
Abstract
Staphylococcus aureus is a clinically important pathogen mainly causing hospital borne infections. These bacterial infections range from mild skin infections to serious health threats like endocarditis, osteomyelitis, and pneumonia. Few strains have developed resistance against antibiotics used to treat S. aureus infections and are termed as Methicillin Resistant S. aureus strains. The pathogen releases Auto Inducing Peptides to establish cell density dependent inter-cell communication, also known as quorum sensing (QS). QS results in the expression of accessory gene regulator system. It causes successful biofilm formation and enhanced expression of toxins. QS mediated biofilm formation provides an additional resistance against the antibiotics used. An innovative therapeutic approach has been studied vastly in last decade to deal with severe infections using specific QS inhibitors (QSIs). This chapter comprehensively describes the QSIs studied to control the infections caused by S. aureus strains.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
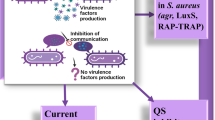
Quorum sensing (QS) is reported to be critical for various human pathogens for example Staphylococcus aureus, Staphylococcus epidermidis, Pseudomonas aeruginosa, Serratia pneumonia, Yersinia pestis, Brucella abortus and Burkholderia pseudomallei (Swift et al. 2001; Williams 2002). The successful establishment of disease is governed through the pathogen’s ability to invade and forms biofilm in the host. This also facilitates the pathogen to avoid antibiotic mediated killing in vivo. There are various methods to prevent the formation and disrupt the pathogenic biofilms in the host for examples nanoparticles, azithromycin, etc (Agarwala et al. 2014; Gui et al. 2014; Wadhwani et al. 2016; Ahiwale et al. 2017). Interfering bacterial QS through QS inhibitors (QSIs) is a novel therapeutic approach to curb the bacterial infection (Kumar et al. 2015). Staphylococcus aureus is the major cause of nosocomial infections in USA and developing countries as well. S. aureus secretes few virulence factors which are under the control of agr operon. The agr operon encodes AgrB (membrane bound peptidase), AgrD (precursor of AIP), AgrC (membrane bound histidine kinase), and AgrA (response regulator). The promoter region P2 governs polycistronic operon agrBDCA while the adjacent promoter P3 encodes mRNA for δ-hemolysin and pleiotropic regulator of other virulence genes (Bronesky et al. 2016). AgrB cleaves AgrD into a thiolactone intermediate which is secreted to undergo subsequent cleavage to yield mature AIP. AIP is then sensed by the receptor histidine kinase AgrC, thus phosphorylating itself and response regulator. The activated response regulator AgrA binds to the P2 and P3 to enhance the expression through these promoters. RNA III encoded RNA which acts as an antisense and interferes with the translation of ‘repressor of toxin’ Rot, an inhibitor of α-hemolysin. Another QS system RAP/TRAP consists of two proteins, which are RAP (RNAIII-activating protein) and TRAP (target RNAIII-activating protein). RAP activates the production of toxins by phosphorylating the histidine amino acid of TRAP, when RAP reaches a certain threshold concentration (Balaban et al. 2001). Emergence of multidrug resistance in S. aureus strains is a serious public health issue (Kalia 2014a, 2015). Therefore, an alternative approach of targeting the QS molecules of bacteria is a viable option to effectively treat the infections (Koul and Kalia 2017) (Fig. 15.1).

Inhibition of Agr and RAP/TRAP systems of S. aureus using quorum sensing inhibitors. Left panel shows Agr inhibition while Right panel shows RAP/TRAP inhibition. Red Dotted Lines indicate specific inhibition steps in the pathways caused by the inhibitors
2 QS Inhibitors in Controlling S. aureus Infections
QSIs are antimicrobial compounds that interfere with the ability of bacteria to communicate in a colony (Kalia and Purohit 2011, Kumar et al. 2013). They should be specific for the protein to be targeted to avoid killing of host and its microbiome. Specific QSIs have been researched for their application in the prophylaxis of S. aureus borne infections. Structurally QSIs can belong to different categories of macromolecules for examples, peptides, sugar, amides or their analogues. Following, we have discussed the inhibitors showing significant potency against the infection caused by methicillin resistant S. aureus (Table 15.1).
3 Savirin Inhibits Growth via Interaction with AgrA
High throughput screening of small molecule inhibitors led to the discovery of the Savirin (S taphylococcus a ureus virulence inhibitor), which specifically inhibits the agr mediated signaling in S. aureus without affecting the growth of skin commensal S. epidermidis. Chemically, Savirin is 3-(4-propan-2-ylphenyl) sulfonyl-1H-triazolo (1,5-a) quinazolin-5-one (Sully et al. 2014). Apart from its molecular weight, the lipophilic nature of savirin makes it an interesting drug candidate for treatment of S. aureus infections (Lipinski et al. 2001). S. aureus possess a two-component system (TCS) which comprises of AgrC, histidine kinase and AgrA, response regulator. The extracellular autoinducing peptide (AIP) binds to the transmembrane protein AgrC, which in turn phosphorylates AgrA. Thus activated AgrA binds to promoter P2 and P3 encodes AgrB, AgrD, AgrC, AgrA and RNAIII respectively. RNA levels of RNAIII increases dramatically upon AgrA binding (Koenig et al. 2004). RNAIII primarily functions as an antisense and is earlier reported to inhibit transcription of repressor of toxins such as rot (Boisset et al. 2007). RNAIII mediated inhibition of rot expression thus increases transcription of downstream virulence factors such as α-hemolysin (Yarwood and Schlievert 2003; Le and Otto 2015). Recently, it was shown that total activity of α-hemolysin was significantly reduced in savirin-treated bacterial supernatants of MRSA isolated from different sites of infection. Since histidine kinase domain of AgrC is conserved in S. aureus and S. epidermidis (a skin commensal), therefore AgrA was selected as a target for drug development using high throughput screening. C-terminal DNA binding domain of AgrA was used to identify the drug candidates using swissdock, an online server. The study reveals that savirin binds to the CTD of AgrA from S. aureus (SA_AgrA) however was unable to interact with AgrA from S. epidermidis (SE_AgrA). DNA binding domain of SA_AgrA and SE_AgrA differs in two positions (229, Tyr to Phe) and (227, His to Asn), which significantly reduces the binding affinity of Savirin (Sully et al. 2014). The crystal structure of LytTR domain of the SA_AgrA was analyzed in a DNA unbound form. At the same time, screening a library of small molecules reveals that the AgrA-DNA interactions might destabilize by targeting an exposed hydrophobic cleft with a small molecule (Leonard et al. 2012). Mechanistic studies involving a novel reporter strain of SA_AgrA activation and electromobility shift assays have demonstrated the efficacy of savirin, both in vivo and in vitro, by inhibiting the binding function of SA_AgrA with DNA in S. aureus. These evidences suggested that savirin impedes the function of SA_AgrA, thus preventing the transcription from agrBDCA promoter P2 and RNAIII promoter P3 and other agr-regulated virulence genes. The clinical isolate of S. aureus was studied for few generations for the emergence of resistance to savirin both in vivo and in vitro. Even the persistent exposure to the drug savirin could not lead to the emergence of resistant strain (Sully et al. 2014). Unlike conventional antibiotics, savirin is highly specific and does not foster stress responses and disrupt membrane integrity (Defoirdt et al. 2013). Moreover, AgrA has similar sequence in all four S. aureus agr groups, making it more desirable therapeutic target (Wang and Muir 2016).
