
Abstract
The pathogenesis of NAFLD is multi-faceted and mechanisms underlying the progression from simple steatosis to NASH have not been fully deciphered. The emerging field of epigenetics, an inheritable phenomenon capable of changing gene expression without altering DNA sequence, unveils a new perspective on the development of NAFLD and subsequent progression to HCC. In fact, numerous studies have highlighted the potential involvement of unhealthy daily habits such as physical inactivity and over-nutrition in the onset and development of NAFLD through epigenetic mechanisms. This chapter will discuss several epigenetic modulations including DNA methylation, histone modifications, functions of non-coding RNAs as well as RNA methylation implicated in the pathogenesis of NAFLD-HCC. On the basis of currently wealthy knowledge of DNA epigenetics, the rapidly growing field of RNA epigenetics will certainly drive forward a new avenue of research direction shedding light on the advancement of better diagnostics, prognostics and therapeutics in the coming era of precision medicine.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Non-alcoholic fatty liver disease
- Non-alcoholic steatohepatitis
- Hepatocellular carcinoma
- Epigenetic modifications
7.1 Introduction
As mentioned in previous sections, non-alcoholic fatty liver disease (NAFLD) is defined as the pathological deposition of triglycerides in hepatocytes due to causes other than excessive alcohol consumption. Non-alcoholic steatohepatitis (NASH), the more severe disease entity of NAFLD, represents the most common liver disease in the Western world and has the capacity to progress to cirrhosis and hepatocellular carcinoma (HCC) [1]. Compared to the high prevalence of NAFLD (20–30%) in Western countries, the prevalence in Asian countries is estimated to be around 5–20% [2]. As with other causes of liver disease, only a minor proportion of patients with NASH progress to advanced fibrosis, cirrhosis and/or HCC [3].
The pathogenesis of NAFLD is multi-faceted and mechanisms underlying the progression from simple steatosis to NASH have not been fully deciphered. According to the double-hit theory attempting to explain the development of NAFLD, the first hit is the accumulation of triglycerides in hepatocytes, accompanied by a second hit describing inflammatory cytokine interplay, mitochondrial dysfunction and oxidative stress causing hepatocellular injury, inflammation and fibrosis [4, 5]. Recent studies devised a new model describing multiple parallel hits in the progression of NAFLD. NAFLD pathogenesis is now commonly described as the excessive deposition of fat in hepatocytes, followed by increase in intracellular fat vacuoles, induction of endoplasmic reticulum and oxidative stress eventually leading to apoptosis of hepatocytes [6].
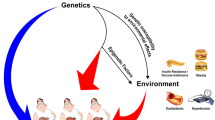
The emerging field of epigenetics, an inheritable phenomenon capable of changing gene expression without altering DNA sequence, unveils a new perspective on the pathogenesis of NAFLD. In fact, numerous studies have highlighted the potential involvement of unhealthy daily habits such as physical inactivity and over-nutrition in the onset and development of NAFLD through epigenetic mechanisms [7, 8]. This chapter will discuss several epigenetic modulations including DNA methylation, histone modifications, functions of non-coding RNAs as well as RNA methylation implicated in the pathogenesis of NAFLD-HCC that might serve as novel diagnostic, prognostic and therapeutic options (Fig. 7.1).

Dysregulated epigenetic modulations including DNA methylation, histone modifications, functions of non-coding RNAs as well as RNA methylation contribute to the pathogenesis of NAFLD-HCC. m6A N6-methyladenosine, lncRNA long non-coding RNA, snRNA small nuclear RNA, snoRNA small nucleolar RNA
7.2 DNA Methylation
The best-known and most intensively studied modification is methylation of cytosine in DNA with a methyl group. DNA methylation is catalyzed by DNA methyltransferases (DNMTs) that transfer a methyl group from S-adenosyl-L-methionine (SAM) to cytosine with guanine as the next nucleotide known as CpG dincleotides, the clustering of which being commonly referred to as CpG islands. Majority of CpG islands are located at the promoter regions of genes and hypermethylation of CpG islands causes gene silencing [9]. On the other hand, the ten-eleven translocation methylcytosine dioxygense (TET) family of enzymes converts the modified DNA base 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC) and this modification has been proposed as the initial step of active demethylation in mammals [10, 11]. Given the central role of DNA methylation in the regulation of gene expression, it comes with no surprise that perturbations to the homeostatic methylation level, due largely to environmental factors, contribute to aberrant gene expressions and trigger various pathological conditions.
