
Abstract
Cardiovascular disease is the number one cause of death in the U.S and results in the loss of approximately one million lives and more than 400 billion U.S. dollars for treatments every year. Recently, tissue engineered blood vessels have been studied and developed as promising replacements for treatment with autologous veins. Here, we summarize the cell sources and methods to make tissue-engineered blood vessels (TEBVs), the recent progress in TEBV related research, and also the recent progress in TEBV related clinical studies.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Cardiovascular disease (CVD) is regarded as the leading cause of mortality and morbidity in America and worldwide (Zhang et al. 2017). According to the 2017 statistic report from the American Heart Association, it was estimated that the deaths caused by CVD in the world were 31.5% of the all global deaths (Benjamin et al. 2017). In addition, about 92.1 million U.S. adults are suffering from some type of CVD, and of great concern nearly half of the population (43.9%) of U.S. adults are expected to get some kind of CVD by 2030 (Benjamin et al. 2017).
The golden standard procedure to treat CVD is to bypass the blocked vessels with an autologous vein harvested from the patient through coronary bypass graft surgery (Seifu et al. 2013). However, a great number of patients still fail to obtain immediate treatment because suitable native vessels are often unavailable due to inherent disease, amputation, or previous harvest. In addition, it was reported that the autologous veins often fail to function after 10–12 years post-implantation (Liu et al. 2011). Thus, other types of grafts such as synthetic grafts or materials were developed to offer a better treatment option. Synthetic materials (Dacron and expanded polytetrafluorethlyne (ePTFE)) are also used to treat the disease. However, limitations such as thrombosis, poor mechanical properties, low patency rate, and long fabrication time significantly impact that use of these synthetic grafts and materials in clinical use (Liu et al. 2011).
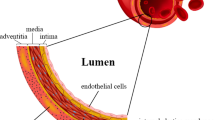
Recently, tissue engineering has become a promising alternative approach to fabricate vascular substitutes to address the shortcomings of current options. The ideal tissue engineered blood vessels (TEBVs) should have similar properties as native blood vessels; for instance: (1) mimic the three layered vessel structures including intima (endothelial cell (EC) layer), media (smooth muscle cell (SMC) layer), and adventitia (fibroblast cell (FB) layer) in a tubular construct, (2) nonthrombogenic and nonimmunogenic, (3) good mechanical stability to withstand physiological blood pressure, and (4) able to vasoconstrict and vasodilate under physiological conditions (Fernandez et al. 2014). For future clinical application, a relatively low cost and short production time should also be considered. To date, significant progress has been made in generating TEBVs in vitro and in vivo. In this book chapter, we summarize the cell sources and methods to make TEBVs, the recent progress in TEBV production, and the recent progress in clinical studies.
2 Fabrication of TEBV Through Tissue Engineering
Tissue engineering involves three independent and crucial factors which contribute to the formation of new vessels: (1) cells, (2) degradable scaffolds in which the cells will be seeded to produce the extracellular matrix and the subsequent in neotissue formation, and (3) humoral and mechanical signals among cells. In this section, we will focus on the first two factors to generate TEBVs (Naito et al. 2011).
2.1 Cell Source for TEBV
As mentioned earlier, the native blood vessels consist of ECs, SMCs, and FBs as internal, middle and outside layers, respectively. Specifically, the EC layer is a single layer of cells that are arranged parallel to the blood flow, while the middle layer consists of concentric layers of SMCs. The fibroblast layer is made of myofibroblasts that are able to produce collagen fibers. So far, TEBVs have been reported to be engineered by using one, two, or all three of these types of cells. The ideal cell source to make the TEBVs is an autologous cell source, which can then divide and differentiate into mature cells. Up to now, several types of cell sources have been used to fabricate TEBVs (Ercolani et al. 2015).
One of the cell sources are autologous SMCs, ECs and FBs taken from a patient’s saphenous vein or mammary artery, as these vessels tend to be more flexible and less prone to thrombosis (Buijtenhuijs et al. 2004). This cell source is advantageous as it significantly reduces the chance of rejection of the cells by the immune system. One study reported the use of autologously sourced fibroblasts to engineer blood vessels that showed similar mechanical (burst pressure) and antithrombogenic properties to natural blood vessels. These properties were achieved by layering cell sheets formed from cultured fibroblasts and seeding the interior of the engineered blood vessels with autologous endothelial cells. This method also avoided the need for synthetic materials or smooth muscle cells in the production of the blood vessels (L’Heureux et al. 2006). Although these cells are preferred for coronary procedures as they are similar to the autologous vein, they are not always available for use because patients with cardiovascular disease may not have sufficient amount of unaffected tissue to be used for the procedure. Other issues include the incompatibility between the size and shape of the source cells with the area in which they would be utilized.
Because of the aforementioned drawbacks of sourcing autologous cells directly from the vein, stem cells have become an alternative cell source to generate TEBVs. One of the advantages of using stem cells in tissue engineering is that they can both regenerate themselves and differentiate into other types of cells. Embryonic stem cells are stem cells taken from human embryos at the blastocyst stage prior to implantation in the uterus and which have the capability to differentiate into any type of cell in the body (Rippon and Bishop 2004). Because of their wide differentiation capability, these cells have been used to produce both endothelial and smooth muscle cells for tissue blood vessel engineering. For example, one study reported the use of embryonic stem cells differentiated into endothelial cells to improve perfusion in the ischemic hindlimb of a mouse (Huang et al. 2010). Another study reported the success of using embryonic stem cell to derive endothelial cells to form stable blood vessels in vivo. However, one challenge associated with using embryonic stem cells to engineer blood vessels is the production of plentiful differentiated cells (Wang et al. 2007). Also, because these cells are usually acquired from a donor, the risk of immune rejection in the affected subject is also present (Thomson et al. 1998).
Another form of stem cells used in blood vessel engineering is adult stem cells. Similar to embryonic stem cells, these cells have the ability to provide an endless supply of themselves through regeneration and also allow for some differentiation. However, the degree of differentiation that is possible from adult stem cells is not as great as that of embryonic stem cells. Adult stem cells are commonly sourced from the marrow of long bones and have already undergone some degree of differentiation (Bishop et al. 2002). Adult stem cells obtained from bone marrow can mature into endothelial cells and blood cells. Mesenchymal stem cells, a type of adult stem cell, can differentiate into bone, adipose, and even muscle and endothelial cells (Krawiec and Vorp 2012). One study noted the successful attachment of smooth muscle cells derived from adult adipose stem cells to a vascular graft scaffold (Harris et al. 2011). One drawback of adult stem cells not seen in embryonic stem cells is their limited ability to differentiate into specific cell types. For example, some adult stem cells may only differentiate into smooth muscle cells while others only differentiate into endothelial cells. Another limitation of adult stem cells is the difficulty to find sufficient numbers of appropriate and unaffected cells in patients with cardiovascular disease (Wang et al. 2014). Despite these two drawbacks, adult stem cells hold one major advantage over embryonic stem cells: the ability to be sourced autologously. As these stem cells can be sourced from the patient’s own body, they hold the benefit of self-regeneration and differentiation without the risk of immunological rejection (Bishop et al. 2002). Induced pluripotent stem cells (hiPSc) have also been used in blood vessel engineering, and seem to solve the issues that come with both adult and embryonic stem cells. While embryonic stem cells have the ability to differentiate into any type of cell (pluripotency), they are difficult to obtain and have the risk of immunologic rejection (Thomson et al. 1998). Adult stem cells can be obtained autologously and limit immunologic rejection, but do not have the range of variability seen in embryonic stem cells (Wang et al. 2014). Induced pluripotent stem cells are obtained from adult cells that are reverted to an embryonic stem cell state. These induced stem cells can be differentiated into a variety of mature cell types, such as smooth muscle cells and endothelial cells. This allows induced pluripotent cells to be sourced autologously. Furthermore, the adult cells they are induced from are also numerous in the body. One study noted the use of aortic fibroblasts to develop induced pluripotent stem cells that were then differentiated into smooth muscle cells to be used in vascular tissue engineering (Wang et al. 2014). Another study noted the use of induced pluripotent stem cells to generate mesenchymal stem cells that improved vascular tissue regeneration upon implantation (Lian et al. 2010).
