
Abstract
Repeat expansion disorders are a group of inherited neuromuscular diseases, which are caused by expansion mutations of repeat sequences in the disease-causing genes. Repeat expansion disorders include a class of diseases caused by repeat expansions in the coding region of the genes, producing mutant proteins with amino acid repeats, mostly the polyglutamine (polyQ) diseases, and another class of diseases caused by repeat expansions in the noncoding regions, producing aberrant RNA with expanded repeats, which are called noncoding repeat expansion diseases. A variety of Drosophila disease models have been established for both types of diseases, and they have made significant contributions toward elucidating the molecular mechanisms of and developing therapies for these neuromuscular diseases.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Repeat expansion diseases
- Drosophila
- Polyglutamine diseases
- Noncoding repeat expansion diseases
- Neurodegenerative diseases
- Spinocerebellar ataxia
- Amyotrophic lateral sclerosis
- RNA foci
- Repeat-associated non-ATG translation
5.1 Introduction
In 1991, expansion mutations of repeat sequences in the genome were discovered to cause human hereditary diseases, namely, a CGG trinucleotide repeat expansion in the fragile X mental retardation 1 (FMR1) gene causing fragile X syndrome (FXS) and a CAG trinucleotide repeat expansion in the androgen receptor (AR) gene in spinal-bulbar muscular atrophy (SBMA) (La Spada et al. 1991; Verkerk et al. 1991). Since these initial findings, more than 23 expansion mutations of 3 or more nucleotide repeats were found to cause various inherited neurological and neuromuscular diseases (Table 5.1) (La Spada and Taylor 2010). These repeat expansion disorders are largely classified into two groups depending on the location of the repeat sequences in the genome, i.e., the coding region or the noncoding region.
In the former group, expanded CAG repeats produce proteins containing an expanded polyglutamine (polyQ) tract, triggering neurodegeneration via toxic gain-of-function mechanisms in Huntington’s disease (HD); spinocerebellar ataxia (SCA) types 1, 2, 3, 6, 7, and 17; dentatorubral-pallidoluysian atrophy; and SBMA, which are collectively called the polyQ diseases (Katsuno et al. 2014; Takeuchi and Nagai 2017). Expansions of the polyQ tract are thought to trigger misfolding and aggregation of these causative proteins, eventually causing neurodegeneration. Expansion mutations of GCN repeats encoding a polyalanine (polyA) tract have also been reported in oculopharyngeal muscular dystrophy (OPMD) and other diseases, which can lead to both gain-of-function and loss-of-function pathogenic mechanisms (Messaed and Rouleau 2009).
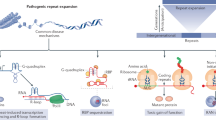
In the latter group, the repeat sequences are located in the noncoding region, such as the 5′-UTR, 3′-UTR, or introns in the genome in FXS and fragile X tremor ataxia syndrome (FXTAS); fragile XE syndrome (FRAXE); myotonic dystrophy (DM) types 1 and 2; Friedreich ataxia; SCA8, 10, 12, 31, 36, and 37; C9orf72-linked amyotrophic lateral sclerosis and frontotemporal dementia (C9-ALS/FTD) ; and Huntington’s disease-like 2 (Table 5.2) (Orr and Zoghbi 2007; Rohilla and Gagnon 2017; Seixas et al. 2017). Since these repeat sequences do not directly encode amino acid sequences in proteins, their pathogenic mechanisms are much more complicated. At least three molecular mechanisms underlying the pathogenesis of these noncoding repeat expansion diseases have been proposed (Nelson et al. 2013; Rohilla and Gagnon 2017). First, loss-of-function of the mutant genes due to silencing of or reduction in gene expression by the repeat expansion mutation has been suggested in FXS, FRAXE, and Friedreich ataxia (Pieretti et al. 1991; Bidichandani et al. 1998). Second, gain-of-function due to aberrant RNAs containing expanded repeats transcribed from the mutant gene have been suggested in most of these diseases, including DM1 and 2, FXTAS, SCA8, 10, 31, and 36, and C9-ALS/FTD . These expanded repeat-containing RNAs were shown to be accumulated as RNA foci in affected tissues and to recruit their corresponding RNA-binding proteins (RBPs), resulting in their loss-of-function (Miller et al. 2000; Mankodi et al. 2001; Jin et al. 2007). Furthermore, a third mechanism has emerged from recent studies, in which expanded repeat RNAs were surprisingly shown to be translated into aberrant repeat polypeptides despite the lack of an initiation codon, via unconventional translation, so-called repeat-associated non-ATG (RAN) translation (Zu et al. 2011; Mori et al. 2013; Ash et al. 2013; Pearson 2011). Subsequent studies demonstrated that these repeat polypeptides produced by RAN translation cause toxicity via gain-of-function mechanisms (Kwon et al. 2014; Mizielinska et al. 2014). However, the molecular mechanisms of RAN translation still remain to be understood, and research toward elucidation of the pathogenic mechanisms of these disorders is still ongoing.
In this chapter, we will introduce a number of studies using a variety of fly models to elucidate the pathogenic mechanisms of these repeat expansion disorders. We will also discuss the advantages of fly models as human disease models for studying pathogenic mechanisms and investigating potential therapies for these disorders.
5.2 Fly Models of Repeat Expansion Disorders
5.2.1 Huntington’s Disease
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder characterized by loss of neurons mainly in the striatum and cortex, leading to progressive motor impairments, cognitive decline, and psychiatric symptoms. HD is caused by an abnormal expansion of CAG repeats encoding the polyQ tract in exon 1 of the huntingtin (Htt) gene. In the polyQ diseases, such as HD, there is a threshold length of polyQ repeats for clinical manifestation of approximately 35 to 40 repeats in general, and longer repeats are associated with earlier age of onset and severity of disease. The abnormal aggregation of mutant proteins into nuclear inclusions (NIs) is also commonly observed in the brains of patients with the polyQ diseases (DiFiglia et al. 1997; Becher et al. 1998; Paulson et al. 1997).