7.2.1 DNA Methylation in Fibrosis and Progression of NASH
S-adenosyl-L-methionine (SAM) is the unique methyl donor for DNA methylation and dietary sources include folate, methionine, betaine and choline [12]. Methyl donor deficient diets have been associated with reduced DNA methylation and disturbed lipid metabolism. For example, folate deficiency has been shown to induce hepatic triglyceride accumulation and alter the expression of genes involved in fatty acid synthesis [13]. Likewise, deficiencies in methionine and choline have been correlated with reduced lipoprotein secretion and increased hepatic triglyceride generation accompanied by NAFLD development [14, 15]. Intriguingly, Tryndyak et al. [16] demonstrated in vivo that low SAM concentration altered the expressions of a series of genes involved in DNA repair, lipid and glucose metabolisms and hepatic fibrosis. This was consistent with a recent observation reporting a significant decrease in serum betaine levels in NASH patients as compared to those with non-alcoholic fatty liver [17], implicating proper dietary intake and maintenance of homeostatic SAM levels are critical for a harmonious hepatic lipid metabolism.
Liver fibrosis is defined by the excessive accumulation of extracellular matrix and scar formation in the context of chronic liver damage [18]. Activation and trans-differentiation of hepatic stellate cell (HSC) in response to various stimuli such as inflammation, from vitamin A storing pericyte to profibrogenic myofibroblastic phenotype, play a key role in the pathogenesis of liver fibrosis [19]. It has been demonstrated that transforming growth factor- β1 (TGF- β1), an inflammatory cytokine secreted by different types of hepatic cells, represented the main fibrogenic cytokine behind HSC activation [20]. Although the underlying molecular mechanisms driving fibrogenesis await further investigations, DNMTs have recently been implicated in the process. In humans there are three DNMT isoforms: DNMT1, DNMT3a and DNMT3b. While DNMT1 recognizes a hemi-methylated site on a new DNA strand during cell division and regenerates the bi-methylated state thereby safeguarding the faithful propagation of methylation patterns in daughter cells, DNMT3a and DNMT3b are central to the regulation of de novo methylation in the absence of cell division [21]. In a mouse model, Pogribny et al. [22] documented a hepatic epigenetic phenotype predetermined individual susceptible to hepatic steatosis in association with altered expressions of DNMT1 and DNMT3a in the liver. DNMT3a has also been shown to enhance HSC activation and liver fibrogenesis via methylation and down-regulation of the GTPase Septin-9 [23]. TET enzymes, responsible for catalyzing the stepwise oxidation of methyl groups on DNA leading eventually to the restoration of the unmodified cytosine residue, have been found to fine-tune the PPARGC1A transcriptional program in liver. Next-generation sequencing further revealed genetic diversity at TET loci was associated with altered 5-hmC levels that might be accountable for the pathogenesis of NAFLD [24]. A recent study elucidating the relationship between methylome and transcriptome in patients with non-alcoholic fatty liver disease revealed differentially methylated genes might distinguish patients with advanced NASH from those with simple steatosis [25]. In the landmark piece of work, 69,247 differentially methylated CpG sites (76% hypomethylated; 24% hypermethylated) were observed in patients with advanced NASH as compared to those with simple steatosis [25]. Aberrant methylation signatures of a plethora of genes have been suggested to predict the progression from NAFLD to NASH. For instance, peroxisome proliferator-activated receptor α (PPAR-α) exhibited significantly higher DNA methylation level in severe NAFLD patients than in mild counterparts [26].