Yet another cell type utilized in vascular tissue engineering is endothelial progenitor cells (EPCs). Progenitor cells are similar to adult stem cells in that they are more specific than embryonic stem cells: they are directed to differentiate and mature into a specific target cell type. Endothelial progenitor cells specifically differentiate and mature into endothelial cells that form the inner lining of blood vessels (Szmitko 2003). These cells are commonly sourced from primitive bone marrow shortly after birth and normally circulate in the blood and aggregate at sites of injury to replenish injured endothelium (Urbich and Dimmeler. 2004). Because of their role in restoring the endothelium, they have been used in the treatment of ischemia in vascular tissue and cancer (Au et al. 2008). One study reported that grafts seeded with EPCs explanted in sheep remained patent for over 4 months, and also contributed to the relaxation of vessels through the release of nitric oxide (Kaushal et al. 2001). Another study noted that widespread vascular networks formed in mice that had been implanted with both mesenchymal and endothelial progenitor cells. Furthermore, these structures remained patent for over 4 weeks (Melero-Martin et al. 2008). Although EPCs hold the advantage of reduced immunologic rejection by being sourced autologously, they also hold some drawbacks. The functionality and sustainability of vessels made from EPCs is not well defined. For example, one study noted that adult peripheral EPCs form unstable vessels that do not last more than 3 weeks, while EPCs from the umbilical cord form functional vessels that remain patent for more than 4 months (Au et al. 2008). Further study on EPCs and their functionality is required before they can be used efficiently in vascular tissue engineering.
2.2 Methods for In Vitro Fabrication of TEBV
Currently, TEBVs can be generally fabricated through the following methods: (1) assembly of cells within scaffolds, (2) self-assembly with cell sheets, (3) 3D-printing.
2.2.1 Assembly of Cells Within Scaffolds
This method includes first seeding the cells in a scaffold, then rolling the cells within the scaffolds into a three dimensional vessel structure using a tubular template, and finally culturing the vessels in static culture or in a bioreactor until the TEBVs are mature. Another approach is to seed the cells into the lumen of a synthetic tubular scaffold, and then culture the cell-seeded scaffold in static or in a bioreactor until the TEBVs are mature (Niklason et al. 1999). Sometimes, cell seeded scaffolds will be implanted in the host to produce the TEBV in vivo, instead of producing the TEBV in vitro.
The scaffolds used in this method can be decellularized or synthetic scaffolds (Zeng et al. 2010). Decellularized scaffolds are always obtained by removing cellular antigenic components from allogeneic tissues cultured in vitro, such as tubular tissues or cell sheets that keep a structured and undamaged extracellular matrix (ECM). Removal of the cells from the tissues can be achieved by physical stirring, enzymatic digestion, and use of a chemical surfactant (Xing et al. 2014). For instance, a TEBV of small diameter was fabricated by seeding ECs or EPCs on a decellularized scaffold derived from porcine SMCs through a biomimetic perfusion system. The seeded EPCs preserved their cobblestone cell morphology and also expressed endothelial cells markers such as CD31, CD144, and von Willibrand factor. Importantly, after the TEBV was implanted into the carotid artery, it remained patent for 30 days and showed little neointimal hyperplasia (Quint et al. 2011).
The advantages of using decellularized scaffolds are: (1) they are non-immunogenic, and (2) decellularized scaffolds can be stored as off-the-shelf products because of their acelluar properties. The disadvantages of decellularized scaffold are that the scaffold degrades very quickly and it is also difficult to modify the scaffold (Fernandez et al. 2014).
Another type of scaffold used is synthetic scaffolds, which are divided into synthetic nondegradable scaffolds and biodegradable scaffolds. Some common nondegradable synthetic scaffolds are expanded polytetrafluoroethylene (ePTFE), Dacron, and polyurethane, which have been used to produce large blood vessel substitutes (McQuade et al. 2009).
One commonly used one is Poly (ε-caprolactone) (PCL). PCL can be degraded slowly by hydrolysis through its ester bond, and is thus usually employed for long-term use. Some other examples are polyglycolic acid (PGA) (Niklason et al. 1999), pol(yglycolic acid)-poly(L-lactic acid) (PGA-PLLA) (Wu et al. 2004), poly (glycerol sebacate) (PGS) (Motlagh et al. 2006), polyhydoxyalkanoates (PHA) (Ma et al. 2010; Cheng et al. 2008), collagen (Shepherd et al. 2008) and silk (Liu et al. 2011). Synthetic and natural degradable scaffolds can be made into tubular scaffolds which mimic the ECM via electrospinning. The electrospinning method involves several steps: (1) the polymer solution is loaded into a syringe and pumped with a syringe pump, (2) the solution is applied with a high DC voltage, which results in a formation of cone shape liquid droplet known as the Taylor cone, (3) a liquid jet of polymer solution is generated from the Taylor cone by the high voltage and then travels to the collector, which has an electrode of the opposite polarity of the polymer, (4) the solid polymer is deposited on the collector as the polymer solvent evaporated during the traveling phase (Hasan et al. 2014). A tubular scaffold can be fabricated by use of a rotating mandrel. After the tubular scaffold is obtained, cells can be seeded onto the scaffold to produce the TEBV. Since the cells are seeded onto the scaffolds, the TEBV is mechanically stable to withstand physiological conditions (Fernandez et al. 2014). In addition, cells seeded onto the scaffold can response to the fluid shear stress, for instance, EC will produce nitric oxide upon stimuli to induce antithrombogenicity. However, the limitations of this method are (1) cells need to be autologous; (2) high cost; (3) need to prepare the TEBV very far ahead before surgery (Fernandez et al. 2014).
2.2.2 Assembly of TEBV Using Cell Sheets
Another way to generate TEBVs is in vitro assembly of cell sheets into tubular structures (L’Heureux et al. 1998, 2006). This method is scaffold-free and involves a method known as cell sheet technology.
The cell sheets can be produced in several ways. The most known method is to culture the cells with medium containing ascorbic acid, as the ascorbic acid will induce the ECM formation. It takes 30 days to obtain the cell sheets with ECM (L’Heureux et al. 1998).
A modified cell sheet method was developed to obtain cell sheets without damage. This method is based on several steps: (1) cells are seeded onto a temperature-responsive culture dish, which is coated with a temperature responsive polymer, Poly(Nisopropylacrylamide) (PIPAAM). (2) The cells are cultured at 37 °C until cell sheet formation. (3) The cell sheets are harvested by placing the culture dish at 25 °C (Yamada et al. 1990). The principle for this technology is that when the culture dish is at 37 °C, the PIPAAM coated surface is hydrophobic, so the cells can attach and proliferate on the surface; when the temperature goes below 32 °C, PIPAAM becomes hydrophilic and the cells cannot adhere to the surface anymore, and thus the intact cell sheet will detach for collection (Okano et al. 1995; Kushida et al. 1999; Matsuura et al. 2014). After the cell sheets are harvested, the cell sheets are rolled into a vascular structure and then cultured until the different layers fuse and a mature vessel forms. The advantage of the cell sheet method is that no scaffold is needed. The number of sheets and the cell layer orientations can also be easily controlled through this approach.
Another way to make the cell layered sheet is known as layer-by-layer technology. Layer-by-layer technology is a common approach used to fabricate multilayer polymeric films using different types of polymer interactions. However, layer-by-layer technology was also applied to build up multilayer cell sheets. For instance, cells coated with a single layer made of gelatin and fibronectin (FN) showed ability to accumulate into ordered cell layer sheets because the FN-gelatin coated surface is like the ECM, which allows the second layer of cells to attach on the first layer of cells, leading to the formation of cell sheet layers. Construction of 3D tissues with blood-capillary networks using human dermal fibroblast cells (hFC) and human umbilical vein endothelial cells (HUVEC) was demonstrated in this study (Nishiguchi et al. 2011). The advantage of using this technology is that the layer number and type and location of the cell can be controlled by altering the seeded cell number and order (Matsusaki et al. 2007). Thus, this method provides a rapid way to fabricate cell layered 3D tissues and shows potential to fabricate cell sheets to assemble TEBVs.