Jackson et al. (1998) first generated fly models of HD, each expressing the exon 1 fragment of the Htt protein with tracts of either 2, 75, or 120 glutamine residues (Httex1-Q2, Q75, or Q120, respectively) in photoreceptor neurons of the eye. Expression of Httex1-Q2 had no effect on the fly eyes, whereas expression of Httex1-Q75 or Q120 caused repeat length- and age-dependent degeneration of photoreceptor neurons. Although the mutant Htt protein accumulated in the cytoplasm and nucleus of the photoreceptor neurons just after eclosion, nuclear accumulation of mutant Htt was observed in aged HD flies, suggesting that accumulation of the mutant Htt protein in the nucleus plays a crucial role in neurodegeneration. Gunawardena et al. (2003) also established HD fly models expressing the exon 1 fragment of the Htt protein with a 93 polyQ tract and showed that overexpression of this mutant Htt causes axonal transport defects accompanied by accumulation of the pathogenic Htt protein. Lee et al. (2004) established other HD fly models expressing the longer 548 amino acids fragment of the Htt protein with a 128 polyQ tract and also reported the disruption of axonal transport and accumulation of aggregates at synapses, indicating that cytoplasmic accumulation of the pathogenic Htt protein leads to neuronal dysfunction. Interestingly, they did not find axonal transport defects in flies expressing an expanded polyQ tract alone, which show only nuclear aggregates. On the other hand, new HD fly models expressing the full-length Htt protein containing a 128 polyQ tract have been established (Romero et al. 2008), and these flies showed behavioral, neurodegenerative, and electrophysiological phenotypes. They found that increased neurotransmission rather than axonal transport defects is at the root of the neurodegeneration caused by full-length mutant Htt during the early stages of pathogenesis (Romero et al. 2008). The results of these studies indicate that pathogenic outcomes can be affected by the protein context of the polyQ proteins.
5.2.2 Spinocerebellar Ataxia Type 1
Spinocerebellar ataxia type 1 (SCA1) is a dominantly inherited ataxia characterized by progressive cerebellar ataxia, dysarthria, dysphagia, and variable neurological symptoms and is caused by an abnormal expansion of the CAG trinucleotide repeat in the coding region of the ataxin-1 gene.
Fernandez-Funez et al. (2000) created a fly model of SCA1 by introducing transgenes encoding the full-length human ataxin-1 with a normal (SCA1-Q30) or expanded (SCA1-Q82) length polyQ repeats. Expression of SCA1-Q82 caused progressive neurodegeneration, as expected, and notably, flies expressing SCA1-Q30 at a high level also showed neurodegenerative phenotypes, indicating that even wild-type ataxin-1 can cause neurodegeneration. Genetic modifier screening using the SCA1 fly models identified several modifiers involved in protein folding/degradation, RNA processing, transcriptional regulation , and cellular detoxification. These findings shed light on a previously unrecognized new pathogenic mechanism of SCA1: the normal function of ataxin-1 could contribute to SCA1 pathogenesis . Subsequent studies also clarified modifiers involved in the signal transduction pathways by genetic interaction analyses using SCA1 fly models, in combination with mammalian-based genetic and proteomic analyses (Chen et al. 2003; Tsuda et al. 2005; Lam et al. 2006; Park et al. 2013).
In addition, genetic interaction between ataxin-1 and ataxin-2 was demonstrated using the SCA1 fly model (Al-Ramahi et al. 2007). The authors showed that wild-type Drosophila ataxin-2 is a major genetic modifier of the phenotypes of SCA1-82Q flies. They also showed that nuclear accumulation of ataxin-2 contributes to mutant ataxin-1-induced toxicity. Altogether, these findings suggest common mechanisms of neurodegeneration in different types of ataxia.
5.2.3 Spinocerebellar Ataxia Type 3
Spinocerebellar ataxia type 3 (SCA3), also known as Machado-Joseph disease (MJD), is the most common dominantly inherited ataxia and is characterized by progressive cerebellar ataxia and variable neurological symptoms. SCA3 is caused by an abnormal expansion of the CAG trinucleotide repeat in the coding region of the ataxin-3 gene.
The first genetically engineered fly models that were established for human neurodegenerative diseases were the SCA3 models (Warrick et al. 1998). These SCA3 fly models express the C-terminal region of the ataxin-3 protein containing normal (MJDtr-Q27) or pathogenic (MJDtr-Q78) length polyQ repeats. Expression of MJDtr-Q78 in the eye led to late-onset cell degeneration and NI formation (Fig. 5.1), similarly to the characteristics observed in SCA3 patients , whereas the expression of MJDtr-Q27 had no effect (Warrick et al. 1998). In a subsequent study, the same group demonstrated that HSP70, a major stress-induced molecular chaperone, suppresses polyQ-induced neurodegeneration in the SCA3 fly model (Warrick et al. 1999). They also showed that the full-length ataxin-3-Q27, which is a polyubiquitin-binding protein with ubiquitin protease activity , suppresses neurodegeneration and delays NI formation in MJDtr-Q78 flies, depending on its ubiquitin-associated activities and proteasome function (Warrick et al. 2005). These results indicate that the physiological function of the host protein plays a crucial role in SCA3 pathogenesis, as well as indicates the potential therapeutic role of ataxin-3 activity for the polyQ diseases. Moreover, Bilen and Bonini (2007) performed a genetic modifier screen using the SCA3 model fly and identified a set of genes that affects protein misfolding. Importantly, some modifiers of the SCA3 flies also modulated toxicity of tau, which is involved in Alzheimer’s disease and frontotemporal dementia, demonstrating common mechanisms of neurodegeneration between distinct neurotoxic proteins. We also showed that the loss of p62/sequestosome 1, which is involved in selective autophagy, delays the degradation of MJDtr-Q78 protein oligomers and exacerbates eye degeneration , indicating that p62 plays a protective role against polyQ-induced neurodegeneration in the SCA3 fly model (Saitoh et al. 2015). Taken together, these results suggest that chaperone activity and the protein-folding pathway play important roles in the pathogenesis of SCA3.

The fly model of SCA3
Expression of MJDtr-Q78 in the eye causes severe eye degeneration as compared to control. Fly genotypes are gmr-Gal4/+ (left) and gmr-Gal4/+; UAS-MJDtr-Q78S/+ (right)
It is widely accepted that mutant ataxin-3 proteins containing an expanded polyQ tract cause neurodegeneration. However, Li et al. (2008) provided evidence for a pathogenic role of CAG repeat RNA in polyQ disease pathogenesis using SCA3 fly models. They performed modifier screening for polyQ-induced neurodegeneration and unexpectedly found that muscleblind, a gene implicated in the RNA toxicity of CUG expansion diseases, enhanced eye degeneration in SCA3 flies. Furthermore, they tested the possible role of RNA toxicity by expressing the CAG repeat in the untranslated region , and found that mRNA expression of an untranslated CAG repeat of pathogenic length induced progressive neuronal dysfunction. These results demonstrate the role of RNA toxicity in the pathogenesis of SCA3.
5.2.4 Spinal and Bulbar Muscular Atrophy
Spinal and bulbar muscular atrophy (SBMA), also known as Kennedy disease, is an adult-onset neurodegenerative disorder with an X-linked recessive inheritance. The disease mainly affects motor neurons and is characterized by slowly progressive limb and bulbar muscle weakness and atrophy and gynecomastia. As described in the Introduction section, SBMA is caused by an abnormal expansion of the CAG repeat encoding a polyQ tract in exon 1 of the AR gene (La Spada et al. 1991).