7.2.2 DNA Methylation in the Progression of HCC
Disruption of DNA methylation has long been recognized as one of the key hallmarks of all cancer types [27]. Typical lesions in cancer include loci-specific de novo hypermethylation at promoter regions of tumour suppressor genes (TSGs) resulting in transcriptional repression of downstream TSGs. Among the plethora of studies reporting changes in DNA methylation pattern in HCC, Villanueva et al. [28] conducted a comprehensive study profiling the DNA methylation landscape in a cohort of 304 patients with HCC treated with surgical resection. Methylome profiling covering 96% of known CpG islands and 485,000 CpG dinucleotides was performed and a methylation signature generated based on 36 methylation probes accurately predicted survival in patients with HCC. While HCC tissue samples displayed general hypomethylation in the intergenic and body regions as compared with normal liver, hypermethylated probes were mainly located in promoter regions [28]. The authors further demonstrated aberrant methylation in established and candidate epidrivers of disease including well-known tumour suppressors such as Ras association domain family member 1 (RASSF1), adenomatous polyposis coli (APC), insulin-like growth factor 2 (IGF2) and NOTCH3, supporting the pivotal role of deregulated DNA methylation in HCC development [28].
The functional relevance of aberrant DNA methylation has been tested in numerous tumour suppressor genes. For instance, sphingomyelin phosphodiesterase 3 (SMPD3) and heavy polypeptide (NEFH) overexpression could inhibit tumour cell proliferation, whereas stable knockdown of the two enhanced cell migration and invasion in vitro and in vitro [29]. Noteworthy, persistent Hepatits B virus (HBV) infection has been shown to stimulate the upregulation of DNMTs, leading to hypermethylation and inactivation of p16 and the subsequent progression of HCC [30]. A recent intriguing study uncovered a role of hypoxia in the process of tumour development. Thienpont et al. [31] observed a direct inhibition of the activity of TET enzymes in a series of cancer cell lines (including HCC) and mouse cells in response to a hypoxic environment. The reduction in activity increased hypermethylation at gene promoters resulting in aberrant gene expressions in various signaling pathways and conferring a selective advantage to cancer cells [31]. Taken together, deregulated DNA methylation will continue to be a hot research area as a more thorough understanding of the underlying mechanisms is crucial to formulating novel prognostic markers and therapeutic targets.
7.3 Histone Modifications
Condensation of 2 m of DNA into a human nucleus is achieved through interaction between DNA and specialized histone proteins to form tightly packed chromatin. The basic level of chromatin packing is known as the nucleosome with each core particle comprising of 147 bp of double stranded DNA wrapped around a complex of eight histone proteins (two copies each of H2A, H2B, H3 and H4). The structure is commonly referred to as “beads on string” with linker DNA being the string and the nucleosome core particle representing the beads. In order to allow chromosomal processes such as gene transcription to occur, the chromatin must be packed lightly (euchromatin) or tightly (heterochromatin) in a finely orchestrated fashion. Indeed, each of the core histones harbours an unstructured N-terminal amino acid tail extension that can be subject to a plethora of posttranslational modifications including acetylation, methylation, phosphorylation, ubiquitination, ribosylation and sumoylation which constitute a crucial determinant of chromatin compactness and accessibility [32].
7.3.1 Histone Acetylation in NAFLD-HCC
Among various types of posttranslational modifications, acetylation of lysine residues at the N-terminus of histone tails has been most extensively investigated [33]. While histone acetylation is catalyzed by histone acetyltransferases (HATs), histone deacetylation is mediated by histone deacetylases (HDACs) [34]. Perturbations to the balance between HAT and HDAC have been reported to alter gene expression profiles in NAFLD [35].