4 Solonamide A and B Act As Antagonist of AgrC
Two of the most important strains of MRSA are “hospital acquired” or HA-MRSA and “community acquired” or CA-MRSA (Gordon and Lowy 2008). In general, HA-MRSA is an opportunistic pathogen unable to infect healthy individuals while in recent years, the most common strain of CA-MRSA, USA300 has emerged as a serious concern due to its capability of infecting healthy individuals (Loughman et al. 2009). Increased resistance to different antibiotics in MRSA has led to the development of new therapeutic strategies. The agr system regulates the expression of virulence gene in S. aureus (Gordon and Lowy 2008). Therefore, anti-virulence therapy has received an appreciable interest for combating S. aureus infections (Wright and Sutherland 2007). Recently, two novel compounds were isolated from the marine bacterium Photobacterium halotolerans (strain S2753) named Solonamides A and B. They impede agr QS of S. aureus and subsequently disrupt the expression of virulence gene. Based on NMR data, solonamides structure was characterized as cyclodepsipeptides consisting of a 3-hydroxy fatty acid and four amino acids (phenylalanine, alanine and two leucines). It was also found that solonamide A contains a 3-hydroxyhexanoic acid (Hha), whereas solonamide B is made up of 3-hydroxyoctanoic acid (Hoa) (Mansson et al. 2011). In vitro, it was shown that signals downstream to the agr sensing system upregulates the expression of α-hemolysin encoded by hla and downregulates the expression of cell surface protein such as protein A encoded by spa at the beginning of the stationary growth phase in S. aureus (Vuong et al. 2000). Northern blot analysis examined the amount of mRNA isolated from strain of CA-MRSA, USA300 and S. aureus 8325–4 after the treatment with solonamide, verified the interference of these compounds in virulence gene expression. Solonamide B minimizes the expression of hla and rnaIII and increases the expression of spa. Whereas solonamide A has been shown to increase the expression of spa however there were minor reduction in hla and rnaIII expression in USA300 and 8325-4 strains (Mansson et al. 2011). The primary host defense in opposition to S. aureus infections are neutrophils and therefore, lysis of neutrophils is crucial for the virulence of these strains. The PSMs and α-hemolysin are two major virulence factors; both are remarkable at killing immune cells and responsible for an increased virulence of CA-MRSA (Bubeck Wardenburg et al. 2007; Wang et al. 2007). It was reported that solonamide B reduces the expression of virulence factors such as phenol soluble modulins, the PSMs and α-hemolysin in USA300 strain. Additionally the toxicity of supernatants was shown to be minimized when tested against human neutrophils. AgrA, the response regulator of agr QS system directly controls the expression of PSMs. Apart from disrupting expression of genes via RNAIII, solonamide B also affects expression of PSMs through AgrA (Nielsen et al. 2014). The QS signal molecules of S. aureus are the cyclic thiolactone peptides generally known as autoinducing peptides (AIPs). AIPs activate the agr QS system and thus controlling the virulence gene expression via the effector molecule RNAIII (Novick and Geisinger 2008). Depending on the strain, there are four distinct types of AIPs in which AIP of one type specifically binds to its cognate receptor agrC (agr signal receptor) but shows antagonistic activity in strains harboring other types of AIPs (George and Muir 2007). It was suggested that solonamides are the competitive inhibitors of the agr system as they have structures similar to the AIPs. Solonamide is a lactone whereas AIP is a thiolactone. However in recent studies, AIP analogues harboring lactone instead of thiolactone have been found to act as competitive inhibitors. It was also found that both solonamides contain hydrophobic phenylalanine and leucine residues that are crucial for the impediment of the agr response (Mayville et al. 1999; Mansson et al. 2011). Moreover, Baldry and colleagues chemically synthesized the solonamide analogues to improve its anti-virulence candidacy (Baldry et al. 2016). These findings suggest that inhibition via solonamides is probable alternative therapeutic approach to treat MRSA infections.
5 Apolipoprotein Act As Sequester of AIP
In recent times serum lipoproteins (LP) have emerged as a molecule having a dual role of contributing to cholesterol homeostasis as well as host innate defense. It has been established that very low levels of serum lipoprotein (hypolipoproteinemia) is related to increased bacterial infection in critically ill patients (Han 2010; Femling et al. 2013). In this respect Apolipoprotein B (apoB100), a 4536 amino acid protein is essential for the formation of these LPs (LDLs, VLDLs, Chylomicrons, etc.). Recent studies have shown that apoB100 disrupts virulence factor expression of S. aureus thus limiting its pathogenesis (Hall et al. 2013). It is done by binding of apoB100 to AIPs and thus disrupting agr mediated virulence. While in human intestinal enterocytes, a truncated form of apoB100 is produced, which is apoB48. It is studied that enteral feeding in critically ill patients leads to reduced risk of infection as compared to parenteral feeding, which suggests the importance of apoB48 in host innate immune response, however the mechanism is unknown (Kattelmann et al. 2006). This led to the development of new quorum quenching inhibitor i.e. apoB48 to control agr mediated S. aureus QS by Bradley and colleague. It was seen that apoB48 and apoB100 antagonizes agr signalling with similar IC50 of 3.5 and 2.3 nM, respectively. The IC50 values were found to below the reported EC50 (28 nM) for activation of agr system via AIP1. This could provide effective protection against S. aureus infections. In vivo studies also showed that exogenous apoB48 treated mice infected with S. aureus USA300 strain had decreased bacterial burden at site of infection as compared to untreated mice. This data makes apoB48 an important inhibitor of agr signalling mediated QS in vivo and providing protection against S. aureus infection (Elmore et al. 2015). Thus apolipoprotein can prove to be a global inhibitor of QS and warrants more research for its use as therapeutic agent.