A fly model of SBMA was generated by introducing a transgene encoding the AR protein with a tract of 52 polyQ (AR-Q52) into flies (Takeyama et al. 2002). Although no obvious phenotype was observed in the photoreceptor neurons of the eyes of these flies, administration of androgen or its antagonists led to marked neurodegeneration accompanied with nuclear translocation of the mutant AR. These findings suggest that ligand binding to polyQ-expanded AR leads to its structural alteration and subsequent nuclear translocation , which eventually leads to neurodegeneration in male SBMA patients (Takeyama et al. 2002). Regarding involvement of native AR functions in the pathogenesis of SMBA, Nedelsky et al. (2010) showed that not only the nuclear translocation of AR but also the DNA-binding activity of AR and recruitment of transcriptional coregulators is necessary for its toxicity. These findings indicate that the native functions of AR play a crucial role in the pathogenesis of SBMA.
5.2.5 Oculopharyngeal Muscular Dystrophy
Oculopharyngeal muscular dystrophy (OPMD) is an adult-onset muscular disorder generally with autosomal dominant traits and is characterized by progressive swallowing difficulties, ptosis, and proximal limb weakness. OPMD is caused by a short expansion of the GCG trinucleotide repeat in the coding region of the nuclear poly(A)-binding protein 1 (PABPN1) gene, which encodes a protein that is involved in the polyadenylation of mRNAs and poly(A) site selection (Brais et al. 1998). Whereas the normal PABPN1 allele has a (GCN)10 repeat encoding a 10 polyalanine (polyA) stretch, OPMD patients carry expanded alleles with (GCG)12–17 repeats, encoding expanded polyA tracts in the N-terminal domain of PABPN1 (Brais et al. 1998).
Chartier et al. (2006) established a fly model of OPMD expressing mutant PABPN1 with a 17 polyA tract in muscle and demonstrated progressive muscle degeneration and nuclear inclusions composed of mutant PABPN1 in these flies, which are reminiscent of the characteristics of human OPMD patients. Notably, in this OPMD fly model, the polyA tract was not sufficient to cause muscle degeneration, and the RNA-binding domain (RRM) of PABPN1 was also required. This suggests that OPMD does not only result from polyA toxicity but also from an intrinsic property of mutant PABPN1 that is dependent on the RRM. The authors also identified several suppressors of the muscular phenotype such as the molecular chaperone HSP70 and the anti-apoptotic protein p35 using the OPMD fly model, demonstrating the protective role of molecular chaperones and involvement of apoptosis in mutant PABPN1-induced muscle degeneration.
Recently, Chartier et al. (2015) found that mRNAs encoding mitochondrial proteins are downregulated starting at the earliest stages of progression in fly and mouse models of OPMD. Since the downregulation of these mRNAs correlates with their shortened poly(A) tails, the authors propose that impaired nuclear polyadenylation is an early defect in OPMD.
5.2.6 Spinocerebellar Ataxia Type 8
Spinocerebellar ataxia type 8 (SCA8) is an adult-onset slowly progressive ataxia with autosomal dominant inheritance, which is associated with an expansion of the CTG repeat in the noncoding region of the ataxin-8 opposite strand gene, and possibly the complementary CAG repeat in the ataxin-8 gene. This was the first example of an expansion mutation of a noncoding trinucleotide repeat in SCA, in contrast to most other repeat expansion mutations occurring in the coding regions in other SCAs (Koob et al. 1999). As the CTG trinucleotide repeat is believed to be located in the noncoding region, toxic gain-of-function mechanisms of repeat RNA are thought to be involved in the pathogenesis of SCA8.
To investigate this possibility, Mutsuddi et al. (2004) generated fly models for SCA8 by expressing 9 (normal) or 112 (expanded ) CTG repeats. Both flies expressing normal and expanded CTG repeats in the eye showed late-onset and progressive eye degeneration . Using these SCA8 fly models, they performed a genetic modifier screen and identified four RBPs that are expressed in neurons.
Later, bidirectional expression of CUG and antisense CAG repeat transcripts were reported in an SCA8 mouse model, as well as in SCA8 patients (Moseley et al. 2006). Most surprisingly, the CAG repeat sequence located in the noncoding region was discovered to be translated into repeat polypeptides in the absence of an initiation ATG codon (Zu et al. 2011) in cell and mouse models of SCA8, as well as in SCA8 patients. This unconventional translation was named repeat-associated non-ATG (RAN) translation. These results suggest that toxic gain-of-function mechanisms at both the protein and RNA levels may contribute to the pathogenesis of SCA8.
5.2.7 Spinocerebellar Ataxia Type 31
Spinocerebellar ataxia type 31 (SCA31) is a late-onset autosomal dominant cerebellar ataxia, which is caused by a complex penta-nucleotide (TGGAA)n repeat insertion in the overlapping intron of the brain expressed, associated with Nedd4 gene and the thymidine kinase 2 gene in the antisense strand (Sato et al. 2009). In the brains of SCA31 patients, RNA foci containing UGGAA repeats were observed (Niimi et al. 2013), supporting a toxic gain-of-function mechanism caused by UGGAA repeat RNA in the pathogenesis of SCA31.
To gain insight into the pathogenic mechanisms of SCA31, we generated SCA31 model flies expressing expanded UGGAA repeats (UGGAAexp) and showed that the expression of UGGAAexp causes neurodegeneration accompanied by the accumulation of UGGAAexp RNA foci and pentapeptide repeat proteins produced by repeat-associated translation, as observed in SCA31 patient brains (Ishiguro et al. 2017). Moreover, the ALS -associated RBPs, TAR DNA-binding protein (TDP-43) , fused in sarcoma (FUS), and heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNPA2/B1) bind to UGGAAexp RNA, alter the structure of UGGAAexp RNA, and suppress UGGAAexp-mediated toxicity . These results demonstrate that these RBPs function as RNA chaperones and regulate repeat-associated translation, suggesting that defects of RNA metabolism associated with RBPs contribute to the pathogenesis of SCA31.
5.2.8 Fragile X Tremor Ataxia Syndrome
Fragile X tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disease characterized by kinetic tremor, gait ataxia, parkinsonism, and dementia. FXTAS is caused by a premutation expansion of CGG repeats (55–200) in the 5′-UTR of the FMR1 gene, which is found in FXS carriers and belongs to the FMR1-related disorders, including FXS and FMR1-related primary ovarian insufficiency.
