interactions with the C-terminal tail of GluA2 (GluR2) or GluA3 (GluR3) do not have any effect. Experiments using genetic knockout mice further support this conclusion. ACC LTP is normal in mice lacking GluA2, whereas it is absent in mice lacking GluA1. Furthermore, a CP-AMPAR antagonist applied at 5 min after the induction of ACC LTP significantly reduced the potentiation. Recent evidence suggests that CP-AMPARs are specifically associated with the PKA-dependent form of LTP and PKA phosphorylation site at serine 845 plays critical roles in ACC LTP [15]. Finally, administration of an inhibitor of PKMζ, zeta inhibitory peptide (ZIP), abolished LTP in the ACC [8] (Fig. 13.1).

Signaling pathways for the induction and expression of two major forms of LTP in the ACC. For NMDA receptor-dependent LTP, neural activity triggered the release of excitatory neurotransmitter glutamate (Glu: filled circles) in the ACC synapses. Activation of glutamate NMDA receptors leads to an increase in postsynaptic Ca2+ in dendritic spines. Both NMDA NR2B and NR2A subunits are important for NMDA receptor functions. Ca2+ serves as an important intracellular signal for triggering a series of biochemical events that contribute to the expression of LTP. Ca2+ binds to CaM and leads to activation of calcium-stimulated AC1 as well as Ca2+-/CaM-dependent protein kinases (PKC, CaMKII, and CaMKIV). Through various protein-kinase-related intracellular signaling pathways, the trafficking of postsynaptic AMPA receptor as well as other synaptic modifications contributes to enhanced synaptic responses. Activation of CaMKIV, a kinase predominantly expressed in the nuclei, will trigger CREB signaling pathways. In addition, activation of AC1 leads to activation of PKA and subsequently CREB as well. For NMDA receptor-independent LTP, neural activity triggered the release of Glu and subsequent activation of presynaptic kainate receptor. Activation of kainate receptor causes the influx of Ca2+ to presynaptic terminal and activation of AC1 as well. Ca2+ binds to CaM and leads to activation of calcium-stimulated AC1. Activation of PKA may contribute to the enhancement of glutamate release
Although there are currently less studies of LTP in the IC, similar synaptic mechanisms are likely to be involved [11, 30].
13.3.1 Presynaptic LTP (Pre-LTP)
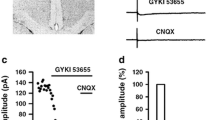
Recent studies show that another form of ACC LTP (called pre-LTP), an NMDA receptor-independent form of LTP, can also be induced by using paired-pulse low-frequency stimulation. The activation of kainate receptors is critical for the induction. In mice lacking the GluK2 subunit, this pre-LTP is blocked. Furthermore, pre-LTP is also blocked by a potent GluK1-selective kainate receptor antagonist, UBP31060. The activation of the cAMP signaling pathway contributes to the induction of pre-LTP in the ACC neurons. Activation of calcium-stimulated adenylyl cyclase subtype 1 (AC1), but not AC8, is selectively required for ACC pre-LTP [6, 7] and IC [22]. Kainate receptor-dependent pre-LTP in the ACC may also involve FMRP signaling [6, 7].
For the expression of LTP, kainate receptor-dependent LTP is expressed by an increase in the probability of release, P(r), as assessed by changes in paired-pulse facilitation. LTP involves the modulation of hyperpolarization-activated cyclic nucleotide-gated (HCN) channels leading to a persistent depolarization of presynaptic terminals, which could account for the increase in P(r). There are four HCN channel subunits (HCN1–4), all of which are expressed in both ACC and thalamus.
13.4 AC1 Acts as a Key Molecule for Triggering LTP
Cyclic adenosine monophosphate (cAMP) is a key intracellular second messenger. The cAMP signaling pathway contributes to learning and memory, chronic pain, emotional fear, and drug abuse. The AC is the enzyme that catalyzes ATP to cAMP. There are two major families of ACs: nine membrane-bound (AC1-9) and one soluble form of AC (sAC). Subtypes of ACs show unique organ and cellular distributions, and the mechanisms leading to the activation of them are different for subtypes of ACs. These observations support possible distinct physiological functions of each AC isoform in biological systems. Among more than ten subunits, AC1 and AC8 are two of the AC subtypes that respond positively to calcium-calmodulin (CaM). AC1 is more sensitive to a calcium increase than AC8 (more than eight times), indicating that AC1 could play more important role in the production of cAMP. Anatomic studies found that AC1 is highly expressed in cingulate neurons and located in most of the layers of the ACC [20]. Genetic studies using mice lacking AC1 show that gene deletion of AC1 selectively impair ACC LTP, while basal excitatory glutamate transmission in the ACC is not affected. LTP induced by TBS or pairing stimulation is abolished in cingulate pyramidal cells of AC1 knockout mice [9]. By using chemical and biochemical screening, selective chemical inhibitors of AC1 have been identified [17]. In consistent with results from genetic knockout mice, pharmacological inhibition of AC1 in ACC neurons abolishes LTP induced by pairing training [3, 17].