2.2.3 Fabrication of TEBV Using 3D-Printing
Recently, three-dimensional (3D) printing has been used to fabricate 3D functional living tissues. 3D printing involves several steps: (1) imaging the damaged tissue and its environment using X-ray, CT, or MRI, (2) the three central design approaches of biomimicry, self-assembly, and mini-tissues are used, (3) materials are chosen (synthetic polymers, natural polymers, and ECM) for the bioprinting process, (4) cell types are chosen for bioprinting, and (5) the chosen cells and materials integrate with bioprinting methods such as inkjet, microextrusion, or laser-assisted printers. 3D printing can be used to produce either a single vascular structure or a vascular network. We will highlight some recent studies in the next section (Murphy and Atala 2014; Kang et al. 2016).
3 Recent Progress in Vascular TEBV
Though successful fabrication of TEBVs has been reported in many studies, several drawbacks of those developed TEBVs, such as long production time, poor mechanical stability, thrombogenicity, and immunogenicity still remain as obstacles that limit clinical use. In addition, methods to generate TEBVs with good cell alignment and contractile SMC, obtain a confluent endothelium on the SMC layer in the lumen, and find alternative cell sources for more native TEBV mimics have been gaining widespread attention in the field. In this section, we will focus on recent representative studies regarding fabrication of TEBVs using the cells, methods, and scaffolds we discussed earlier to solve those drawbacks.
In the recent related studies, several groups worked on developing technology to produce mechanically stable TEBVs with a shorter maturation time. For instance, functional and mechanically robust tissue engineered blood vessels known as nTEVM were developed using human fibroblast-derived ECM seeded with SMCs via combining cell sheet technology and decellularization (Fig. 8.1). Specifically, the decellularized matrix scaffolds were obtained by culturing dermal fibroblasts (DF) or saphenous vein fibroblasts (SVF) for 3 weeks in the medium with ascorbic acid, and then decellularizing the fibroblast sheets. Then, human umbilical artery SMCs were seeded on the decellularized matrix scaffolds and cultured for 1 week. Finally, the SMC cell sheets were wrapped around a 4.5 mm cylindrical mandrel and cultured for 3 weeks to obtain a tubular construct. This approach significantly decreased the TEBV production time by 1–2 weeks compared to the method that directly cultured SMC sheets to make the tissue engineered blood vessels (sTEVM) (Fig. 8.4). In addition, based on the immunostaining, the nTEVM were positive for SMC markers such as α-SM-actin and calponin (Fig. 8.2). Thus, the nTEVM demonstrated vascular contraction. The burst pressure of the nTEVM was also significantly higher (~2000 mm Hg) than that of native blood vessels (Bourget et al. 2012).

Schematic view of the processes and timeline required for the production of the vascular constructs. (a) Human fibroblasts are isolated either from skin or saphenous vein biopsies and smooth muscle cells are isolated from the media of an umbilical artery. (b) The tissue sheets are peeled off from the flask and decellularized to produce a dMS or rolled to produce a sTEVM. (c) Production of the nTEVM; the dMS are seeded with smooth muscle cells and the sheets are rolled around a tubular mandrel. (d) Timeline of the experiment comparing the time required for nTEVM versus sTEVM fabrication, showing that the use of a dMS reduces the production of a vascular construct by 1–2 weeks. (Reused with permission Bourget et al. 2012)

Vascular constructs were stained for SMC markers (a, c, e) a-SM-actin (red), and (b, d, f) calponin (red). nTEVM constructs produced from (a, b) dermal fibroblast, or (c, d) saphenous vein fibroblasts seeded with SMCs stained positive for both of these markers. (e, f) similar results were obtained for the sTEVM. Scale bar = 100 mm. (Reused with permission Bourget et al. 2012)
Another continuous related study was reported recently. In this study, an off-the-shelf tissue-engineered fibroblast-derived vascular scaffold (FDVS) was fabricated using a similar method to what we described earlier; then, the fibroblast sheets were rolled into a tubular structure using a mandrel, followed by decellularization and endothelialization. The EC seeded tubular endothelialized construct was perfused in a bioreactor for 1 week to obtain a TEBV. It took around 8 weeks to culture and produce the decellularized scaffold, and an additional 4 weeks for culture and endothelialization. The burst pressure of the vascular construct seeded with endothelial cells cultured in the bioreactor was 1498 ± 187 mmHg, indicating good mechanical integrity of the TEBV. Also, the decellularized scaffold could be stored for up to 1 month. The advantages of this study are that it allowed preparation of the scaffold in advance and storage until needed for production the TEBV for the patients, which significantly shifted a large part of the online production time (TEBV production) to the off-line phase time, and thus a reduction of the total online time to make the EC seeded TEBV from 11 weeks to 4 weeks (Tondreau et al., 2015).
In addition to the off-the-shelf scaffold for TEBV production described above using dermal fibroblast cells, other “off-the-shelf “grafts with burst pressure around 4000 mm Hg were developed by injecting fibroblast-seeded fibrin gel into a tubular mold and then culturing them for 2 weeks in static culture followed by 3 weeks in a bioreactor (Syedain et al. 2014). After the grafts were decellularized, the grafts were highly stable and could be stored at 4 °C. The grafts were implanted into the femoral artery of sheep for up to 24 weeks. Throughout the in vivo study, all grafts were patent without any mineralization after 24 weeks. No significant differences were observed in graft thickness, stiffness, and collagen concentration compared to native vessels, and complete endothelialization occurred at 24 weeks. Later, the off-the-shelf tissue engineered acellular vascular grafts were implanted in three growing lambs aged between 8 weeks and 50 weeks; the in vivo performance of the TEBVs was evaluated up to 44 weeks. The results demonstrated that the grafts were capable of growth after implantation; furthermore, the lambs’ weight increased by 366% after implantation (Syedain et al. 2016).
Different cell sources to produce TEBVs have attracted much attention recently. For instance, due to the similarity of DF to the human adipose-derived stromal cells (ASC) and the potential to increase TEBV patency, the possibility of using ASC as a novel source to produce TEBVs was investigated. Due to the limitations of obtaining sufficient contractile mature SMCs, ASCs were also studied as a cell source for functional SMCs (Rodriguez et al. 2006). The results demonstrated that ACS can produce mechanically stable TEBVs with sufficient endothelialization within 10 weeks (Fig. 8.3). Importantly, ACS-derived TEBVs can withstand physiological blood pressure and had better compliance than fibroblast-derived TEBVs (DF-TEBV) but similar burst pressures, elastic moduli, failure strains, and suture strengths as DF-TEBVs (Tondreau et al. 2015).

Top view confocal imaging of the endothelium on DF (a) and ASC (b) sheets as well as on the luminal surface of DF–TEVS (c) and ASC–TEVS (d). EC on top of DF and ASC cell sheets (a and b) were submitted to mechanical stimulation on a GyroTwisterTM 3-D shaker plate at 35 rpm for 6 days. These dynamic culture conditions induced the spindled shape of EC and their alignment parallel to the flow. Inversely, no mechanical stimulation was applied to EC inside the DF– and ASC–TEVS and they displayed a polygonal cobblestone-like shape. Labeling for PECAM is shown in red in panel a–b and in green in panels c–d. All nuclei were counterstained with Hoechst (in blue). Representative micrographs are presented (n = 3). Scale bars: 100 lm. (e). Schematic view of the tissue-engineered vascular substitutes (TEVS) production method. Mature TEVS were produced in 10 weeks. Typically, the first step consists in the isolation and amplification of dermal fibroblasts (DF) from a skin biopsy or adipose-derived stromal cells (ASC) from a lipoaspiration procedure. For this study, this step was performed beforehand and banked cells were used. Cell sheets of DF and ASC were produced by culturing cells for 3 weeks with ascorbate. Individual cell sheets were then rolled around 4.7-mm mandrels to form the TEVS. DF- and ASC-TEVS were maintained in culture with ascorbate for a maturation period of 5 weeks before histological and mechanical characterization. (Reproduced with permission Vallières et al. 2015)
Additionally, hiPSCs have been shown to differentiate into contractile SMCs on nanofiber scaffolds through transforming growth factor β1 (TGF-β1) treatment. The hiPSCs-derived SMCs showed high expression of SMC markers, such as α-SMA, CNN1, and SM22a (Wang et al. 2014). Furthermore, vascular smooth muscle cells (VSMCs) obtained from hiPSC differentiation were able to assemble into TEBVs using PLGA grafts. The VSMC TEBVs were generated by culturing the VSMC-seeded PLGA grafts in a bioreactor for 9 weeks. Importantly, the VSMC-based TEBVs remained complete and patent after the grafts were implanted into rat aorta for 2 weeks and could integrate into the native vessels of the host. This study demonstrated that hiPSC-TEBV might offer a chance to treat patients with dysfunctional vascular cells (Gui et al. 2016).