To investigate whether premutation alleles of FMR1 lead to neurodegeneration in vivo, Jin et al. (2003) established FXTAS fly models expressing 60 or 90 CGG repeats. They showed that expression of premutation CGG repeats alone is sufficient to cause neurodegeneration in a dose- and repeat length-dependent manner, suggesting RNA-mediated neurodegeneration in these fly models. In their following study, the authors screened for CGG repeat RNA-binding proteins from mouse brain lysates and identified Pur α and hnRNP A2/B1 as RBPs binding to CGG repeat RNA. They further showed that Pur α suppresses neurodegeneration caused by CGG repeat RNA in the FXTAS fly models, indicating that Pur α plays an important role in the pathogenesis of FXTAS (Jin et al. 2007). Sofola et al. (2007b) also identified RBPs such as hnRNP A2/B1 and CUG-binding protein 1 (CUGBP1) that bind to the CGG repeat and suppresses its toxicity in the FXTAS fly models. These results suggest sequestration of RBPs by CGG repeat RNA as one of the pathogenic mechanisms of FXTAS.
They also reported that co-expression of CGG repeat RNA together with CCG repeat RNA, whose expansion in the FMR2 gene causes another type of X-linked mental retardation, FRAXE, decreases their independent toxicities with each other, by reducing their transcript levels through the RNAi pathway (Sofola et al. 2007a). Furthermore, Sellier et al. (2013) found that the double-stranded RNA-binding protein DGCR8 binds to CGG repeats and is sequestered in CGG RNA aggregates together with its partner, DROSHA, resulting in a reduction in microRNA processing . These results suggest that alteration of the microRNA-processing machinery is involved in the pathogenic mechanisms in FXTAS.
Intriguingly, Todd et al. (2013) demonstrated that CGG repeats work as a template for RAN translation to produce polyglycine-containing proteins, which accumulate in ubiquitin-positive inclusions in the FXTAS fly models and FXTAS patient brains. Moreover, CGG repeat toxicity is suppressed by eliminating RAN translation and is enhanced by increased polyglycine production via ATG-initiated translation, indicating that RAN translation, which produces aberrant polypeptides, is involved in the neurodegeneration in FXTAS.
5.2.9 Myotonic Dystrophy Type 1
Myotonic dystrophy type 1 (DM1) is an autosomal dominant muscular dystrophy characterized by myotonia and muscular dystrophy , together with multisystem impairments, including cataracts, hypogonadism, endocrine dysfunction, heart defects, and cognitive decline. DM1 is the most common muscular dystrophy affected in adulthood, but it also appears as a congenital form. DM1 is caused by an abnormal expansion of CTG repeats in the 3′-UTR of the dystrophia myotonica protein kinase gene (Mahadevan et al. 1992; Brook et al. 1992). In DM1 patients, expanded CUG repeat-containing RNA accumulates as RNA foci in the nucleus of affected tissues and recruit two major RBPs, muscleblind like splicing regulator 1 (MBNL1) and CUGBP1, which bind to the CUG repeat RNA, resulting in their misregulation and alteration of RNA metabolism (Philips et al. 1998; Miller et al. 2000; Timchenko 2013).
To provide further insight into the pathogenic mechanisms of DM1, Houseley et al. (2005) generated DM1 fly models expressing expanded (162), intermediate (48, 56), or normal (11) CTG repeats in the 3′-UTR of a GFP reporter gene. In muscle cells, expanded CUG repeats formed RNA foci and colocalized with muscleblind, which is the Drosophila ortholog of human MBNL1, whereas normal and intermediate CTG repeats did not. However, no pathological phenotype, such as locomotor impairment, shortened life span, or muscular pathology, was detectable in this fly model. Further investigation was conducted by creating a more severe fly disease model with a larger number (480) of interrupted CUG (iCUG) repeats (de Haro et al. 2006). Expressions of this expanded iCUG repeat caused eye and muscle degeneration and the accumulation of expanded iCUG transcripts in nuclear RNA foci. Moreover, expression of MBNL1 was found to suppress expanded iCUG-induced toxicity, whereas expression of CUGBP1 worsened the iCUG-induced toxicity in these DM1 fly models (de Haro et al. 2006). Using this DM1 fly model, de Haro et al. (2013) further identified smaug, which is the Drosophila ortholog of human Smaug1/Samd4A, a translational repressor, as a suppressor of iCUG repeat-induced toxicity. Smaug was found to physically and genetically interact with CUGBP1 and suppresses iCUG-induced myopathy via restoration of the translational activity of CUGBP1 (de Haro et al. 2013).
5.2.10 C9orf72-Linked Amyotrophic Lateral Sclerosis and Frontotemporal Dementia
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease predominantly affecting upper and lower motor neurons, resulting in muscle weakness and atrophy, bulbar dysfunction, and eventual respiratory impairment. Frontotemporal dementia (FTD) is a neurodegenerative dementia characterized by cognitive impairment together with behavioral and personality changes. Since the discovery of TDP-43 as a key molecule aggregated in the pathological inclusions of both diseases, these intractable neurodegenerative diseases have been considered to belong to the same disease spectrum with overlapping genetic and neuropathological features (ALS/FTD) (Ling et al. 2013). In 2011, an abnormal expansion of a GGGGCC repeat in the first intron of the C9orf72 gene was identified as the most common genetic mutation of ALS /FTD (C9-ALS/FTD) (Renton et al. 2011; DeJesus-Hernandez et al. 2011). Three hypotheses in the pathogenesis of C9-ALS/FTD have been proposed so far, as follows: loss-of-function of the C9ORF72 protein, toxic gain-of-function of expanded GGGGCC repeat RNAs, and toxic gain-of-function of dipeptide repeat (DPR) proteins generated from expanded repeat RNAs by RAN translation (Ling et al. 2013). However, an FTD patient homozygous for the C9orf72 GGGGCC repeat expansion mutation was reported to demonstrate clinical and pathological features that fit within the range of those of heterozygous patients (Fratta et al. 2013). This fact, together with the lack of C9orf72-coding mutations in ALS patients, excludes the possibility of a loss-of-function mechanism in C9-ALS/FTD (Harms et al. 2013). Moreover, knockout mice for the C9orf72 gene demonstrate immunological defects, but no or mild neurological dysfunction (Koppers et al. 2015; Atanasio et al. 2016; Jiang et al. 2016; Sudria-Lopez et al. 2016). Thus, loss of C9orf72 function may not play a key role in the pathogenesis of C9-ALS/FTD.