Our recent data also found that AC1 is essential for the induction of late-phase LTP (L-LTP) in the ACC synapses [3]. While in wild-type mice, TBS induced L-LTP that lasted for at least 3–6 h. TBS failed to induce any significant potentiation in ACC slices of AC1 KO mice. Since AC1 is a neuronal selective form of ACs, it is likely that AC1 activity may also contribute to LTP in other pain-related cortical areas such as the prefrontal cortex (PFC), insular cortex, and somatosensory cortex. It has been reported that AC1 activity is required for injury-activated immediate early gene activity in these cortical areas. LTP has been reported in the PFC, somatosensory cortex and IC. Recent studies have reported that AC1 contributes to LTP in the IC [30].
13.5 Requirement of AC1 for Behavioral Sensitization and Spinal Enhancement
Behavioral studies using AC1 knockout (KO) mice have demonstrated that AC1 contributes to chronic pain [20]. Behavioral responses to peripheral injection of two inflammatory stimuli, formalin and complete Freund’s adjuvant, were reduced or abolished in AC1 KO mice. However, wild-type and AC1 KO mice were indistinguishable in behavioral tests of acute pain. Using activity-dependent immediate early genes as markers, AC1 is also found to contribute to inflammation-induced activation of CREB. In addition to sensory inputs from the skin, behavioral nociceptive responses in animal models of muscle pain or chronic muscle inflammatory pain were significantly reduced in AC1 KO mice.
The possible roles of AC1 in chronic pain-related plasticity are also reported at areas outside of the cortex. In the spinal cord, AC1 activity is found to be critical for 5-HT-induced spinal facilitation and synaptic plasticity. Application of a low dose of serotonin (5-HT) alone or a co-application of forskolin with 5-HT produced long-lasting facilitation of excitatory synaptic transmission between primary afferent fibers and dorsal horn neurons. This enhancement requires the recruitment or trafficking of functional postsynaptic AMPA receptors. AC1 is required for 5-HT-induced enhancement using AC1 KO mice. In addition, AC1 also contributes to the activation of the extracellular signal-regulated kinase (Erk) in spinal cord dorsal horn neurons, suggesting that AC1 may link upstream signals to long-term gene regulation and protein synthesis that are linked to synaptic plasticity. Finally, AC1 activity is also found to be required for spinal LTP induced by pairing protocol in spinal dorsal horn neurons [27].
13.6 Discovery of AC1 Inhibitor NB001
Considering the important roles of AC1 in injury-related synaptic plasticity as well as behavioral responses in animal models of chronic pain, it is critical to develop a selective inhibitor for AC1. Using chemical design and screening experiments, a selective inhibitor for AC1, NB001, has been identified. NB001 shows its selective inhibition for AC1 in both human embryonic kidney (HEK) 293 cells, in which AC1 was stably expressed, and in adult mouse neurons. In addition, NB001 produces a dose-dependent inhibition of cAMP production in adult mouse ACC slices, suggesting that NB001 is effective in whole animals. By contrast, NB001 did not significantly affect other isoforms of ACs such as AC5-8 activity at effective inhibiting doses for AC1.
Results from electrophysiological experiments using AC1 KO mice or NB001 are quite similar. NB001 inhibited sensory-related LTP in ACC and spinal cord dorsal horn. Postsynaptic application of NB001 completely blocked the induction of LTP in ACC pyramidal neurons of adult mice. Furthermore, NB001 also prevented the induction of LTP in spinal cord dorsal horn neurons. These findings strongly indicate that AC1 may contribute to chronic pain by playing important roles in injury-related LTP in ACC and spinal cord. Finally, LTP in central synapses contains at least two different major forms: early-phase LTP (E-LTP) and L-LTP. Recent studies using a multiple channel recording system found that NB001 produced powerful inhibition of L-LTP that lasted at least 3 h after the induction.