Moreover, hiPSCs can also differentiate into endothelial cells. For example, functional and durable blood vessels were successfully fabricated in vivo using endothelial cells and mesenchymal precursor cells derived from hiPSCs. Importantly, the endothelial cells derived from hiPSCs could form stable functional blood vessels and lasted for 280 days in an in vivo mouse model. These results indicate the potential of autologous hiPSC-derived vascular precursors to treat vascular disease (Samuel et al. 2013).
Besides hiPSCs, a bone marrow hMSC-based TEBV was developed by wrapping aligning hEPC-seeded hMSC sheets in a layer-by-layer fashion around a mandrel. It was reported to take around only 5 weeks to obtain a mature hMSC-based TEBV with hEPCs and a burst pressure around 342 mmHg, which is higher than that of human physiological vascular vein pressure (below 200 mmHg). This TEBV can mimic the native porcine femoral vein, as the TEBV diameter decreases upon additional of phenylephrine; also, the NO release amount upon drug treatment was similar to that of native porcine femoral vein (Jung et al. 2015).
In addition to improving the TEBV properties and fabrication process by use of a decellularized scaffold and alternative cell resources, a method combining pNIPAm-assisted cell sheet technology and electrospun PCL aligned nanofiber micropatterned scaffold was reported. The most important advantages of using this method were that: (1) the SMCs could differentiate into contractile human aortic smooth muscle cells (AoSMCs) for use in the TEBV, and (2) PCL can enhance the cell strength sheet in order to make the sheet easy to harvest, as PCL serves plays a similar role as internal elastic lamina in blood vessels (IEL) (Rayatpisheh et al. 2014).
Another study reported that TEBVs made using electrospun sulfated silk fibroin nanofibers demonstrated improved anticoagulant activities due to the sulfuric group incorporated into the silk fibroin nanofibers; also, the porcine vascular ECs and SMCs could attach and proliferate on the silk scaffold and expressed genes for specific cellular phenotype, such as SM-MHC2, α-SM actin, and collagen type I for SMCs, and CD146, vWF, and VE-C gene makers for ECs (Liu et al. 2011). Moreover, an alternative method to increase the scaffold anti-thrombogenicity included coating the scaffold with heparin, as heparin is a highly sulfated polysaccharide. It was reported that heparin coated decellularized vessels seeded with EPCs demonstrated improved anti-thrombogenicity and also inhibited neointimal hyperplasia after the grafts were implanted into dogs for 3 months. The EPCs might also contribute to the decrease in thrombogenicity (Zhou et al. 2012 2011). Interestingly, by combining the highly tubular interconnected porous SF scaffolds coated with heparin, the neovascularization in vivo was significantly improved (Zhu et al. 2014).
For synthetic nondegradable scaffolds in recent years, several studies focused on in vivo evaluation of the use of polyurethane as a scaffold for vascular prostheses. Polyurethane scaffolds were found to be non-cytotoxic and have good mechanical stability. The HUVEC-seeded polyurethane scaffolds were implanted into the abdominal area of rats for 7 weeks, 14 weeks, 3 months, and 6 months. A 95% patency rate of the polyurethane vascular scaffolds and host cell growth were observed (Grasl et al. 2010; Bergmeister et al. 2012). In addition, the in vivo performance, ability to form the vascular wall, and pro-inflammatory properties of TEBVs made using bacterially-synthesized cellulose (BC) as a scaffold were evaluated in a sheep model. However, the TEBVs only showed a burst strength around 800 mm Hg. Vascular wall-like structure formation comprised of vascular SMC and endothelial cells was observed in the scaffold, and the patency rate was around 50% (Scherner et al. 2014).
Besides the progress in TEBV production, some recent studies focused on generating tissue engineered blood vessel models of specific disease. For instance, a Hutchison-Gilford Progeria Syndrome (HGPS) model was developed using TEBVs made with Human iPSC-derived SMCs. Briefly, fibroblasts were first obtained from a HGPS patient; then, the fibroblast were converted into hiPSCs and then differentiated into HGPS-iSMCs. The TEBVs made with HGPS-iSMCs demonstrated HGPS phenotype, such as higher amounts of calcification and thicker walls, but decreased cellularity compared to the TEBVs made from normal healthy cells and MSC-derived TEBVs (Fig. 8.4) (Atchison et al. 2017). Another study reported building up a TEBV atherosclerosis model that consisted of LDL or TNF-α-treated EC and SMC, and LDL treated monocytes. The TEBV was fabricated by seeding the cells within a tubular PGA-P4HB composite scaffold and culturing the TEBV in a bioreactor. The total TEBV fabrication time was around 5 weeks. After the endothelium activation, monocytes were circulated into the system and the attachment of monocytes to the activated endothelium and monocyte migration through the endothelium cell layer were observed (Fig. 8.5) (Robert et al. 2013).

Progeria disease characterization of TEBVs fabricated from MSC or iPSC-derived SMC TEBVs from healthy and Progeria patients. (a) Histochemical analysis of HGPS iSMC, normal iSMC, and MSC TEBVs at week 4 with Alizarin Red staining (Scale bar, 200 μm). (b) Quantification of A, total area positive for Alizarin Red. (c) Representative images of immunofluorescence staining with fibronectin antibodies at week 4 of perfusion on TEBVs fabricated from HGPS iSMCs, normal iSMCs, and MSCs and seeded with hCB-ECs in the lumen (Scale bar, 50 μm). (d) Histochemical analysis of MSC, normal iSMC and HGPS iSMC TEBVs at week 4 with TUNEL staining. Red arrows indicate TUNEL positive cells and black arrows indicate TUNEL negative cells (Scale bar, 200 μm). (e) Histochemical analysis of HGPS iSMC, normal iSMC, and MSC TEBVs at week 4 with H&E (Scale bar, 200 μm). (f) The average thickness of MSC, normal iSMC and HGPS iSMC TEBVs at week 1 and week 4 based on H&E images in E. n = 3 TEBVs for each TEBV cell type. *P < 0.05, **P < 0.01, #P < 0.000. (Reused with permission Atchison et al. 2017)

(a, d) After pre-treatment in the absence or the presence of 3 h TNFa (10 ng/ml) (b, d) or 24 hours LDL (20 mg/ml) (c–i) 16106 fluorescently labelled monocytes (white) per ml were injected into the circulation loop and circulated for 24 h. Tissues were analyzed by confocal microscopy and after cryosectionning. In addition, monocytes remaining in the circulation were counted. More monocytes (white arrows) adhered after pre-treatment with TNFa (b) or LDL (c) compared to the not stimulated (a). Less monocyte remained into the circulation after TNFa or LDL pre-treatment compared to the absence of stimuli (d). Monocytes adhesion and migration in the tissue was further analyzed by microscopy of cryosections after LDL pre-treatment. Microscopic observations demonstrated adhesion and migration of monocytes through the endothelium (dash line) (e, g) and accumulation of monocytes into the tissue (f, h–i). Bars represent 200 mm (a–c, e–f) and 20 mm (g–i). (Reused with permission Robert et al. 2013)
As mentioned earlier, 3D-printing also has been used to make vascular structures. One study reported the development of 3D vascular networking by directly 3D bioprinting cell-resonpsive bio-ink that contains gelatin methacryloyl (GelMA), sodium alginate, and 4-arm poly(ethylene glycol)-tetra-acrylate (PEGTA). A stable structure was obtained by first crosslinking alginate with Ca2+ and then photocrosslinking the GelMA and PEGTA using UV light (Fig. 8.6). The cells could be encapsulated in the stable structures by mixing the cells with the polymer solution before any crosslinking procedures. The developed blend bio-ink also allowed the endothelial and stem cells to spread and proliferate after encapsulation, which resulted in the formation of perfusable vessels with organized structures (Fig. 8.7). The advantages of this reported method were that the size and diameter of the vascular tube made by this method are changeable, and a vascular network can be fabricated through this method. Also, this method was simple, as it only involved a multilayered coaxial nozzle device and cell-laden blend bio-ink (Jia et al. 2016). Another example was demonstrated by Chen et al. They developed a new platform for rapid construction of vascularized tissues through 3D microscale continuous optical bioprinting using HUVECs and 10 T1/2 cells, gelatin methacrylate, hyaluronic acid and photoinitiator lithium phenyl-2,4,6 trimethylbenzoylphosphinate (Fig. 8.8). The prevascularized tissues printed by this method showed good cell viability and HUVEC network formation in vivo after a 2 week implantation period (Zhu et al. 2017).