6 Non Cognate AIP
QS in S. aureus is controlled by the chromosome locus named agr (Accessory Gene Regulator). It is an operon system, genes of which encodes for and also sense a small peptide autoinducer named AIP (Autoinducing Peptide) (Novick and Geisinger 2008). AIP consists of 7–9 amino acid residues and harbours a five membered ring wherein the C-terminal forms the thiolactone bond with cysteine (central position). This arrangement is crucial for AIP’s activity (Ji et al. 1997; Mayville et al. 1999; McDowell et al. 2001). A conserved hydrophobic patch in the C-terminus and few specific contacts aid in binding of AIP to its cognate receptor, AgrC via the hexahelical transmembrane (TM) sensor domain. Thus resulting in activation of downstream signalling cascade (Lyon et al. 2002; Wright et al. 2004; Geisinger et al. 2008). The agr locus possesses polymorphism within a single species. This polymorphism is due to the variability in the regions of RNAII, AgrB, AgrD and AgrC, giving rise to four allelic variants of S. aureus. This hypervariability guides the generation of four different types of AIPs (I-IV) on the basis of the strain (Ji et al. 1997; Jarraud et al. 2000). Generally, only the cognate interaction of AIP with AgrC guides the expression of agr operon whilst the non-cognate interactions of the same lead to the inhibition of the expression, thus causing the inhibition of QS. Owing to this property of inhibition of QS by non-cognate AIPs Lyon and McDowell research groups independently designed hybrid AIPs by altering length or amino acid sequence, by introducing truncations and structural substitutions. The hybrid AIPs thus created have the property to act as universal inhibitors of all the AgrC and thus outcompeting all types of AIP (Lyon et al. 2000, 2002; McDowell et al. 2001). Based on the Structure activity relationship (SAR) studies conducted on AIP-I, II and III, a few important points have been revealed. Modifications of these can convert the AIPs to global agr inhibitors, for instance, a 16-membered macrocycle important for binding. Playing with the size and stoichiometry of this ring is deleterious to AIP activity (McDowell et al. 2001; Johnson et al. 2015). Second, C-terminal end of AIPs have hydrophobic residues which are important for effective binding to AgrC. Point mutations on alanine at these particular positions destroy the potency of the AIPs (McDowell et al. 2001; Tal-Gan et al. 2013b). Lastly, structural modification of AIPs plays a detrimental role in its activity. Owing to this the second residue within the macrocycle and the exocyclic tail are required for AgrC activation. Modification and truncations of these sites lead to loss of its potency (Tal-Gan et al. 2013a). Owing to its peptidic backbone and its consequent higher immunogenicity and lack of stability in vivo, elaborate research is underway to make the peptidomimetics corresponding to these AIPs. For this purpose, modifications in AIP-III by replacement of amino acid residues with corresponding peptoids or N-methyl mimics has produced new QSIs (Tal-Gan et al. 2014). Further research is needed to completely turn them into peptidomimetics, which would help to bring them in clinical trials.
Recently a group of scientists created a focussed library of 63 peptidomimetic by using standard Fmoc Solid Phase Peptide Synthesis (SPPS) method for evaluating AgrC inhibition in four groups of S. aureus. These were the simplified peptidomimetics of the previously reported truncated native AIP, t-AIP-II (Lyon et al. 2002; George et al. 2008). Out of these, three peptidomimetics namely n7FF, n8FF, and n7OFF inhibited AgrC activity in the clinically relevant group I: S. aureus strain with potencies similar to that of the parent peptide minus their shortcomings like solubility and stability (Vasquez et al. 2017). However, further research is required to test these non cognate AIPs as therapeutic agents to control infections by methicillin resistant S. aureus strains.
7 Analogues of Signal Molecules
Acyl homoserine lactone (AHL) is a class of QS molecule produced by gram negative bacteria and shows polymorphisms even in the same genera (Huma et al. 2011; Kalia 2014b). Two AHL compounds are produced by P. aeruginosa, which are short chain N-butanoyl-L-homoserine lactone (C4-HSL) and long chain N-(3-oxododecanoyl)-L-homoserine lactone (3-oxo-C12-HSL). These compounds regulate virulence and the generation of secondary metabolites. However only 3-oxo-C12-HSL acts on gram positive bacteria by inhibiting their growth. The 3-oxo-C12-HSL is earlier reported to have a killing effect on S. aureus (Kaufmann et al. 2005; Qazi et al. 2006). While at subinhibitory concentrations it hinders the release of S. aureus exotoxins (α-hemolysin, δ-hemolysin and toxic shock syndrome toxin) and thus acts as a quorum quenching agents (Qazi et al. 2006; Kalia et al. 2011). 3-oxo-C12-HSL undergoes intramolecular changes to give acid product 3-(1-hydroxydecylidene)-5-(2-hydroxyethyl)pyrrolidine-2,4-dione [(S)-5-hydroxyethyl-3-decanoyltetramic acid;8 5-HE-C10-TMA, 5] (Kaufmann et al. 2005). This belongs to TMA family of compounds which have antibacterial activity. Lately Murray and colleagues designed a series of 3-oxo-C12-HSL, TMA, and TOA analogues. This was done by bringing about systematic modifications on the parent compound 3-oxo-12-HSL focusing on (I) homoserine lactone, (II) 3-oxo substituent, (III) acyl side chain and (IV) amide structural units. HSL analogue namely 3-oxo-C12-HSL 1 having modifications in the homoserine lactone ring inhibited AgrC with an IC50 of 22 ± 6 μM. TMA analogues (namely 3–13) created by varying the 3-acyl chain length 3–8, stereochemistry 9, and substitution at the 5-position of the heterocyclic ring 12 and 13 were tested for their inhibitory activity against agr. It was observed that compound 4 5-HE-C8-TMA has good inhibitory activity (42 ± 13 μM) against agr. It also fully abolished the expression of agr-mediated exotoxin α-hemolysin at 100 μM. This makes it a good candidate for future therapeutics however research should be focused on increasing its stability (Murray et al. 2014). Next in line are the TOA compounds (namely 14–18) synthesized by bringing about variations in TMA structure wherein the ring nitrogen was replaced by oxygen. Upon evaluation of these TOAs against S. aureus growth and agr inhibition, it was found that C-14 TOA 17 was the most effective having an IC50 of 3 ± 1 μM which is approximately 8 times lower than the MIC (25 μM). Another compound C-12 TOA 16 was found to be most potent than any other compound in preventing AIP mediated activation of AgrC by maintaining allosteric interaction with AgrC. Finally C-14 TOA 17 also reduced S. aureus colonization of human nasal passage. C-14 TOA 17 also showed its potency in mouse model system without any toxicity to host (Murray et al. 2014). Recently Zapotoczna and colleagues tested antibacterial and anti-biofilm potential along with a new sulphur-containing analogue (3-tetradecanoylthiotetronic acid; C14-TTA) towards MRSA and MSSA strains of S. aureus. Their potential clinical use as catheter lock solution was also examined using in vitro and in vivo models of IVC infection (Zapotoczna et al. 2015). Evaluation of biofilm killing activity of these compounds 5HE-C14-TMA killed over 50% of both MSSA and MRSA biofilms at 128 μg/ml with full abolishment at 512–1024 μg/ml. Similar results were obtained in in vivo rat model for IVC infections. However the efficacy of C14-TOA and C14-TTA were far less in killing MSSA and MRSA biofilms. Taking into account of all these observations 5HE-C14-TMA proves to be a compound of therapeutic value against S. aureus biofilms (Zapotoczna et al. 2017).