13.7 NB001 Is Analgesic in Different Animal Models of Chronic Pain
Behavioral studies using AC1 KO mice demonstrate that behavioral allodynia in animal models of neuropathic pain and inflammatory pain was significantly reduced [20, 27]. To confirm the requirement of AC1 activity in these behavioral responses, the effects of NB001 [17, 27] on behavioral allodynia in animal models of neuropathic pain induced by nerve ligation have been examined. Administration of NB001 (1–5 mg/kg, i.p.) produced significant analgesic effects in different animal models of chronic pain. By contrast, application of NB001 at different dosages did not cause any abnormal behaviors in animals. Animals treated with NB001 showed calm and normal motor functions. Furthermore, emotional responses were also normal after NB001 treatment.
In addition, intraperitoneal injection of NB001 also reduced spontaneous pain in an animal model of IBS as well as cancer pain [23, 4]. These findings suggest that NB001 may serve as a novel analgesic to treat bone cancer pain and visceral pain.
13.8 Conclusion and Future Directions
In summary, integrative experimental approaches have demonstrated that cortical synapses that receive sensory painful information are highly plastic. Major forms of synaptic plasticity discovered in central synapses, including LTP and LTD, are also found in pain-related cortical areas such as ACC and IC. Investigation of cortical LTP mechanisms has provided molecular insights for the induction and expression of chronic pain in the central synapses. Among several key signaling molecules, AC1 has been found to be a key signaling protein for triggering chronic pain-related central plasticity. Inhibiting AC1 activity by using a selective inhibitor NB001 produces analgesic effects in different animal models of chronic pain. In addition to LTP, long-term depression (LTD) has been also reported in the ACC and IC [10, 19]. Peripheral injury causes loss of LTD, offering a new mechanism for cortical excitation. The use of tree shrew, a species with more advanced cortical structures, will facilitate translational research of cortical plasticity (see [12]).
References
Bliss TVP, Collingridge GL, Kaang B-K, Zhuo M (2016) Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat Rev Neurosci 17:485–496
Bushnell MC, Čeko M, Low LA (2013) Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci 14:502–511
Chen T, O’Den G, Song Q, Koga K, Zhang M-M, Zhuo M (2014) Adenylyl cyclase subtype 1 is essential for late-phase long term potentiation and spatial propagation of synaptic responses in the anterior cingulate cortex of adult mice. Mol Pain 10:65
Kang W-b, Yang Q, Guo Y-y, Wang L, Wang D-s, Cheng Q, X-m L, Tang J, Zhao J-N, Liu G, Zhuo M, Zhao M-G (2016) Analgesic effects of adenylyl cyclase inhibitor NB001 on bone cancer pain in a mouse model. Mol Pain 12:1744806916652409
Ko H-G et al (2018) Rapid turnover of cortical NCAM1 regulates synaptic reorganization after peripheral nerve injury. Cell Rep 22:748–759
Koga K, Descalzi G, Chen T, Ko H-G, Lu J, Li S, Son J, Kim T, Kwak C, Huganir Richard L, M-g Z, Kaang B-K, Collingridge Graham L, Zhuo M (2015a) Coexistence of two forms of LTP in ACC provides a synaptic mechanism for the interactions between anxiety and chronic pain. Neuron 85:377–389
Koga K, Liu MG, Qiu S, Song Q, O’Den G, Chen T, Zhuo M (2015b) Impaired presynaptic long-term potentiation in the anterior cingulate cortex of Fmr1 knock-out mice. J Neurosci 35:2033–2043
Li XYKH, Chen T, Descalzi G, Koga K, Wang H, Kim SS, Shang Y, Kwak C, Park SW, Shim J, Lee K, Collingridge GL, Kaang BK, Zhuo M (2010) Alleviating neuropathic pain hypersensitivity by inhibiting PKMζ in the anterior cingulate cortex. Science 330:1400–1404
Liauw J, Wu L-J, Zhuo M (2005) Calcium-stimulated adenylyl Cyclases required for long-term potentiation in the anterior cingulate cortex. J Neurophysiol 94:878–882
Liu M-G, Zhuo M (2014) Loss of long-term depression in the insular cortex after tail amputation in adult mice. Mol Pain 10:1–1