7.3.1.1 Histone Acetyltransferases (HATs)
Histone acetyltransferases (HATs) acetylate conserved amino acid residues on histone proteins by transferring an acetyl group from acetyl-CoA to form ε-N-acetyllysine enabling enhanced gene expression. HATs can be divided into different classes depending on their subcellular localization [36]. Type A HATs are mainly located in the nucleus including Gcn5-related N-acetyltransferases (GNATs), p300/CBP and TAFII250, whereas type B HATs function predominantly in the cytoplasm [36]. In particular, p300/CBP has been shown to be involved in NF-κB dependent inflammatory pathways [37]. Inhibition of hepatic p300 was further suggested to be beneficial for treating hepatic steatosis in obesity and type 2 diabetes [38]. On the contrary, a recent report demonstrated p300/CBP-associated factor inhibited the growth of HCC cells by promoting autophagy, suggesting restoration of the specific HAT might prove to be a novel therapeutic strategy of HCC treatment [39].
7.3.1.2 Histone Deacetylases (HDACs)
Histone deacetylases (HDACs) remove acetyl groups from ε-N-acetyl lysine residues on histone, a process that is essential for tight wrapping between histones and DNA, as well as subsequent inhibition of gene transcription. HDAC superfamily is sub-divided into four classes: I, II, III (also referred to as Sirtuins or SIRTs) and IV on the basis of varying structure, enzymatic function and subcellular localization. Not surprisingly, dysregulations of HDACs have been implicated in the progression of NAFLD. Disruption of the circadian clock by HDAC3, a member of class I HDACs, resulted in perturbation to hepatic lipid metabolism and obesity [40]. Another member HDAC6 has been documented to function as a tumour suppressor in HCC and suppression of which by induction of miR-221 accompanied by activation of downstream oncogenic pathways contributed to liver tumorigenesis [41].
Silent information regulator 2 proteins (Sirtuins or SIRTs) belong to class III HDACs. Seven members have been identified in human so far (SIRT1-7) with different subcellular localizations. While some are present predominantly in the nucleus, others display cytoplasmic (SIRT1,2) and mitochondrial (SIRT3,4,5) localizations [42]. Research on mammals has been focused on SIRT1, which acts as a potent protector from a wide array of pathological conditions such as diabetes, liver steatosis and various types of cancer [43]. Although overexpression of SIRT1 appeared to offer protection against DNA damage and metabolic derangement induced by high fat diet [44], recent studies highlighted up-regulation of SIRT1 facilitated HCC metastasis and self-renewal of liver cancer stem cells [45, 46]. Similarly, SIRT2 overexpression has also been demonstrated in HCC promoting epithelial-mesenchymal transition and an aggressive phenotype [47]. Another member of the SIRT family of HDACs, SIRT3, represents the primary mitochondrial deacetylase that is indispensable for the maintenance of mitochondrial integrity and metabolism during oxidative stress [48]. In a mouse model fed a high fat diet, Hirschey et al. observed down-regulation of SIRT3 and mice lacking SIRT3 exhibited exacerbated obesity, insulin resistance, hyperlipidemia and steatohepatitis supporting a role of SIRT3 in safeguarding metabolic homeostasis [49]. Studies looking into the potential roles of other SIRT members in the development of liver diseases are expanding.
7.3.2 Histone Methylation in NAFLD-HCC
Though less well studied as compared to DNA methylation, histone methylation can be associated with transcriptional activation or repression. Histone methyltransferases mediate the transfer of methyl groups from S-adenosyl-L-methionine (SAM) to lysine or arginine residues of H3 or H4 histones. Common sites of methylation that have been reported to be involved in gene activation include H3K4, H3K48 and H3K79, whereas H3K9 and H3K27 are associated with gene inactivation [50]. Recent investigations demonstrated participation of histone methyltransferases in the development of diseases. For instance, Fei et al. recently reported the H3K9 methyltransferase SETDB1 was overexpressed in HCC and regulated tumour cell growth via di-methylation of p53 [51].