(a) Schematic diagram showing two independent crosslinking processes of the bioink, where alginate, GelMA, and PEGTA are ionically and covalently crosslinked, respectively, upon exposure to CaCl2 solution and UV light. (b) Schematics showing the procedure of bioprinting perfusable hollow tubes with the cell-encapsulating blend bioink and subsequent vascular formation. (c) The designed multilayered coaxial nozzles and schematic diagram showing fabrication of perfusable hollow tubes with constant diameters and changeable sizes. (Reused with permission Jia et al. 2016)

(a) Schematics and corresponding fluorescence micrographs of the bioprinted tubular constructs with different aspect ratios of internal grids (I) and numbers of layers (II). (b) Confocal micrographs showing a uniform 3D structure composed of 10 layers of bioprinted tubes (containing green fluorescent beads), which were perfused with red fluorescentmicrobeads inside the lumens. (c) Fluorescence photographs before (inset) and after injection with red fluorescent microbeads into the lumen of the single, continuous bioprinted tube. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.) (Reused with permission Jia et al. 2016)

3D bioprinting of the prevascularized tissue constructs. (a) Schematic of the bioprinting platform. (b) Bioprinted acellular construct featuring intended channels with gradient widths. (c) Bioprinted cellular construct with HUVECs and 10T1/2 (50:1) encapsulated in the intended channels. (d–f) Fluorescent images demonstrating the bioprinting of heterogeneous cell-laden tissue constructs with uniform channel width. HUVECs (red) are encapsulated in the intended channels and HepG2 (green) are encapsulated in the surrounding area. (g–i) Fluorescent images demonstrating the bioprinting of heterogeneous cell-laden tissue constructs with gradient channel widths. Scale bars, 250 μm. (Reused with permission Zhu et al. 2017)
4 Recent Progress in Clinical Studies of TEBV
While the creation of a TEBV in the laboratory setting is a major milestone in the progress towards blood vessel replacement, the ultimate goal is a fully functional, biocompatible, bioresponsive TEBV that is readily available and can be quickly utilized in the clinical environment. Researchers, in conjunction with medical doctors, have been working towards this goal for years now with varying levels of success (Fig. 8.9) (Drews et al. 2017). Here, we will focus on the most progress and advances within the recent years.

Gross image of a TEBV 13 years after implantation. The appearance is similar to native vein. LPA, left pulmonary artery; RPA, right pulmonary artery; SVC, superior vena cava; TEV, tissue-engineered vessel. (Reused with permission Drews et al. 2017)
4.1 Clinical Study Results
Many patients could benefit from a viable TEBV, such as those who must undergo arterial bypass surgery, who need grafts for dialysis access, and who have congenital cardiovascular defects, just to name a few (Patterson et al. 2012). This has been widely recognized for several decades, first in the 1980s with the first successful creation of living blood vessels in vitro by Weinberg and Bell, then with the first successful clinical application of a TEBV in 1999, with continuing progress up to the present day (Weinberg and Bell 1986). These were monumental steps toward the dream of fully autologous TEBV in clinical practice.
For example, Shinoka et al. conducted the first clinical trial in children with congenital heart disease in Japan in which 25 TEBV grafts were implanted between 2001 and 2004, with follow up that ranged from 4.3 to most recently 10.3 years. At the 5.8 year follow up, all grafts were patent, and there was no graft-related mortality, no evidence of formation of aneurysms in the grafts, no graft infection or rupture, and no ectopic calcification observed in the grafts (Hibino et al. 2010). Admittedly, after a mean follow-up time of 10.3 years there was some graft stenosis noted in 28% of the patients, but they either stabilized or were resolved after angioplasty and stenting procedures. Nevertheless, on the whole, grafts remained patent and functional, even years after the implantation procedure (Fig. 8.10) (Hibino et al. 2010; Drews et al. 2017). Furthermore, even at the 10.3 year follow up, there was no evidence of aneurysm formation, graft rupture, or calcification (Drews et al. 2017). In 2011, Shinoka et el began the first Food and Drug Administration-approved clinical trial in the United States investigating the use of TEBV in children with congenital heart defects; while the study is ongoing and results are to date unpublished, the trends have been similar to the Japanese study cohort. These exciting results demonstrate the feasibility and clinical potential of TEBV.

Postoperative growth of a TEBV. A TEBV was implanted in a 5-year-old patient undergoing a Fontan procedure. Angiography 2 years (a) and 11 years (b) after implantation demonstrate growth, with length of the graft increased from 43.4 to 60.4 cm. (Reused with permission Drews et al. 2017)
A second example is by the L’Heureux group, who implanted autologous TEBV fabricated utilizing sheet-based tissue engineering for hemodialysis access (L’Heureux et al. 2007a, b; McAllister et al. 2009). Cells were extracted from ten patients, grown into sheets of fibroblasts, wrapped around stainless steel mandrels, and allowed to mature and fuse together over a 10 week period; afterwards, they were endothelialized and preconditioned to in vivo flows and pressures (McAllister et al. 2009; L’Heureux et al. 2007a, b). The whole process took between 6 and 9 months, with an average period of 7.5 months. Cumulatively speaking, 78% of the grafts maintained primary patency 1 month after implantation, and 60% maintained patency at 3 and 6 months (McAllister et al. 2009). Thus, the L’Heureux group demonstrated successful TEBV implantation in an extremely difficult patient population (end-stage renal disease with at least one previous hemodialysis access graft failure), high arteriovenous hemodynamic flow loads and pressures, and repeated graft puncture for hemodialysis (L’Heureux et al. 2007a, b; McAllister et al. 2009).
The L’Heureux group also attempted the first human use of a nonliving, allogeneic, completely biological TEBV for hemodialysis access, which they reported in 2014 (Wystrychowski et al. 2014). These particular grafts were composed of extracellular matrix and fibroblasts which were dehydrated and devitalized prior to long term (6–9 months) storage. Three patients then received the allogeneic grafts as brachial-axillary arteriovenous shunts for hemodialysis access (Fig. 8.11). The grafts showed no signs of degradation, graft-related infection, or aneurysm formation; they also demonstrated hemodialysis functionality; however, problems with graft stenosis and thrombosis remained (Wystrychowski et al. 2014). Nevertheless, this study demonstrated the potential of nonliving, allogeneic, off the shelf grafts for human use: grafts maintained requisite mechanical strength and did not cause immune response, major milestones in TEBV development. Thus, this study represents a critical advance in the field, as it would be ideal for grafts to be available off the shelf without concern of degradation or loss of function during the storage time.