8 RNAIII-Inhibiting Peptide (RIP) Binds to TRAP
The key feature in pathogenesis of S. aureus is the regulation of toxin production. S. aureus produces different toxins during its proliferation that can cause severe disease. At the initiation of growth, when the population of S. aureus is scarce, various molecules required for adhesion such as protein A, fibronectin binding-proteins and fibrinogen binding-proteins are expressed and help bacteria to colonize and attach to host cells. Whereas at early stationary phase of growth, bacteria are in greater density, produce toxic molecules such as hemolysins, enterotoxins and Toxic Shock Syndrome Toxin-1 (TSST-1) that help the bacteria to spread, survive and initiate the infection (Lowy 1998). There are two QS mechanisms in S. aureus which regulates the production of toxin molecules in greater densities and adhesion molecules expression in lesser densities. The first one is RAP/TRAP QS system, made up of two components, RAP and TRAP (mentioned in introduction). RAP is a protein that activates the production of toxins by phosphorylating the histidine amino acid of TRAP, when RAP reaches a certain threshold concentration (Balaban et al. 2001). With an unknown mechanism, phosphorylation of TRAP causes increased cell attachment to the host and activation of agr QS system. The chromosomal locus, agr encodes RNAII and RNAIII transcripts. RNAII transcript encodes AgrA, AgrD, AgrC and AgrB, where propeptide AgrD is processed, and secreted in the form of an autoinducer AIP with the help of transmembrane protein, AgrB. In the mid exponential phase of growth, agr is activated which results in AIP secretion. The secreted AIP molecules then bind to the AgrC and causes AgrC phosphorylation. In turn, AgrA is activated which leads to RNAIII production. RNAIII upregulates the expression of toxins and downregulates the expression of cell surface proteins (Bronesky et al. 2016). In addition, AIP reduces the phosphorylation of TRAP and thus, leading to decreased cell adhesion (Balaban et al. 2001). RNAIII- inhibiting peptide (RIP) is a heptapeptide that can attenuate the virulence of S. aureus. YSPXTNF-NH2 was identified as a sequence of RIP (Balaban et al. 1998). RIP acts as a competitor of RAP on activating TRAP and thus inhibits its phosphorylation, which leads to attenuation of transcription from RNAII and RNAIII promoters and thus inhibiting toxin production. Synthetic analogues of RIP, YSPWTNF was made and shown to effectively inhibits the RNAIII synthesis in vitro and reduces the S. aureus infections caused by different strains in vivo, including osteomylitis,cellulitis, mastitis, septic arthritis and keratitis. Theoretically, RIP would lead to increase bacterial adhesion as it inhibits the RNAIII synthesis and RNAIII function is to decrease the cell surface adhesion molecules. But, by using atomic force and fluorescence microscopy, it was shown that RIP decreases attachment of bacterial cells to mammalian cells (HEP2) and to polystyrene. Thus, RIP can be used as a better therapeutic candidate for S. aureus infections (Gov et al. 2001). S. aureus infections connected to biofilm formation are commonly linked with the implantated medical devices (Costerton et al. 1999). After the removal of devices, the predominant species found on biofilms are S. aureus (Marr 2000). Biofilm is the structure formed due to QS or cell-cell communication and highly resistant to antibiotics. A novel way to treat biofilm related S. aureus infections is to use RNAIII inhibiting peptide, which disrupts the QS system and decreases bacterial adhesion. In an experiment, RIP was applied systematically and locally in a vascular-graft rat model, suggested that RIP completely inhibits the antibiotic- resistant S. aureus infections (Dell’Acqua et al. 2004). Therefore, RIP can thus be used as a coating material for various medical devices to be used during medical procedure. Moreover, antibiotics such as carbapenems (imipenem) and cephalosporins (cefazolin) in combination with RIP, inhibits the infection completely (Giacometti et al. 2003). Therefore, RIP can inhibit QS regulated toxin production and biofilm formation.