Liu M-G, Kang SJ, Shi T-Y, Koga K, Zhang M-M, Collingridge GL, Kaang B-K, Zhuo M (2013) Long-term potentiation of synaptic transmission in the adult mouse insular cortex: multielectrode array recordings. J Neurophysiol 110:505–521
Lu J-S, Yue F, Liu X, Chen T, Zhuo M (2016) Characterization of the anterior cingulate cortex in adult tree shrew. Mol Pain 12:1744806916684515
Qiu S, Zhang M, Liu Y, Guo Y, Zhao H, Song Q, Zhao M, Huganir RL, Luo J, Xu H, Zhuo M (2014) GluA1 phosphorylation contributes to postsynaptic amplification of neuropathic pain in the insular cortex. J Neurosci 34:13505–13515
Qiu SCT, Koga K, Guo YY, Xu H, Song Q, Wang JJ, Descalzi G, Kaang BK, Luo JH, Zhuo M, Zhao MG (2013) An increase in synaptic NMDA receptors in the insular cortex contributes to neuropathic pain. Sci Signal 6:ra34–ra34
Song Q, Zheng H-W, Li X-H, Huganir RL, Kuner T, Zhuo M, Chen T (2017) Selective phosphorylation of AMPA receptor contributes to the network of long-term potentiation in the anterior cingulate cortex. J Neurosci 37:8534–8548
Tang J, Ko S, Ding H-K, Qiu C-S, Calejesan AA, Zhuo M (2005) Pavlovian fear memory induced by activation in the anterior cingulate cortex. Mol Pain 1:6–6
Wang H, Xu H, Wu L-J, Kim SS, Chen T, Koga K, Descalzi G, Gong B, Vadakkan KI, Zhang X, Kaang B-K, Zhuo M (2011) Identification of an adenylyl cyclase inhibitor for treating neuropathic and inflammatory pain. Sci Transl Med 3:65ra63–65ra63
Wei F, Zhuo M (2001) Potentiation of sensory responses in the anterior cingulate cortex following digit amputation in the anaesthetised rat. J Physiol 532:823–833
Wei F, Li P, Zhuo M (1999) Loss of synaptic depression in mammalian anterior cingulate cortex after amputation. J Neurosci 19:9346–9354
Wei FQC, Kim SJ, Muglia L, Maas JW Jr, Pineda VV, Xu HM, Chen ZF, Storm DR, Muglia LJ, Zhuo M (2002) Genetic elimination of behavioral sensitization in mice lacking calmodulin-stimulated adenylyl cyclases. Neuron 36:713–726
Xu H, Wu LJ, Wang H, Zhang X, Vadakkan KI, Kim SS, Steenland HW, Zhuo M (2008) Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J Neurosci 28:7445–7453
Yamanaka M, Matsuura T, Pan H, Zhuo M (2017) Calcium-stimulated adenylyl cyclase subtype 1 (AC1) contributes to LTP in the insular cortex of adult mice. Heliyon 3:e00338
Zhang M-M, Liu S-B, Chen T, Koga K, Zhang T, Li Y-Q, Zhuo M (2014) Effects of NB001 and gabapentin on irritable bowel syndrome-induced behavioral anxiety and spontaneous pain. Mol Brain 7:47–47
Zhuo M (2002) Glutamate receptors and persistent pain: targeting forebrain NR2B subunits. Drug Discov Today 7:259–267
Zhuo M (2008) Cortical excitation and chronic pain. Trends Neurosci 31:199–207
Zhuo M (2011) Cortical plasticity as a new endpoint measurement for chronic pain. Mol Pain 7:54–54
Zhuo M (2012) Targeting neuronal adenylyl cyclase for the treatment of chronic pain. Drug Discov Today 17:573–582
Zhuo M (2014) Long-term potentiation in the anterior cingulate cortex and chronic pain. Philos Trans R Soc B: Biol Sci 369:20130146
Zhuo M (2016a) Neural mechanisms underlying anxiety-chronic pain interactions. Trends Neurosci 39:136–145
Zhuo M (2016b) Contribution of synaptic plasticity in the insular cortex to chronic pain. Neuroscience 338:220–229
Acknowledgment
I would like to thank Melissa Lepp for the help of the citation and reading of the manuscript. This work was supported by grants from the EJLB-CIHR Michael Smith Chair in Neurosciences and Mental Health, Canada Research Chair, Canadian Institute for Health Research operating grants (CIHR66975, 84256) and project grant (PJT-148648), Azrieli Neurodevelopmental Research Program, and Brain Canada (MZ).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Zhuo, M. (2018). Cortical LTP: A Synaptic Model for Chronic Pain. In: Shyu, BC., Tominaga, M. (eds) Advances in Pain Research: Mechanisms and Modulation of Chronic Pain. Advances in Experimental Medicine and Biology, vol 1099. Springer, Singapore. https://doi.org/10.1007/978-981-13-1756-9_13
Download citation
DOI: https://doi.org/10.1007/978-981-13-1756-9_13
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-1755-2
Online ISBN: 978-981-13-1756-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)