Graft of patient 1. (a) Preimplantation hematoxylin and eosin staining of the de-vitalized graft showed the same organization as a living graft, but with dense nuclear remnants in the outer layers. (b) Macroscopic view shows the implanted. (Reused with permission Wystrychowski et al. 2014)
4.2 Challenges
Despite decades of research, autologous vein and artery grafts remain the clinical gold standard today. Significant challenges remain before TEBV can be adopted for widespread clinical use. These challenges include: (1) similar burst pressure strength (>1700 mm Hg), (2) stable vessel diameter over long time periods, (3) biocompatibility, including non-thrombogenicity, non-inflammatory, maintenance of vessel patency, and structural stability of the TEBV, (4) demonstrated feasibility and consistency in the manufacturing process on a large scale, and (5) the prohibitively long time is takes to create an autologous TEBV (L’Heureux et al. 2007a, b). Furthermore, tissue engineering-based approaches must demonstrate improvements in efficacy and quality of life above synthetic material and native vein approaches that are common clinical practice today. Aside from the purely clinical aspect, challenges also exist in the regulatory, reimbursement, and cost-effectiveness areas for TEBV.
One of the greatest ongoing challenges to the clinical application of TEBV is postoperative vessel stenosis. Shinoka et al. reported asymptomatic graft narrowing (stenosis) in 24% (six) of patients who received a TEBV at 5.8 year follow up; four of these six patients underwent successful balloon angioplasty treatment (REF: late term results). The number of patients with TEBV stenosis increased to 28% (seven) by 10.3 years of follow up (Drews et al. 2017). While this treatment cohort was relatively small, these still represent unacceptable rates of postoperative vessel stenosis; thus, research is ongoing to find ways to limit and prevent stenosis after TEBV implantation.
L’Heureux also encountered challenges with both the autologous and allogeneic TEBV studies. This included instances of aneurysm formation within the graft, thrombosis, graft dilation, stenosis, and strong acute immune response (McAllister et al. 2009; Wystrychowski et al. 2014). Unacceptably long production times (7.5 months) also hamper widespread adoption of autologous graft use (McAllister et al. 2009). While L’Heureux et al. bypassed common challenges of TEBV (infection, poor mechanical strength, use of synthetic or exogenous materials, chemical modification leading to inflammation, etc.), many challenges remain.
5 Summary and Future Perspective
Although the concept, fabrication, and implementation of TEBVs have progressed quickly since the original idea more than 30 years ago, a great deal of research and work still remains. TEBVs clearly offer distinct advantages over synthetic grafts, which suffer from limitations such as thrombosis, poor patency, and inadequate mechanical properties; however, more progress is needed before TEBVs can replace autologous grafts, the current gold standard.
Recent advances in cell technology, such as the development of iPSCs, offer a variety of potential cell sources for use in TEBVs. Autologous fully differentiated cells, stem cells, induced pluripotent stem cells, and progenitor cells are all potential sources for use in fabricating TEBVs. Moreover, a variety of methods to create the TEBVs currently exist, including (but not limited to) cell assembly, self-assembly with cell sheets, 3D bioprinting, and layer-by-layer technology. Undoubtedly, as scientific progress is made, better and higher-precision methods will be developed and utilized.
Importantly, TEBVs have already been utilized both in animal models as well as within the clinic for human use. Significant reductions in production and maturation time, off-the-shelf TEBV scaffolds, and improvements in cell sources have all contributed to these recent advances, utilizations, and in vivo studies with TEBVs. Although major milestones have been accomplished, including successful long-term TEBV implantation in humans for patients with congenital heart disease and hemodialysis access, many challenges remain to be overcome before TEBVs will be accepted for widespread clinical use. Despite these challenges, TEBVs have a bright future, and there is little doubt that advances in science and technology will lead to improvements in TEBVs and subsequent pervasive use within the clinic.
References
Atchison L, Zhang H, Cao K, Truskey GA (2017) A tissue engineered blood vessel model of Hutchinson-Gilford progeria syndrome using human iPSC-derived smooth muscle cells. Sci Rep 7(1):8168–8180. https://doi.org/10.1038/s41598-017-08632-4
Au P, Daheron LM, Duda DG, Cohen KS, Tyrrell JA, Lanning RM, Fukumura D, Scadden DT, Jain RK (2008) Differential in vivo potential of endothelial progenitor cells from human umbilical cord blood and adult peripheral blood to form functional long-lasting vessels. Blood 111(3):1302–1305. https://doi.org/10.1182/blood-2007-06-094318
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C (2017) Heart disease and stroke statistics—2017 update: a report from the American Heart Association. Circulation 135(10):e146–e603. https://doi.org/10.1161/CIR.0000000000000485
Bergmeister H, Grasl C, Walter I, Plasenzotti R, Stoiber M, Schreiber C, Losert U, Weigel G, Schima H (2012) Electrospun small-diameter polyurethane vascular grafts: ingrowth and differentiation of vascular-specific host cells. Artif Organs 36(1):54–61. https://doi.org/10.1111/j.1525-1594.2011.01297.x
Bishop AE, Buttery LDK, Polak JM (2002) Embryonic stem cells. J Pathol 197(4):424–429
Bourget J-M, Gauvin R, Larouche D, Lavoie A, Labbé R, Auger FA, Germain L (2012) Human fibroblast-derived ECM as a scaffold for vascular tissue engineering. Biomaterials 33(36):9205–9213. https://doi.org/10.1016/j.biomaterials.2012.09.015
Buijtenhuijs P, Buttafoco L, Poot AA, Daamen WF, Van Kuppevelt TH, Dijkstra PJ, De Vos RA, Sterk LMT, Geelkerken BR, Feijen J (2004) Tissue engineering of blood vessels: characterization of smooth-muscle cells for culturing on collagen-and-elastin-based scaffolds. Biotechnol Appl Biochem 39(2):141–149. https://doi.org/10.1042/BA20030105
Cheng S-T, Chen Z-F, Chen G-Q (2008) The expression of cross-linked elastin by rabbit blood vessel smooth muscle cells cultured in polyhydroxyalkanoate scaffolds. Biomaterials 29(31):4187–4194. https://doi.org/10.1016/j.biomaterials.2008.07.022
Drews JD, Miyachi H, Shinoka T (2017) Tissue-engineered vascular grafts for congenital cardiac disease: clinical experience and current status. Trends Cardiovasc Med 27:521. https://doi.org/10.1016/j.tcm.2017.06.013
Ercolani E, Del Gaudio C, Bianco A (2015) Vascular tissue engineering of small-diameter blood vessels: reviewing the electrospinning approach. J Tissue Eng Regen Med 9(8):861–888. https://doi.org/10.1002/term.1697
Fernandez CE, Achneck HE, Reichert WM, Truskey GA (2014) Biological and engineering design considerations for vascular tissue engineered blood vessels (TEBVs). Curr Opin Chem Eng 3:83–90. https://doi.org/10.1016/j.coche.2013.12.001