9 Non-peptide Analogues of RIP
Hamamelitannin (HAM), condensed tannin is a natural product obtained from the bark of the plant witch hazel (Hamamelis virginiana). It is the ester of D-hamamelose (2-hydroxymethyl-D-ribose) with 2 molecules of gallic acid (2′, 5-di-O-galloyl-Dhamamelose). Because gallic acid contains three phenolic functional groups, it is considered a polyphenol. Owing to studies on HAM in last decade, it emerged as a candidate of QSI of drug resistant Staphylococcal infection. It works by acting as non-peptide analogue of RIP and thus hinders biofilm formation. Non-peptide analogue of RIP also block the production of RNAIII in vitro as well as in vivo by blocking TRAP phosphorylation and thus affects TRAP mediated agr expression (Gov et al. 2004; Kiran et al. 2008). A recent study conducted by Brackman and colleagues showed that HAM increased the antibiotic susceptibility of S. aureus biofilms. It was observed that HAM in combination with vancomycin resulted in enhanced killing of S. aureus Mu50 biofilm cells compared to vancomycin alone in in vitro models. Similar results were observed for a combination of HAM with clindamycin. The in vivo effect of combined treatment was seen in C. elegans model system. HAM and vancomycin together significantly (p < 0.01) increased the survival of S. aureus Mu50 infected C. elegans model system (Brackman et al. 2011). They further elaborated their study to give the mechanistic view about the action of HAM by showing that this increase in susceptibility towards antibiotics is via affecting peptidoglycan biosynthesis and exogenous DNA (eDNA) release. Combintion of HAM with other antibiotics such as vancomycin, cefazolin, cefalonium, cephalexin, cefoxitin, daptomycin, linezolid, tobramycin or fusidic acid also significantly increase the killing of biofilm cells for various S. aureus strains. Mutations in gene belonging to QS and RNA sequencing studies showed that HAM has specificity towards TRAP receptor (Brackman et al. 2016).
However the structure of HAM makes it very polar affecting its bioavailability. It is also more prone to oxidation and glucoronidation because of its aromatic hydroxy functional moieties. Formation of ester linkages in vivo also raise an issue related to its stability (Vermote et al. 2016). Based on these observations, Vermote and group worked on making analogues of HAM by improving its stability. Three modifications were made in the HAM structure. These were modification or elimination of the aromatic hydroxy groups, replacement of the ester groups with isosteric linker moieties and lastly removal of the anomeric hydroxy group. This resulted in developing of rigid and structurally well-defined tetrahydrofuran core (position 5). Further changes led to the development of 58 analogues of HAM. Out of these the ortho chloro derivative i.e. 38 came out to be the most potent analogue of HAM. The compound 38 in combination with vancomycin resulted in enhanced killing of S. aureus Mu50 biofilm cells. Also it had better stability in vivo and displayed no cytotoxicity towards host cells. Thus giving 38 a better hand over HAM for therapeutic use (Vermote et al. 2016). In a latest study by same group more analogues were created by making changes at C-2′ position and conducting Structure Activity Relationship (SAR) based studies. This led to the generation of 52 analogues of HAM focussing on benzamides with different substituents at different positions. Three derivatives namely 10u, 15 and 25 showed promising results when tested for disruption of S. aureus biofilm cells in vitro and their susceptibility to vancomycin on these biofilm cells. These products warrant more study for their therapeutic use (Vermote et al. 2017).
10 Future Directions
Hospital borne infections are a nuisance to the medical industry. Staphylococcus aureus is the causative agent of diseases like endocarditis, osteomyelitis, and pneumonia. Using small molecule inhibitors to combat the infections is therapeutically effective approach in case of various pathogens like Bacillus anthracis (Dhasmana et al. 2014). As discussed in this review, various small molecule inhibitors have been tested against S. aureus which have proved their efficacy in various in vitro as well as in vivo model systems. However bacterial colonization takes places during the initial phases of disease establishment and hence the implication of QSIs becomes limiting. It is important to take precautionary measures in case of medical devices. These potent QSIs could be used as a coating material on these medical devices, which would help in reducing nosocomial infections by MRSA strains. Currently, there are fewer studies testing these inhibitors on various medical devices and this field should be explored further.
References
Agarwala M, Choudhury B, Yadav RN (2014) Comparative study of antibiofilm activity of copper oxide and iron oxide nanoparticles against multidrug resistant biofilm forming uropathogens. Indian J Microbiol 54:365–368. https://doi.org/10.1007/s12088-014-0462-z
Ahiwale SS, Bankar AV, Tagunde S, Kapadnis BP (2017) A bacteriophage mediated gold nanoparticle synthesis and their anti-biofilm activity. Indian J Microbiol 57:188–194. https://doi.org/10.1007/s12088-017-0640-x
Balaban N, Goldkorn T, Nhan RT, Dang LB, Scott S, Ridgley RM, Rasooly A, Wright SC, Larrick JW, Rasooly R, Carlson JR (1998) Autoinducer of virulence as a target for vaccine and therapy against Staphylococcus aureus. Science 280:438–440. https://doi.org/10.1126/science.280.5362.438
Balaban N, Goldkorn T, Gov Y, Hirshberg M, Koyfman N, Matthews HR, Nhan RT, Singh B, Uziel O (2001) Regulation of Staphylococcus aureus pathogenesis via target of RNAIII-activating protein (TRAP). J Biol Chem 276:2658–2667. https://doi.org/10.1074/jbc.M005446200