Although Drosophila do not have an ortholog of the C9orf72 gene, fly models were employed to explore the toxic gain-of-function mechanisms in the pathogenesis of C9-ALS /FTD. The first C9-ALS/FTD fly model was established by expressing expanded 30 GGGGCC repeats with a CTCGAG interruption (iGGGGCC). Flies expressing iGGGGCC repeats in the eye caused eye degeneration, and those in motor neurons demonstrated motor dysfunction with aging (Xu et al. 2013). To distinguish the toxic gain-of-function mechanisms between expanded repeat RNAs themselves and DPR proteins produced by RAN translation, Mizielinska et al. (2014) generated three C9-ALS /FTD fly models, as follows: (1) flies expressing expanded pure GGGGCC repeats that produce both expanded RNAs and DPR proteins, (2) RNA-only flies expressing stop codon-interrupted expanded GGGGCC repeats that only produce expanded RNAs, and (3) DPR protein-only flies expressing non-GGGGCC RNAs with alternative codons that only produce DPR proteins . They found that flies expressing pure GGGGCC repeats showed neurodegenerative phenotypes, such as rough eye and decreased life span, whereas RNA-only flies showed no apparent phenotype, despite RNA foci formation in both pure GGGGCC repeat and interrupted repeat RNA-only flies. These findings suggest that expanded GGGGCC repeats cause neurotoxicity through the DPR proteins, and RNA foci may not be a direct cause of neurodegeneration in these fly models. The authors further investigated whether expression of the DPR protein alone is sufficient to induce toxicity using DPR protein-only flies. They found that only poly-GR and poly-PR proteins cause eye degeneration , whereas poly-GA and poly-PA proteins do not, indicating that arginine-containing DPR proteins are the major cause of neurodegeneration in C9-ALS/FTD fly models (Mizielinska et al. 2014). Tran et al. (2015) reported a new C9-ALS /FTD fly model expressing 160 GGGGCC repeats flanked by human intronic and exonic sequences. Spliced intronic 160 GGGGCC repeat RNA formed RNA foci in the nucleus of neurons but resulted in low levels of DPRs and no neurodegeneration. These results also indicate that the accumulation of RNA foci is not sufficient to drive neurodegeneration, and the sequences flanking the GGGGCC repeats may modulate RAN translation.
Toward elucidation of the molecular mechanisms underlying the pathogenesis of C9-ALS/FTD, several groups have performed genetic modifier screening using C9-ALS/FTD fly models. Zhang et al. (2015) identified Ran GTPase-activating protein (RanGAP), which is a key regulator of nucleocytoplasmic transport, and showed a genetic interaction between GGGGCC repeats and the nucleocytoplasmic transport machinery. Freibaum et al. (2015) performed genetic modifier screening using flies expressing GGGGCC repeats and GFP in frame to monitor RAN translation and identified 18 genes involved in the nuclear pore complex and nucleocytoplasmic transport. Boeynaems et al. (2016) also discovered genes encoding components of the nuclear pore complex, importins, exportins, Ran-GTP regulators, and arginine methylases as modifiers of C9-ALS /FTD flies. These findings provide evidence that nucleocytoplasmic transport contributes to the pathogenesis of C9-ALS/FTD.
5.3 Perspectives
As introduced above, a number of studies on repeat expansion disorders have been performed using fly models and have contributed toward elucidating the molecular mechanisms of these diseases. In particular, by taking advantage of fly models in rapid and efficient genetic analyses, various modifier genes have been identified by genetic screening , providing insight into the pathogenic mechanisms of these disorders.
The other remarkable advantage of fly models is their short generation cycle, which is useful for research on intergenerational repeat instability. Repeat instabilities are commonly observed in most of the repeat expansion disorders, and further elongation of expanded repeats in the next generation often results in earlier onset and more severe disease phenotypes, which is called anticipation (Mirkin 2007; Orr and Zoghbi 2007). Such elongation of expanded repeats is thought to occur during meiosis in germline cells, whereas repeat instability during mitosis is also known to cause somatic mosaicism (Pearson et al. 2005; Kovtun and McMurray 2008). Jung and Bonini (2007) used a fly model of SCA3 expressing an expanded CAG repeat to clarify the mechanisms underlying repeat instability. They found that repeat instability was enhanced by transcription and was modulated by Rad2/XPG, which is involved in DNA repair mechanisms. Furthermore, repeat instability was increased in SCA3 flies by the loss of CREB-binding protein, which is a histone acetyltransferase, and treatment with trichostatin A, a histone deacetylase (HDAC) inhibitor suppressed this repeat instability. These results clearly indicate the usefulness of fly models to study the mechanisms of repeat instability, which is thought to underlie the fundamental etiology of repeat expansion disorders.
In addition, several studies have shown the usefulness of fly models for the identification of potential drug targets. Using HD fly models, Steffan et al. (2001) first identified HDAC inhibitors, which increase the acetylation levels of histones, as therapeutic candidates for HD. They showed that the administration of sodium butyrate and suberoylanilide hydroxamic acid (SAHA) to HD flies by feeding suppressed neurodegeneration. Based on these findings, the therapeutic potential of HDAC inhibitors for HD were further explored in mouse models, and the therapeutic effects of SAHA were indeed replicated in a HD mouse model (Hockly et al. 2003). Several molecules targeting the misfolding and aggregation of polyQ proteins, such as polyglutamine binding peptide 1 (QBP1), Congo red, and methylene blue, have also been analyzed using fly models for their therapeutic potential (Nagai et al. 2003; Apostol et al. 2003; Sontag et al. 2012).
Although fly models have significantly contributed to extend our knowledge of repeat expansion disorders as mentioned above, we need to recognize the limitations of fly models in studying human diseases, due to the many differences between flies and humans, such as in their development, physiology, metabolism, nervous system, etc. Nevertheless, considering their rapid generation cycle, cost-effectiveness, and advantages in genetic analyses, fly models are powerful tools for studying human diseases (McGurk et al. 2015; Koon and Chan 2017).
References
Al-Ramahi I, Perez AM, Lim J, Zhang M, Sorensen R, de Haro M, Branco J, Pulst SM, Zoghbi HY, Botas J. dAtaxin-2 mediates expanded Ataxin-1-induced neurodegeneration in a Drosophila model of SCA1. PLoS Genet. 2007;3(12):e234. https://doi.org/10.1371/journal.pgen.0030234.
Apostol BL, Kazantsev A, Raffioni S, Illes K, Pallos J, Bodai L, Slepko N, Bear JE, Gertler FB, Hersch S, Housman DE, Marsh JL, Thompson LM. A cell-based assay for aggregation inhibitors as therapeutics of polyglutamine-repeat disease and validation in Drosophila. Proc Natl Acad Sci U S A. 2003;100(10):5950–5. https://doi.org/10.1073/pnas.2628045100.
Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW III, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77(4):639–46. https://doi.org/10.1016/j.neuron.2013.02.004.
Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y, Fury W, Burfeind P, Zamfirova R, Warshaw G, Orengo J, Oyejide A, Fralish M, Auerbach W, Poueymirou W, Freudenberg J, Gong G, Zambrowicz B, Valenzuela D, Yancopoulos G, Murphy A, Thurston G, Lai KM. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016;6:23204. https://doi.org/10.1038/srep23204.
Becher MW, Kotzuk JA, Sharp AH, Davies SW, Bates GP, Price DL, Ross CA. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis. 1998;4(6):387–97. https://doi.org/10.1006/nbdi.1998.0168.
Bidichandani SI, Ashizawa T, Patel PI. The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am J Hum Genet. 1998;62(1):111–21. https://doi.org/10.1086/301680.