Grasl C, Bergmeister H, Stoiber M, Schima H, Weigel G (2010) Electrospun polyurethane vascular grafts: in vitro mechanical behavior and endothelial adhesion molecule expression. J Biomed Mater Res A 93(2):716–723. https://doi.org/10.1002/jbm.a.32584
Gui L, Dash BC, Luo J, Qin L, Zhao L, Yamamoto K, Hashimoto T, Wu H, Dardik A, Tellides G (2016) Implantable tissue-engineered blood vessels from human induced pluripotent stem cells. Biomaterials 102:120–129. https://doi.org/10.1016/j.biomaterials.2016.06.010
Harris LJ, Abdollahi H, Zhang P, McIlhenny S, Tulenko TN, DiMuzio PJ (2011) Differentiation of adult stem cells into smooth muscle for vascular tissue engineering. J Surg Res 168(2):306–314. https://doi.org/10.1016/j.jss.2009.08.001
Hasan A, Memic A, Annabi N, Hossain M, Paul A, Dokmeci MR, Dehghani F, Khademhosseini A (2014) Electrospun scaffolds for tissue engineering of vascular grafts. Acta Biomater 10(1):11–25. https://doi.org/10.1016/j.actbio.2013.08.022
Hibino N, McGillicuddy E, Matsumura G, Ichihara Y, Naito Y, Breuer C, Shinoka T (2010) Late-term results of tissue-engineered vascular grafts in humans. J Thorac Cardiovasc Surg 139(2):431–436. e432. https://doi.org/10.1016/j.jtcvs.2009.09.057
Huang NF, Niiyama H, Peter C, De A, Natkunam Y, Fleissner F, Li Z, Rollins MD, Wu JC, Gambhir SS (2010) Embryonic stem cell–derived endothelial cells engraft into the ischemic hindlimb and restore perfusion. Arterioscler Thromb Vasc Biol 30(5):984–991. https://doi.org/10.1161/ATVBAHA
Jia W, Gungor-Ozkerim PS, Zhang YS, Yue K, Zhu K, Liu W, Pi Q, Byambaa B, Dokmeci MR, Shin SR (2016) Direct 3D bioprinting of perfusable vascular constructs using a blend bioink. Biomaterials 106:58–68. https://doi.org/10.1016/j.biomaterials
Jung Y, Ji H, Chen Z, Chan HF, Atchison L, Klitzman B, Truskey G, Leong KW (2015) Scaffold-free, human mesenchymal stem cell-based tissue engineered blood vessels. Sci Rep 5:15116. https://doi.org/10.1038/srep15116
Kang H-W, Lee SJ, Ko IK, Kengla C, Yoo JJ, Atala A (2016) A 3D bioprinting system to produce human-scale tissue constructs with structural integrity. Nat Biotechnol 34(3):312–319. https://doi.org/10.1038/nbt.3413
Kaushal S, Amiel GE, Guleserian KJ, Shapira OM, Perry T, Sutherland FW, Rabkin E, Moran AM, Schoen FJ, Atala A (2001) Functional small-diameter neovessels created using endothelial progenitor cells expanded ex vivo. Nat Med 7(9):1035–1040. https://doi.org/10.1038/nm0901-1035
Krawiec JT, Vorp DA (2012) Adult stem cell-based tissue engineered blood vessels: a review. Biomaterials 33(12):3388–3400. https://doi.org/10.1016/j.biomaterials
Kushida A, Yamato M, Konno C, Kikuchi A, Sakurai Y, Okano T (1999) Decrease in culture temperature releases monolayer endothelial cell sheets together with deposited fibronectin matrix from temperature-responsive culture surfaces. J Biomed Mater Res A 45(4):355–362. https://doi.org/10.1002/(SICI)1097-4636(19990615)45:4<355::AID-JBM10>3.0.CO;2-7
L’Heureux N, Pâquet S, Labbé R, Germain L, Auger FA (1998) A completely biological tissue-engineered human blood vessel. FASEB J 12(1):47–56
L’Heureux N, Dusserre N, Konig G, Victor B, Keire P, Wight TN, Chronos NA, Kyles AE, Gregory CR, Hoyt G (2006) Human tissue engineered blood vessel for adult arterial revascularization. Nat Med 12(3):361. https://doi.org/10.1038/nm1364
L’Heureux N, Dusserre N, Marini A, Garrido S, De La Fuente L, McAllister T (2007a) Technology insight: the evolution of tissue-engineered vascular grafts--from research to clinical practice. Nat Clin Pract Cardiovasc Med 4(7):389. https://doi.org/10.1038/ncpcardio0930
L’Heureux N, McAllister TN, de la Fuente LM (2007b) Tissue-engineered blood vessel for adult arterial revascularization. N Engl J Med 357(14):1451–1453. https://doi.org/10.1056/NEJMc071536
Lian Q, Zhang Y, Zhang J, Zhang HK, Wu X, Zhang Y, Lam FF-Y, Kang S, Xia JC, Lai W-H (2010) Functional mesenchymal stem cells derived from human induced pluripotent stem cells attenuate limb ischemia in mice. Circulation 121(9):1113–1123. https://doi.org/10.1161/CIRCULATIONAHA.109.898312
Liu H, Li X, Zhou G, Fan H, Fan Y (2011) Electrospun sulfated silk fibroin nanofibrous scaffolds for vascular tissue engineering. Biomaterials 32(15):3784–3793. https://doi.org/10.1016/j.biomaterials.2011.02.002
Ma H, Hu J, Ma PX (2010) Polymer scaffolds for small-diameter vascular tissue engineering. Adv Funct Mater 20(17):2833–2841. https://doi.org/10.1002/adfm.201000922
Matsusaki M, Kadowaki K, Nakahara Y, Akashi M (2007) Fabrication of cellular multilayers with nanometer-sized extracellular matrix films. Angew Chem 119(25):4773–4776. https://doi.org/10.1002/anie.200701089
Matsuura K, Utoh R, Nagase K, Okano T (2014) Cell sheet approach for tissue engineering and regenerative medicine. J Control Release 190:228–239. https://doi.org/10.1016/j.jconrel.2014.05.024
McAllister TN, Maruszewski M, Garrido SA, Wystrychowski W, Dusserre N, Marini A, Zagalski K, Fiorillo A, Avila H, Manglano X (2009) Effectiveness of haemodialysis access with an autologous tissue-engineered vascular graft: a multicentre cohort study. Lancet 373(9673):1440–1446. https://doi.org/10.1016/S0140-6736(09)60248-8
McQuade K, Gable D, Hohman S, Pearl G, Theune B (2009) Randomized comparison of ePTFE/nitinol self-expanding stent graft vs prosthetic femoralpopliteal bypass in the treatment of superficial femoral artery occlusive disease. J Vasc Surg 49(1):109–116 e9
Melero-Martin JM, De Obaldia ME, Kang S-Y, Khan ZA, Yuan L, Oettgen P, Bischoff J (2008) Engineering robust and functional vascular networks in vivo with human adult and cord blood–derived progenitor cells. Circ Res 103(2):194–202. https://doi.org/10.1161/CIRCRESAHA.108.178590
Motlagh D, Yang J, Lui KY, Webb AR, Ameer GA (2006) Hemocompatibility evaluation of poly (glycerol-sebacate) in vitro for vascular tissue engineering. Biomaterials 27(24):4315–4324
Murphy SV, Atala A (2014) 3D bioprinting of tissues and organs. Nat Biotechnol 32(8):773–785
Naito Y, Shinoka T, Duncan D, Hibino N, Solomon D, Cleary M, Rathore A, Fein C, Church S, Breuer C (2011) Vascular tissue engineering: towards the next generation vascular grafts. Adv Drug Deliv Rev 63(4):312–323. https://doi.org/10.1016/j.addr.2011.03.001
Niklason L, Gao J, Abbott W, Hirschi K, Houser S, Marini R, Langer R (1999) Functional arteries grown in vitro. Science 284(5413):489–493. https://doi.org/10.1126/science.284.5413.489
Nishiguchi A, Yoshida H, Matsusaki M, Akashi M (2011) Rapid construction of three-dimensional multilayered tissues with endothelial tube networks by the cell-accumulation technique. Adv Mater 23(31):3506–3510. https://doi.org/10.1002/adma.201101787
Okano T, Yamada N, Okuhara M, Sakai H, Sakurai Y (1995) Mechanism of cell detachment from temperature-modulated, hydrophilic-hydrophobic polymer surfaces. Biomaterials 16(4):297–303. https://doi.org/10.1016/0142-9612(95)93257-E
Patterson JT, Gilliland T, Maxfield MW, Church S, Naito Y, Shinoka T, Breuer CK (2012) Tissue-engineered vascular grafts for use in the treatment of congenital heart disease: from the bench to the clinic and back again. Regen Med 7(3):409–419. https://doi.org/10.2217/rme.12.12