Baldry M, Kitir B, Frøkiær H, Christensen SB, Taverne N, Meijerink M, Franzyk H, Olsen CA, Wells JM, Ingmer H (2016) The agr inhibitors solonamide B and analogues alter immune responses to Staphylococccus aureus but do not exhibit adverse effects on immune cell functions. PLoS One 11:e0145618. https://doi.org/10.1371/journal.pone.0145618
Boisset S, Geissmann T, Huntzinger E, Fechter P, Bendridi N, Possedko M, Chevalier C, Helfer AC, Benito Y, Jacquier A, Gaspin C, Vandenesch F, Romby P (2007) Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev 21:1353–1366. https://doi.org/10.1101/gad.423507
Brackman G, Cos P, Maes L, Nelis HJ, Coenye T (2011) Quorum sensing inhibitors increase the susceptibility of bacterial biofilms to antibiotics in vitro and in vivo. Antimicrob Agents Chemother 55:2655–2661. https://doi.org/10.1128/aac.00045-11
Brackman G, Breyne K, De Rycke R, Vermote A, Van Nieuwerburgh F, Meyer E, Van Calenbergh S, Coenye T (2016) The quorum sensing inhibitor hamamelitannin increases antibiotic susceptibility of Staphylococcus aureus biofilms by affecting peptidoglycan biosynthesis and eDNA release. Sci Rep 6:20321. https://doi.org/10.1038/srep20321
Bronesky D, Wu Z, Marzi S, Walter P, Geissmann T, Moreau K, Vandenesch F, Caldelari I, Romby P (2016) Staphylococcus aureus RNAIII and its regulon link quorum sensing, stress responses, metabolic adaptation and regulation of virulence gene expression. J Clin Invest 70:299–316. https://doi.org/10.1146/annurev-micro-102215-095708
Bubeck Wardenburg J, Bae T, Otto M, Deleo FR, Schneewind O (2007) Poring over pores: alpha-hemolysin and Panton-valentine leukocidin in Staphylococcus aureus pneumonia. Nat Med 13:1405–1406. https://doi.org/10.1038/nm1207-1405
Costerton JW, Stewart PS, Greenberg EP (1999) Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322. https://doi.org/10.1126/science.284.5418.1318
Defoirdt T, Brackman G, Coenye T (2013) Quorum sensing inhibitors: how strong is the evidence? Trends Microbiol 21:619–624. https://doi.org/10.1016/j.tim.2013.09.006
Dell’Acqua G, Giacometti A, Cirioni O, Ghiselli R, Saba V, Scalise G, Gov Y, Balaban N (2004) Suppression of drug-resistant staphylococcal infections by the quorum-sensing inhibitor RNAIII-inhibiting peptide. J Infect Dis 190:318–320. https://doi.org/10.1086/386546
Dhasmana N, Singh LK, Bhaduri A, Misra R, Singh Y (2014) Recent developments in anti-dotes against anthrax. Recent Pat Antiinfect Drug Discov 9:83–96
Elmore BO, Triplett KD, Hall PR (2015) Apolipoprotein B48, the structural component of chylomicrons, is sufficient to antagonize Staphylococcus aureus quorum-sensing. PLoS One 10:e0125027. https://doi.org/10.1371/journal.pone.0125027
Femling JK, West SD, Hauswald EK, Gresham HD, Hall PR (2013) Nosocomial infections after severe trauma are associated with lower apolipoproteins B and AII. J Trauma Acute Care Surg 74:1067–1073. https://doi.org/10.1097/TA.0b013e3182826be0
Geisinger E, George EA, Muir TW, Novick RP (2008) Identification of ligand specificity determinants in AgrC, the Staphylococcus aureus quorum-sensing receptor. J Biol Chem 283:8930–8938. https://doi.org/10.1074/jbc.M710227200
George EA, Muir TW (2007) Molecular mechanisms of agr quorum sensing in virulent staphylococci. Chembiochem 8:847–855. https://doi.org/10.1002/cbic.200700023
George EA, Novick RP, Muir TW (2008) Cyclic peptide inhibitors of staphylococcal virulence prepared by Fmoc-based thiolactone peptide synthesis. J Am Chem Soc 130:4914–4924. https://doi.org/10.1021/ja711126e
Giacometti A, Cirioni O, Gov Y, Ghiselli R, Del Prete MS, Mocchegiani F, Saba V, Orlando F, Scalise G, Balaban N, Dell’Acqua G (2003) RNA III inhibiting peptide inhibits in vivo biofilm formation by drug-resistant Staphylococcus aureus. Antimicrob Agents Chemother 47:1979–1983. https://doi.org/10.1128/AAC.47.6.1979-1983.2003
Gordon RJ, Lowy FD (2008) Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis 46:350–359. https://doi.org/10.1086/533591
Gov Y, Bitler A, Dell’Acqua G, Torres JV, Balaban N (2001) RNAIII inhibiting peptide (RIP), a global inhibitor of Staphylococcus aureus pathogenesis: structure and function analysis. Peptides 22:1609–1620. https://doi.org/10.1016/S0196-9781(01)00496-X
Gov Y, Borovok I, Korem M, Singh VK, Jayaswal RK, Wilkinson BJ, Rich SM, Balaban N (2004) Quorum sensing in staphylococci is regulated via phosphorylation of three conserved histidine residues. J Biol Chem 279:14665–14672. https://doi.org/10.1074/jbc.M311106200
Gui Z, Wang H, Ding T, Zhu W, Zhuang X, Chu W (2014) Azithromycin reduces the production of α-hemolysin and biofilm formation in Staphylococcus aureus. Indian J Microbiol 54:114–117. https://doi.org/10.1007/s12088-013-0438-4
Hall PR, Elmore BO, Spang CH, Alexander SM, Manifold-Wheeler BC, Castleman MJ, Daly SM, Peterson MM, Sully EK, Femling JK, Otto M, Horswill AR, Timmins GS, Gresham HD (2013) Nox2 modification of LDL is essential for optimal apolipoprotein B-mediated control of agr type III Staphylococcus aureus quorum-sensing. PLoS Pathog 9:e1003166. https://doi.org/10.1371/journal.ppat.1003166
Han R (2010) Plasma lipoproteins are important components of the immune system. Microbiol Immunol 54:246–253. https://doi.org/10.1111/j.1348-0421.2010.00203.x
Huma N, Shankar P, Kushwah J, Bhushan A, Joshi J, Mukherjee T, Raju SC, Purohit HJ, Kalia VC (2011) Diversity and polymorphism in AHL-lactonase gene (aiiA) of Bacillus. J Microbiol Biotechnol 21:1001–1011. https://doi.org/10.4014/jmb.1105.05056
Jarraud S, Lyon GJ, Figueiredo AMS, Gerard L, Vandenesch F, Etienne J, Muir TW, Novick RP (2000) Exfoliatin-producing strains define a fourth agr specificity group in Staphylococcus aureus. J Bacteriol 182:6517–6522. https://doi.org/10.1128/JB.182.22.6517-6522.2000
Ji G, Beavis R, Novick RP (1997) Bacterial interference caused by autoinducing peptide variants. Science 276:2027–2030. https://doi.org/10.1126/science.276.5321.2027