Bilen J, Bonini NM. Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet. 2007;3(10):1950–64. https://doi.org/10.1371/journal.pgen.0030177.
Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovičić A, De Baets G, Scheveneels W, Steyaert J, Cuijt I, Verstrepen KJ, Callaerts P, Rousseau F, Schymkowitz J, Cruts M, Van Broeckhoven C, Van Damme P, Gitler AD, Robberecht W, Van Den Bosch L. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep. 2016;6:20877. https://doi.org/10.1038/srep20877.
Brais B, Bouchard JP, Xie YG, Rochefort DL, Chrètien N, Tomé FM, Lafrenière RG, Rommens JM, Uyama E, Nohira O, Blumen S, Korczyn AD, Heutink P, Mathieu J, Duranceau A, Codère F, Fardeau M, Rouleau GA. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998;18(2):164–7. https://doi.org/10.1038/ng0298-164.
Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68(4):799–808.
Chartier A, Benoit BA, Simonelig M. A Drosophila model of oculopharyngeal muscular dystrophy reveals intrinsic toxicity of PABPN1. EMBO J. 2006;25(10):2253–62. https://doi.org/10.1038/sj.emboj.7601117.
Chartier A, Klein P, Pierson S, Barbezier N, Gidaro T, Casas F, Carberry S, Dowling P, Maynadier L, Bellec M, Oloko M, Jardel C, Moritz B, Dickson G, Mouly V, Ohlendieck K, Butler-Browne G, Trollet C, Simonelig M. Mitochondrial dysfunction reveals the role of mRNA poly(A) tail regulation in oculopharyngeal muscular dystrophy pathogenesis. PLoS Genet. 2015;11(3):e1005092. https://doi.org/10.1371/journal.pgen.1005092.
Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, Aitken A, Skoulakis EM, Orr HT, Botas J, Zoghbi HY. Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell. 2003;113(4):457–68.
de Haro M, Al-Ramahi I, De Gouyon B, Ukani L, Rosa A, Faustino NA, Ashizawa T, Cooper TA, Botas J. MBNL1 and CUGBP1 modify expanded CUG-induced toxicity in a Drosophila model of myotonic dystrophy type 1. Hum Mol Genet. 2006;15(13):2138–45. https://doi.org/10.1093/hmg/ddl137.
de Haro M, Al-Ramahi I, Jones KR, Holth JK, Timchenko LT, Botas J. Smaug/SAMD4A restores translational activity of CUGBP1 and suppresses CUG-induced myopathy. PLoS Genet. 2013;9(4):e1003445. https://doi.org/10.1371/journal.pgen.1003445.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56. https://doi.org/10.1016/j.neuron.2011.09.011.
DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277(5334):1990–3.
Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, McCall A, Canal I, Orr HT, Zoghbi HY, Botas J. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408(6808):101–6. https://doi.org/10.1038/35040584.
Fratta P, Poulter M, Lashley T, Rohrer JD, Polke JM, Beck J, Ryan N, Hensman D, Mizielinska S, Waite AJ, Lai MC, Gendron TF, Petrucelli L, Fisher EM, Revesz T, Warren JD, Collinge J, Isaacs AM, Mead S. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol. 2013;126(3):401–9. https://doi.org/10.1007/s00401-013-1147-0.
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB, Taylor JP. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525(7567):129–33. https://doi.org/10.1038/nature14974.
Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40(1):25–40.
Harms MB, Cady J, Zaidman C, Cooper P, Bali T, Allred P, Cruchaga C, Baughn M, Libby RT, Pestronk A, Goate A, Ravits J, Baloh RH. Lack of C9ORF72 coding mutations supports a gain of function for repeat expansions in amyotrophic lateral sclerosis. Neurobiol Aging. 2013;34(9):2234 e2213–39. https://doi.org/10.1016/j.neurobiolaging.2013.03.006.
Hockly E, Richon VM, Woodman B, Smith DL, Zhou XB, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PAS, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci U S A. 2003;100(4):2041–6. https://doi.org/10.1073/pnas.0437870100.
Houseley JM, Wang Z, Brock GJ, Soloway J, Artero R, Perez-Alonso M, O’Dell KM, Monckton DG. Myotonic dystrophy associated expanded CUG repeat muscleblind positive ribonuclear foci are not toxic to Drosophila. Hum Mol Genet. 2005;14(6):873–83. https://doi.org/10.1093/hmg/ddi080.
Ishiguro T, Sato N, Ueyama M, Fujikake N, Sellier C, Kanegami A, Tokuda E, Zamiri B, Gall-Duncan T, Mirceta M, Furukawa Y, Yokota T, Wada K, Taylor JP, Pearson CE, Charlet-Berguerand N, Mizusawa H, Nagai Y, Ishikawa K. Regulatory role of RNA chaperone TDP-43 for RNA misfolding and repeat-associated translation in SCA31. Neuron. 2017;94(1):108–+. https://doi.org/10.1016/j.neuron.2017.02.046.
Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, Faber PW, MacDonald ME, Zipursky SL. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron. 1998;21(3):633–42.
Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, Stauffer JE, Jafar-Nejad P, Drenner K, Schulte D, Chun S, Sun S, Ling SC, Myers B, Engelhardt J, Katz M, Baughn M, Platoshyn O, Marsala M, Watt A, Heyser CJ, Ard MC, De Muynck L, Daughrity LM, Swing DA, Tessarollo L, Jung CJ, Delpoux A, Utzschneider DT, Hedrick SM, de Jong PJ, Edbauer D, Van Damme P, Petrucelli L, Shaw CE, Bennett CF, Da Cruz S, Ravits J, Rigo F, Cleveland DW, Lagier-Tourenne C. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90(3):535–50. https://doi.org/10.1016/j.neuron.2016.04.006.
Jin P, Zarnescu DC, Zhang F, Pearson CE, Lucchesi JC, Moses K, Warren ST. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron. 2003;39(5):739–47.
Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, Liu H, Feng Y, Warren ST. Pur α binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron. 2007;55(4):556–64. https://doi.org/10.1016/j.neuron.2007.07.020.
Jung J, Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science. 2007;315(5820):1857–9. https://doi.org/10.1126/science.1139517.
Katsuno M, Watanabe H, Yamamoto M, Sobue G. Potential therapeutic targets in polyglutamine-mediated diseases. Expert Rev Neurother. 2014;14(10):1215–28. https://doi.org/10.1586/14737175.2014.956727.
Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, Ranum LP. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat Genet. 1999;21(4):379–84. https://doi.org/10.1038/7710.
Koon AC, Chan HY. Drosophila melanogaster as a model organism to study RNA toxicity of repeat expansion-associated neurodegenerative and neuromuscular diseases. Front Cell Neurosci. 2017;11:70. https://doi.org/10.3389/fncel.2017.00070.
Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CA, Vieira de Sa R, Schellevis RD, Waite AJ, Blake DJ, Veldink JH, van den Berg LH, Pasterkamp RJ. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol. 2015;78(3):426–38. https://doi.org/10.1002/ana.24453.
Kovtun IV, McMurray CT. Features of trinucleotide repeat instability in vivo. Cell Res. 2008;18(1):198–213. https://doi.org/10.1038/cr.2008.5.
Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, Xie Y, McKnight SL. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345(6201):1139–45. https://doi.org/10.1126/science.1254917.
La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11(4):247–58. https://doi.org/10.1038/nrg2748.
La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352(6330):77–9. https://doi.org/10.1038/352077a0.
Lam YC, Bowman AB, Jafar-Nejad P, Lim J, Richman R, Fryer JD, Hyun ED, Duvick LA, Orr HT, Botas J, Zoghbi HY. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127(7):1335–47. https://doi.org/10.1016/j.cell.2006.11.038.
Lee WCM, Yoshihara M, Littleton JT. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc Natl Acad Sci U S A. 2004;101(9):3224–9. https://doi.org/10.1073/Pnas.0400243101.
Li LB, Yu Z, Teng X, Bonini NM. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature. 2008;453(7198):1107–11. https://doi.org/10.1038/nature06909.
Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–38. https://doi.org/10.1016/j.neuron.2013.07.033.
Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O’Hoy K, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255(5049):1253–5.
Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, Henderson D, Schalling M, Swanson MS, Thornton CA. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet. 2001;10(19):2165–70.
McGurk L, Berson A, Bonini NM. Drosophila as an in vivo model for human neurodegenerative disease. Genetics. 2015;201(2):377–402. https://doi.org/10.1534/genetics.115.179457.
Messaed C, Rouleau GA. Molecular mechanisms underlying polyalanine diseases. Neurobiol Dis. 2009;34(3):397–405. https://doi.org/10.1016/j.nbd.2009.02.013.
Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS. Recruitment of human muscleblind proteins to (CUG) (n) expansions associated with myotonic dystrophy. EMBO J. 2000;19(17):4439–48. https://doi.org/10.1093/emboj/19.17.4439.
Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447(7147):932–40. https://doi.org/10.1038/nature05977.
Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IOC, Pietrzyk J, Cleverley K, Nicoll AJ, Pickering-Brown S, Dols J, Cabecinha M, Hendrich O, Fratta P, Fisher EMC, Partridge L, Isaacs AM. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345(6201):1192–4. https://doi.org/10.1126/science.1256800.
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335–8. https://doi.org/10.1126/science.1232927.
Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, Chen G, Weatherspoon MR, Clark HB, Ebner TJ, Day JW, Ranum LP. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet. 2006;38(7):758–69. https://doi.org/10.1038/ng1827.
Mutsuddi M, Marshall CM, Benzow KA, Koob MD, Rebay I. The spinocerebellar ataxia 8 noncoding RNA causes neurodegeneration and associates with staufen in Drosophila. Curr Biol: CB. 2004;14(4):302–8. https://doi.org/10.1016/j.cub.2004.01.034.
Nagai Y, Fujikake N, Ohno K, Higashiyama H, Popiel HA, Rahadian J, Yamaguchi M, Strittmatter WJ, Burke JR, Toda T. Prevention of polyglutamine oligomerization and neurodegeneration by the peptide inhibitor QBP1 in Drosophila. Hum Mol Genet. 2003;12(11):1253–9. https://doi.org/10.1093/hmg/ddg144.
Nedelsky NB, Pennuto M, Smith RB, Palazzolo I, Moore J, Nie Z, Neale G, Taylor JP. Native functions of the androgen receptor are essential to pathogenesis in a Drosophila model of spinobulbar muscular atrophy. Neuron. 2010;67(6):936–52. https://doi.org/10.1016/j.neuron.2010.08.034.
Nelson DL, Orr HT, Warren ST. The unstable repeats-three evolving faces of neurological disease. Neuron. 2013;77(5):825–43. https://doi.org/10.1016/j.neuron.2013.02.022.
Niimi Y, Takahashi M, Sugawara E, Umeda S, Obayashi M, Sato N, Ishiguro T, Higashi M, Eishi Y, Mizusawa H, Ishikawa K. Abnormal RNA structures (RNA foci) containing a penta-nucleotide repeat (UGGAA)n in the Purkinje cell nucleus is associated with spinocerebellar ataxia type 31 pathogenesis. Neuropathol Off J Jpn Soc Neuropathol. 2013;33(6):600–11. https://doi.org/10.1111/neup.12032.
Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. https://doi.org/10.1146/annurev.neuro.29.051605.113042.
Park J, Al-Ramahi I, Tan Q, Mollema N, Diaz-Garcia JR, Gallego-Flores T, Lu HC, Lagalwar S, Duvick L, Kang H, Lee Y, Jafar-Nejad P, Sayegh LS, Richman R, Liu X, Gao Y, Shaw CA, Arthur JSC, Orr HT, Westbrook TF, Botas J, Zoghbi HY. RAS-MAPK-MSK1 pathway modulates ataxin 1 protein levels and toxicity in SCA1. Nature. 2013;498(7454):325–31. https://doi.org/10.1038/nature12204.
Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, Vig P, Mandel JL, Fischbeck KH, Pittman RN. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19(2):333–44.
Pearson CE. Repeat associated non-ATG translation initiation: one DNA, two transcripts, seven reading frames, potentially nine toxic entities! PLoS Genet. 2011;7(3):e1002018. https://doi.org/10.1371/journal.pgen.1002018.
Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6(10):729–42. https://doi.org/10.1038/nrg1689.
Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280(5364):737–41.
Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66(4):817–22.
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Hölttä-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chiò A, Restagno G, Borghero G, Sabatelli M, The ITALSGEN Consortium, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68. https://doi.org/10.1016/j.neuron.2011.09.010.
Rohilla KJ, Gagnon KT. RNA biology of disease-associated microsatellite repeat expansions. Acta Neuropathol Commun. 2017;5:63. https://doi.org/10.1186/s40478-017-0468-y.
Romero E, Cha GH, Verstreken P, Ly CV, Hughes RE, Bellen HJ, Botas J. Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron. 2008;57(1):27–40. https://doi.org/10.1016/j.neuron.2007.11.025.
Saitoh Y, Fujikake N, Okamoto Y, Popiel HA, Hatanaka Y, Ueyama M, Suzuki M, Gaumer S, Murata M, Wada K, Nagai Y. p62 plays a protective role in the autophagic degradation of polyglutamine protein oligomers in polyglutamine disease model flies. J Biol Chem. 2015;290(3):1442–53. https://doi.org/10.1074/jbc.M114.590281.
Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, Takahashi M, Matsuura T, Flanigan KM, Iwasaki S, Ishino F, Saito Y, Murayama S, Yoshida M, Hashizume Y, Takahashi Y, Tsuji S, Shimizu N, Toda T, Ishikawa K, Mizusawa H. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing; (TGGAA)n. Am J Hum Genet. 2009;85(5):544–57. https://doi.org/10.1016/j.ajhg.2009.09.019.
Seixas AI, Loureiro JR, Costa C, Ordóñez-Ugalde A, Marcelino H, Oliveira CL, Loureiro JL, Dhingra A, Brandão E, Cruz VT, Timóteo A, Quintáns B, Rouleau GA, Rizzu P, Carracedo A, Bessa J, Heutink P, Sequeiros J, Sobrido MJ, Coutinho P, Silveira I. A pentanucleotide ATTTC repeat insertion in the non-coding region of DAB1, mapping to SCA37, causes spinocerebellar ataxia. Am J Hum Genet. 2017;101(1):87–103. https://doi.org/10.1016/j.ajhg.2017.06.007.
Sellier C, Freyermuth F, Tabet R, Tran T, He F, Ruffenach F, Alunni V, Moine H, Thibault C, Page A, Tassone F, Willemsen R, Disney MD, Hagerman PJ, Todd PK, Charlet-Berguerand N. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep. 2013;3(3):869–80. https://doi.org/10.1016/j.celrep.2013.02.004.
Sofola OA, Jin P, Botas J, Nelson DL. Argonaute-2-dependent rescue of a Drosophila model of FXTAS by FRAXE premutation repeat. Hum Mol Genet. 2007a;16(19):2326–32. https://doi.org/10.1093/hmg/ddm186.
Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron. 2007b;55(4):565–71. https://doi.org/10.1016/j.neuron.2007.07.021.
Sontag EM, Lotz GP, Agrawal N, Tran A, Aron R, Yang G, Necula M, Lau A, Finkbeiner S, Glabe C, Marsh JL, Muchowski PJ, Thompson LM. Methylene blue modulates huntingtin aggregation intermediates and is protective in Huntington’s disease models. J Neurosci. 2012;32(32):11109–19. https://doi.org/10.1523/JNEUROSCI.0895-12.2012.
Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413(6857):739–43. https://doi.org/10.1038/35099568.
Sudria-Lopez E, Koppers M, de Wit M, van der Meer C, Westeneng HJ, Zundel CA, Youssef SA, Harkema L, de Bruin A, Veldink JH, van den Berg LH, Pasterkamp RJ. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol. 2016;132(1):145–7. https://doi.org/10.1007/s00401-016-1581-x.
Takeuchi T, Nagai Y. Protein misfolding and aggregation as a therapeutic target for polyglutamine diseases. Brain Sci. 2017;7(10). https://doi.org/10.3390/brainsci7100128.
Takeyama K, Ito S, Yamamoto A, Tanimoto H, Furutani T, Kanuka H, Miura M, Tabata T, Kato S. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron. 2002;35(5):855–64.
Timchenko L. Molecular mechanisms of muscle atrophy in myotonic dystrophies. Int J Biochem Cell Biol. 2013;45(10):2280–7. https://doi.org/10.1016/j.biocel.2013.06.010.
Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V, Elenitoba-Johnson K, Vonsattel JP, Louis ED, Sutton MA, Taylor JP, Mills RE, Charlet-Berguerand N, Paulson HL. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron. 2013;78(3):440–55. https://doi.org/10.1016/j.neuron.2013.03.026.
Tran H, Almeida S, Moore J, Gendron TF, Chalasani U, Lu Y, Du X, Nickerson JA, Petrucelli L, Weng Z, Gao FB. Differential toxicity of nuclear RNA foci versus dipeptide repeat proteins in a Drosophila model of C9ORF72 FTD/ALS. Neuron. 2015;87(6):1207–14. https://doi.org/10.1016/j.neuron.2015.09.015.
Tsuda H, Jafar-Nejad H, Patel AJ, Sun Y, Chen HK, Rose MF, Venken KJ, Botas J, Orr HT, Bellen HJ, Zoghbi HY. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/senseless proteins. Cell. 2005;122(4):633–44. https://doi.org/10.1016/j.cell.2005.06.012.
Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65(5):905–14.
Warrick JM, Paulson HL, Gray-Board GL, Bui QT, Fischbeck KH, Pittman RN, Bonini NM. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell. 1998;93(6):939–49.
Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23(4):425–8. https://doi.org/10.1038/70532.
Warrick JM, Morabito LM, Bilen J, Gordesky-Gold B, Faust LZ, Paulson HL, Bonini NM. Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol Cell. 2005;18(1):37–48. https://doi.org/10.1016/j.molcel.2005.02.030.
Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS, Jin P. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A. 2013;110(19):7778–83. https://doi.org/10.1073/pnas.1219643110.
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525(7567):56–61. https://doi.org/10.1038/nature14973.
Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LP. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A. 2011;108(1):260–5. https://doi.org/10.1073/pnas.1013343108.
Acknowledgments
We thank Dr. H. Akiko Popiel for excellent editing of the manuscript and lab members for their helpful discussions. This work was supported in part by Grant-in-Aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control) (17H05699 to Y.N.) and Strategic Research Program for Brain Sciences (Integrated research on neuropsychiatric disorders) (11013026 to Y.N.) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; by Grants-in-Aid for Scientific Research (B) (16H05325 to Y.N.), Challenging Exploratory Research (24659438 to Y.N.), Scientific Research (C) (15K09331 to M.U.), and Young Scientists (B) (25860733 to M.U.) from the Japan Society for the Promotion of Science, Japan; by Health Labour Sciences Research Grant for Research on Development of New Drugs (H24-Soyakusogo-Ippan-002 to Y.N.) from the Ministry of Health, Labour and Welfare, Japan; by grants for Practical Research Projects for Rare/Intractable Diseases (15Aek0109018h0002, 17ek0109222h0001, 18ek0109316h0001 to Y.N.) and Strategic Research Program for Brain Sciences (Integrative Research on Depression, Dementia and Development disorders) (16dm0107061h0001 to Y.N.) from Japan Agency for Medical Research and Development, Japan; by Intramural Research Grants for Neurological and Psychiatric Disorders (27-7, 27-9, 30-9 to Y.N.) from the National Center of Neurology and Psychiatry; by an IBC Grant to Y.N. from the Japan Amyotrophic Lateral Sclerosis Association; and by a grant from the Takeda Science Foundation (to M.U.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Ueyama, M., Nagai, Y. (2018). Repeat Expansion Disease Models. In: Yamaguchi, M. (eds) Drosophila Models for Human Diseases. Advances in Experimental Medicine and Biology, vol 1076. Springer, Singapore. https://doi.org/10.1007/978-981-13-0529-0_5
Download citation
DOI: https://doi.org/10.1007/978-981-13-0529-0_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-0528-3
Online ISBN: 978-981-13-0529-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)