Quint C, Kondo Y, Manson RJ, Lawson JH, Dardik A, Niklason LE (2011) Decellularized tissue-engineered blood vessel as an arterial conduit. Proc Natl Acad Sci 108(22):9214–9219. https://doi.org/10.1073/pnas.1019506108
Rayatpisheh S, Heath DE, Shakouri A, Rujitanaroj P-O, Chew SY, Chan-Park MB (2014) Combining cell sheet technology and electrospun scaffolding for engineered tubular, aligned, and contractile blood vessels. Biomaterials 35(9):2713–2719
Rippon H, Bishop A (2004) Embryonic stem cells. Cell Prolif 37(1):23–34. https://doi.org/10.1111/j.1365-2184.2004.00298.x
Robert J, Weber B, Frese L, Emmert MY, Schmidt D, von Eckardstein A, Rohrer L, Hoerstrup SP (2013) A three-dimensional engineered artery model for in vitro atherosclerosis research. PLoS One 8(11):e79821. https://doi.org/10.1371/journal.pone.0079821
Rodríguez LV, Alfonso Z, Zhang R, Leung J, Wu B, Ignarro LJ (2006) Clonogenic multipotent stem cells in human adipose tissue differentiate into functional smooth muscle cells. Proc Natl Acad Sci 103(32):12167–12172. https://doi.org/10.1073/pnas.0604850103
Samuel R, Daheron L, Liao S, Vardam T, Kamoun WS, Batista A, Buecker C, Schäfer R, Han X, Au P (2013) Generation of functionally competent and durable engineered blood vessels from human induced pluripotent stem cells. Proc Natl Acad Sci 110(31):12774–12779. https://doi.org/10.1073/pnas.1310675110
Scherner M, Reutter S, Klemm D, Sterner-Kock A, Guschlbauer M, Richter T, Langebartels G, Madershahian N, Wahlers T, Wippermann J (2014) In vivo application of tissue-engineered blood vessels of bacterial cellulose as small arterial substitutes: proof of concept? J Surg Res 189(2):340–347. https://doi.org/10.1016/j.jss.2014.02.011
Seifu DG, Purnama A, Mequanint K, Mantovani D (2013) Small-diameter vascular tissue engineering. Nat Rev Cardiol 10(7):410–421. https://doi.org/10.1038/nrcardio.2013.77
Shepherd BR, Jay SM, Saltzman WM, Tellides G, Pober JS (2008) Human aortic smooth muscle cells promote arteriole formation by coengrafted endothelial cells. Tissue Eng A 15(1):165–173. https://doi.org/10.1089/ten.tea.2008.0010
Syedain ZH, Meier LA, Lahti MT, Johnson SL, Tranquillo RT (2014) Implantation of completely biological engineered grafts following decellularization into the sheep femoral artery. Tissue Eng A 20(11–12):1726–1734. https://doi.org/10.1089/ten.TEA.2013.0550
Syedain Z, Reimer J, Lahti M, Berry J, Johnson S, Tranquillo RT (2016) Tissue engineering of acellular vascular grafts capable of somatic growth in young lambs. Nat Commun 7:12951
Szmitko PE (2003) Endothelial progenitor cells: new hope for a broken heart. Circulation 107(24):3093–3100
Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM (1998) Embryonic stem cell lines derived from human blastocysts. Science 282(5391):1145–1147. https://doi.org/10.1126/science.282.5391.1145
Tondreau MY, Laterreur V, Gauvin R, Vallières K, Bourget J-M, Lacroix D, Tremblay C, Germain L, Ruel J, Auger FA (2015) Mechanical properties of endothelialized fibroblast-derived vascular scaffolds stimulated in a bioreactor. Acta Biomater 18:176–185. https://doi.org/10.1016/j.actbio.2015.02.026
Urbich C, Dimmeler S (2004) Endothelial progenitor cells. Circ Res 95(4):343–353. https://doi.org/10.1161/01.RES.0000137877.89448.78
Vallières K, Laterreur V, Tondreau MY, Ruel J, Germain L, Fradette J, Auger FA (2015) Human adipose-derived stromal cells for the production of completely autologous self-assembled tissue-engineered vascular substitutes. Acta Biomater 24:209–219
Wang ZZ, Au P, Chen T, Shao Y, Daheron LM, Bai H, Arzigian M, Fukumura D, Jain RK, Scadden DT (2007) Endothelial cells derived from human embryonic stem cells form durable blood vessels in vivo. Nat Biotechnol 25(3):317. https://doi.org/10.1038/nbt1287
Wang Y, Hu J, Jiao J, Liu Z, Zhou Z, Zhao C, Chang L-J, Chen YE, Ma PX, Yang B (2014) Engineering vascular tissue with functional smooth muscle cells derived from human iPS cells and nanofibrous scaffolds. Biomaterials 35(32):8960–8969. https://doi.org/10.1016/j.biomaterials.2014.07.011
Weinberg CB, Bell E (1986) A blood vessel model constructed from collagen and cultured vascular cells. Science 231:397–401. https://doi.org/10.1126/science.2934816
Wu X, Rabkin-Aikawa E, Guleserian KJ, Perry TE, Masuda Y, Sutherland FW, Schoen FJ, Mayer JE, Bischoff J (2004) Tissue-engineered microvessels on three-dimensional biodegradable scaffolds using human endothelial progenitor cells. Am J Phys Heart Circ Phys 287(2):H480–H487. https://doi.org/10.1152/ajpheart.01232.2003
Wystrychowski W, McAllister TN, Zagalski K, Dusserre N, Cierpka L, L’Heureux N (2014) First human use of an allogeneic tissue-engineered vascular graft for hemodialysis access. J Vasc Surg 60(5):1353–1357
Xing Q, Yates K, Tahtinen M, Shearier E, Qian Z, Zhao F (2014) Decellularization of fibroblast cell sheets for natural extracellular matrix scaffold preparation. Tissue Eng Part C Methods 21(1):77–87. https://doi.org/10.1089/ten.TEC.2013.0666
Yamada N, Okano T, Sakai H, Karikusa F, Sawasaki Y, Sakurai Y (1990) Thermo-responsive polymeric surfaces; control of attachment and detachment of cultured cells. Macromol Rapid Commun 11(11):571–576. https://doi.org/10.1002/marc.1990.030111109
Zeng W, Yuan W, Li L, Mi J, Xu S, Wen C, Zhou Z, Sun J, Ying D, Yang M (2010) The promotion of endothelial progenitor cells recruitment by nerve growth factors in tissue-engineered blood vessels. Biomaterials 31(7):1636–1645. https://doi.org/10.1016/j.biomaterials.2009.11.037
Zhang J, Huang H, Ju R, Chen K, Li S, Wang W, Yan Y (2017) In vivo biocompatibility and hemocompatibility of a polytetrafluoroethylene small diameter vascular graft modified with sulfonated silk fibroin. Am J Surg 213(1):87–93. https://doi.org/10.1016/j.amjsurg.2016.04.005
Zhou M, Liu Z, Liu C, Jiang X, Wei Z, Qiao W, Ran F, Wang W, Qiao T, Liu C (2012) Tissue engineering of small-diameter vascular grafts by endothelial progenitor cells seeding heparin-coated decellularized scaffolds. J Biomed Mater Res B Appl Biomater 100(1):111–120. https://doi.org/10.1002/jbm.b.31928
Zhu M, Wang K, Mei J, Li C, Zhang J, Zheng W, An D, Xiao N, Zhao Q, Kong D (2014) Fabrication of highly interconnected porous silk fibroin scaffolds for potential use as vascular grafts. Acta Biomater 10(5):2014. https://doi.org/10.1016/j.actbio.2014.01.022
Zhu W, Xin Q, Zhu J, Ma X, Patel S, Liu J, Wang P, Lai CSE, Gou M, Xu Y, Zhang K, Chen S (2017) Direct 3D bioprinting of prevascularized tissue constructs with complex microarchitecture. Biomaterials 124:106–115
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Chen, J., Alexander, G.C., Bobba, P.S., Jun, HW. (2018). Recent Progress in Vascular Tissue-Engineered Blood Vessels. In: Noh, I. (eds) Biomimetic Medical Materials. Advances in Experimental Medicine and Biology, vol 1064. Springer, Singapore. https://doi.org/10.1007/978-981-13-0445-3_8
Download citation
DOI: https://doi.org/10.1007/978-981-13-0445-3_8
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-0444-6
Online ISBN: 978-981-13-0445-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)