Johnson JG, Wang BY, Debelouchina GT, Novick RP, Muir TW (2015) Increasing AIP macrocycle size reveals key features of agr activation in Staphylococcus aureus. Chem Bio Chem 16:1093–1100. https://doi.org/10.1002/cbic.201500006
Kalia VC (2014a) Microbes, antimicrobials and resistance: the battle goes on. Indian J Microbiol 54:1–2. https://doi.org/10.1007/s12088-013-0443-7
Kalia VC (2014b) In search of versatile organisms for quorum-sensing inhibitors: acyl homoserine lactones (AHL)-acylase and AHL-lactonase. FEMS Microbiol Letts 359:143. https://doi.org/10.1111/1574-6968.12585
Kalia VC, Purohit HJ (2011) Quenching the quorum sensing system: potential antibacterial drug targets. Critical Rev Microbiol 37:121–140. https://doi.org/10.3109/1040841X.2010.532479
Kalia VC, Raju SC, Purohit HJ (2011) Genomic analysis reveals versatile organisms for quorum quenching enzymes: acyl-homoserine lactone-acylase and –lactonase. Open Microbiol J 5:1–13. https://doi.org/10.2174/1874285801105010001
Kalia VC (2015) Microbes: the most friendly beings? In: Quorum sensing vs quorum quenching: a battle with no end in sight. Springer, New Delhi, pp 1–5. http://dx.doi.org/10.1007/978-81-322-1982-8_1
Kattelmann KK, Hise M, Russell M, Charney P, Stokes M, Compher C (2006) Preliminary evidence for a medical nutrition therapy protocol: enteral feedings for critically ill patients. J Am Diet Assoc 106:1226–1241. https://doi.org/10.1016/j.jada.2006.05.320
Kaufmann GF, Sartorio R, Lee SY, Rogers CJ, Meijler MM, Moss JA, Clapham B, Brogan AP, Dickerson TJ, Janda KD (2005) Revisiting quorum sensing: discovery of additional chemical and biological functions for 3-oxo-N-acylhomoserine lactones. Proc Natl Acad Sci U S A 102:309–314. https://doi.org/10.1073/pnas.0408639102
Kiran MD, Adikesavan NV, Cirioni O, Giacometti A, Silvestri C, Scalise G, Ghiselli R, Saba V, Orlando F, Shoham M, Balaban N (2008) Discovery of a quorum-sensing inhibitor of drug resistant staphylococcal infections by structure-based virtual screening. Mol Pharmacol 73:1578–1586. https://doi.org/10.1124/mol.107.044164
Koenig RL, Ray JL, Maleki SJ, Smeltzer MS, Hurlburt BK (2004) Staphylococcus aureus AgrA binding to the RNAIII-agr regulatory region. J Bacteriol 186:7549–7555. https://doi.org/10.1128/JB.186.22.7549-7555.2004
Koul S, Kalia VC (2017) Multiplicity of quorum quenching enzymes: a potential mechanism to limit quorum sensing bacterial population. Indian J Microbiol 57:100–108. https://doi.org/10.1007/s12088-016-0633-1
Kumar P, Patel SKS, Lee J-K, Kalia VC (2013) Extending the limits of Bacillus for novel biotechnological applications. Biotechnol Adv 31(8):1543–1561
Kumar P, Koul S, Patel SKS, Lee JK, Kalia VC (2015) Heterologous expression of quorum sensing inhibitory genes in diverse organisms. In: Quorum sensing vs quorum quenching: a battle with no end in sight. Springer, New Delhi, pp 343–356. http://dx.doi.org/10.1007/978-81-322-1982-8_28
Le KY, Otto M (2015) Quorum-sensing regulation in staphylococci-an overview. Front Microbiol 6:1174. https://doi.org/10.3389/fmicb.2015.01174
Leonard PG, Bezar IF, Sidote DJ, Stock AM (2012) Identification of a hydrophobic cleft in the LytTR domain of AgrA as a locus for small molecule interactions that inhibit DNA binding. Biochemistry 51:10035–10043. https://doi.org/10.1021/bi3011785
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26. https://doi.org/10.1016/S0169-409X(00)00129-0
Loughman JA, Fritz SA, Storch GA, Hunstad DA (2009) Virulence gene expression in human community acquired Staphylococcus aureus infection. J Infect Dis 199:294–301. https://doi.org/10.1086/595982
Lowy FD (1998) Staphylococcus aureus infections. N Engl J Med 339:520–532. https://doi.org/10.1056/NEJM199808203390806
Lyon GJ, Mayville P, Muir TW, Novick RP (2000) Rational design of a global inhibitor of the virulence response in Staphylococcus aureus, based in part on localization of the site of inhibition to the receptor-histidine kinase AgrC. Proc Natl Acad Sci U S A 97:13330–13335. https://doi.org/10.1073/pnas.97.24.13330
Lyon GJ, Wright JS, Muir TW, Novick RP (2002) Key determinants of receptor activation in the agr autoinducing peptides of Staphylococcus aureus. Biochemistry 41:10095–11104. https://doi.org/10.1021/bi026049u
Mansson M, Nielsen A, Kjærulff L, Gotfredsen CH, Wietz M, Ingmer H, Gram L, Larsen TO (2011) Inhibition of virulence gene expression in Staphylococcus aureus by novel depsipeptides from a marine photobacterium. Mar Drugs 9:2537–2552. https://doi.org/10.3390/md9122537
Marr KA (2000) Staphylococcus aureus bacteremia in patients undergoing hemodialysis. Semin Dial 13:23–29. https://doi.org/10.1046/j.1525-139x.2000.00009.x
Mayville P, Ji G, Beavis R, Yang H, Goger M, Novick RP, Muir TW (1999) Structure-activity analysis of synthetic autoinducing thiolactone peptides from Staphylococcus aureus responsible for virulence. Proc Natl Acad Sci U S A 96:1218–1223. https://doi.org/10.1073/pnas.96.4.1218
McDowell P, Affas Z, Reynolds C, Holden MT, Wood SJ, Saint S, Cockayne A, Hill PJ, Dodd CE, Bycroft BW, Chan WC, Williams P (2001) Structure, activity and evolution of the group I thiolactone peptide quorum-sensing system of Staphylococcus aureus. Mol Microbiol 41:503–512. https://doi.org/10.1046/j.1365-2958.2001.02539.x
Murray EJ, Crowley RC, Truman A, Clarke SR, Cottam JA, Jadhav GP, Steele VR, O’Shea P, Lindholm C, Cockayne A, Chhabra SR, Chan WC, Williams P (2014) Targeting Staphylococcus aureus quorum sensing with nonpeptidic small molecule inhibitors. J Med Chem 57:2813–2819. https://doi.org/10.1021/jm500215s
Nielsen A, Månsson M, Bojer MS, Gram L, Larsen TO, Novick RP, Frees D, Frøkiær H, Ingmer H (2014) Solonamide B inhibits quorum sensing and reduces Staphylococcus aureus mediated killing of human neutrophils. PLoS One 9:e84992. https://doi.org/10.1371/journal.pone.0084992
Novick RP, Geisinger E (2008) Quorum sensing in staphylococci. Annu Rev Genet 42:541–564. https://doi.org/10.1146/annurev.genet.42.110807.091640
Qazi S, Middleton B, Muharram SH, Cockayne A, Hill P, O’Shea P, Chhabra SR, Cámara M, Williams P (2006) N-acylhomoserine lactones antagonize virulence gene expression and quorum sensing in Staphylococcus aureus. Infect Immun 74:910–919. https://doi.org/10.1128/IAI.74.2.910–919.2006
Sully EK, Malachowa N, Elmore BO, Alexander SM, Femling JK, Gray BM, DeLeo FR, Otto M, Cheung AL, Edwards BS, Sklar LA, Horswill AR, Hall PR, Gresham HD (2014) Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance. PLoS Pathog 10:e1004174. https://doi.org/10.1371/journal.ppat.1004174
Swift S, Downie JA, Whithead N, Barnard AML, Salmond GPC, Williams P (2001) Quorum sensing as a population density dependent determinant of bacterial physiology. Adv Microb Physiol 45:199–270. https://doi.org/10.1016/S0065-2911(01)45005-3
Tal-Gan Y, Ivancic M, Cornilescu G, Cornilescu CC, Blackwell HE (2013a) Structural characterization of native autoinducing peptides and abiotic analogues reveals key features essential for activation and inhibition of an AgrC quorum sensing receptor in Staphylococcus aureus. J Am Chem Soc 135:18436–18444. https://doi.org/10.1021/ja407533e
Tal-Gan Y, Stacy DM, Foegen MK, Koenig DW, Blackwell HE (2013b) Highly potent inhibitors of quorum sensing in Staphylococcus aureus revealed through a systematic synthetic study of the group-III autoinducing peptide. J Am Chem Soc 135:7869–7882. https://doi.org/10.1021/ja3112115
Tal-Gan Y, Stacy DM, Blackwell HE (2014) N-methyl and peptoid scans of an autoinducing peptide reveal new structural features required for inhibition and activation of AgrC quorum sensing receptors in Staphylococcus aureus. Chem Commun 50:3000–3003. https://doi.org/10.1039/c4cc00117f
Vasquez JK, Tal-Gan Y, Cornilescu G, Tyler KA, Blackwell HE (2017) Simplified AIP-II peptidomimetics are potent inhibitors of Staphylococcus aureus AgrC quorum sensing receptors. Chem Bio Chem 18:413–423. https://doi.org/10.1002/cbic.201600516
Vermote A, Brackman G, Risseeuw MDP, Vanhoutte B, Cos P, Van Hecke K, Breyne K, Meyer E, Coenye T, Van Calenbergh S (2016) Hamamelitannin analogues that modulate quorum sensing as potentiators of antibiotics against Staphylococcus aureus. Angew Chem Int Ed 55:6551–6555. https://doi.org/10.1002/anie.201601973
Vermote A, Brackman G, Risseeuw MDP, Cappoen D, Cos P, Coenye T, Van Calenbergh S (2017) Novel potentiators for vancomycin in the treatment of biofilm-related MRSA infections via a mix and match approach. ACS Med Chem Lett 8:38–42. https://doi.org/10.1021/acsmedchemlett.6b00315
Vuong C, Götz F, Otto M (2000) Construction and characterization of an agr deletion mutant of Staphylococcus epidermidis. Infect Immun 68:1048–1053. https://doi.org/10.1128/IAI.68.3.1048-1053.2000
Wadhwani SA, Shedbalkar UU, Singh R, Vashisth P, Pruthi V, Chopade BA (2016) Kinetics of synthesis of gold nanoparticles by Acinetobacter sp. SW30 isolated from environment. Indian J Microbiol 56:439–444. https://doi.org/10.1007/s12088-016-0598-0
Wang B, Muir TW (2016) Regulation of virulence in Staphylococcus aureus: molecular mechanisms and remaining puzzles. Cell Chem Biol 23:214–224. https://doi.org/10.1016/j.chembiol.2016.01.004
Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, Kennedy AD, Dorward DW, Klebanoff SJ, Peschel A, DeLeo FR, Otto M (2007) Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med 13:1510–1514. https://doi.org/10.1038/nm1656
Williams P (2002) Quorum sensing: an emerging target for antibacterial chemotherapy? Expert Opin Ther Targets 6:257–274. https://doi.org/10.1517/14728222.6.3.257
Wright JS 3rd, Lyon GJ, George EA, Muir TW, Novick RP (2004) Hydrophobic interactions drive ligand-receptor recognition for activation and inhibition of staphylococcal quorum sensing. Proc Natl Acad Sci U S A 101:16168–16173. https://doi.org/10.1073/pnas.0404039101
Wright GD, Sutherland AD (2007) New strategies for combating multidrug-resistant bacteria. Trends Mol Med 13:260–267. https://doi.org/10.1016/j.molmed.2007.04.004
Yarwood JM, Schlievert PM (2003) Quorum sensing in Staphylococcus infections. J Clin Invest 112:1620–1625. https://doi.org/10.1172/JCI20442
Zapotoczna M, McCarthy H, Rudkin JK, O’Gara JP, O’Neill E (2015) An essential role for coagulase in Staphylococcus aureus biofilm development reveals new therapeutic possibilities for device-related infections. J Infect Dis 212:1883–1893. https://doi.org/10.1093/infdis/jiv319
Zapotoczna M, Murray EJ, Hogan S, O’Gara JP, Chhabra S, Chan WC, O’Neil E, Williams P (2017) 5-Hydroxyethyl-3-tetradecanoyltetramic acid represents a novel treatment for intravascular catheter infections due to Staphylococcus aureus. J Antimicrob Chemother 72:744–753. https://doi.org/10.1093/jac/dkw482
Acknowledgements
This work is supported by J C Bose Fellowship (SERB) to YS and Research Grant by University of Delhi. NK is UGC-SRF. HG is Masters of Science in Zoology from University of Delhi. ND is Shyama Prasad Mukherjee Fellow (CSIR-SRF) and Fulbright-Nehru Doctoral Fellow (2015–16) at NIAID NIH.
Author Information
The authors declare no competing financial interests. Correspondence and requests for materials should be addressed to YS (ysinghdu@gmail.com).
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Kumar, N., Gupta, H., Dhasmana, N., Singh, Y. (2018). Combating Staphylococcal Infections Through Quorum Sensing Inhibitors. In: Kalia, V. (eds) Biotechnological Applications of Quorum Sensing Inhibitors. Springer, Singapore. https://doi.org/10.1007/978-981-10-9026-4_15
Download citation
DOI: https://doi.org/10.1007/978-981-10-9026-4_15
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-9025-7
Online ISBN: 978-981-10-9026-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)