7.3.3 Histone Ribosylation in NAFLD-HCC
Adenosine diphosphate (ADP)-ribosylation refers to the addition of one or more ADP-ribose moieties from nicotinamide adenine dinucleotide (NAD) to acceptor proteins. The reaction is a reversible posttranslational modification catalyzed by two classes of enzymes: mono-ADP-ribosyltransferases and poly (ADP-ribose) polymerase (PARP) [52]. PARP is involved in a broad range of cellular functions including gene regulation, DNA damage repair, cell signaling as well as apoptosis [53, 54]. As with other types of modifications, aberrant PARP expression has been documented in various types of cancer including HCC. Poly-ADP-ribosylation and PARP expression were found to be significantly upregulated in human HCC when compared to adjacent non-tumour tissues [55]. Since then the potential of PARP as a therapeutic target for cancer has been intensively studied. In combination with DHMEQ (a novel inhibitor of NF-κB), the PARP inhibitor Olaparib has recently been shown to exert synergistic anti-tumour effects on HCC cells [56].
7.3.4 Histone Sumoylation in NAFLD-HCC
Sumoylation describes the covalent attachment of small ubiquitin-related modifier (SUMO) proteins to acceptor proteins. Four SUMO family members, SUMO-1 to SUMO-4, have been identified so far. Though SUMO-1 exhibits 18% sequence identity with ubiquitin and the two share similar three-dimensional structures, sumoylated proteins are not designated for degradation [57]. Indeed, sumoylation is commonly involved in various cellular processes including intracellular trafficking, transcriptional regulation, response to oxidative stress and cell cycle progression [58]. Sumoylation is also a dynamic process catalyzed by SUMO-specific activating (E1), conjugating (E2) and ligating (E3) enzymes [59] and can be reversed by the family of SUMO-specific proteases (SENPs) [60]. In addition to mediating transcriptional repression through recruitment of histone deacetylases and heterochromatin protein 1, sumoylation has been implicated in tumorigenesis [61]. Recently, upregulation of one of the SUMO-specific proteases, SENP5, has been observed in HCC to promote tumorigenesis in vitro and in vivo via de-sumoylation and regulation of DNA damage response [62]. SUMO1 has also been demonstrated to possess oncogenic properties in HCC by promoting p65 nuclear translocation and regulating NF-κB activity [63].
7.4 Non-coding RNAs (ncRNAs)
Non-coding RNAs (ncRNAs) constitute a significant proportion of the transcribed genome that is not destined to be translated into proteins. ncRNAs comprise highly abundant RNAs including transfer RNAs (tRNAs), ribosomal RNAs (rRNAs), microRNAs (miRNAs), small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), extracellular RNAs (exRNAs) and long ncRNAs (lncRNAs) [64]. The plethora of ncRNAs play crucial roles in a broad spectrum of biological processes while dysregulations of which contribute to the development of various diseases.
7.4.1 miRNAs
7.4.1.1 Definition
Ever since the discovery of lin-4 in the nematode Caenorhabditis elegans (C.elegans) in 1993, members of the novel class of small non-coding single strand regulatory RNAs, the microRNA (miRNAs) family, have been expanding drawing the attention of research focus [65]. miRNAs are each comprised of approximately 22 nucleotides and are found in a diverse array of organisms ranging from prokaryotes, eukaryotes to viruses. miRNAs can be either encoded by specific genes or located in the introns or exons of protein-coding genes and expressed as a by-product [65]. They play crucial roles in a wide spectrum of cellular and physiological functions, including cell proliferation, cell death, metabolism, haematopoiesis, and chromatin modification by modulating the expression of target genes [66].
7.4.1.2 Biogenesis and Mechanism of Action
Biogenesis of miRNAs in vertebrate initiates with the generation of a long primary miRNA (pri-miRNA) which is transcribed mostly by RNA polymerases type II (Pol-II). Each pri-miRNA is then processed into a hairpin-shaped precursor miRNA (pre-miRNA) of approximately 60–70 nucleotides by Drosha-like RNase III endonucleases. The pre-miRNA is subsequently transported out of nucleus into cytoplasm by Exportin-5 and Ran-GTP, and is then cleaved by Dicer-like RNase III endonuclease to form the mature miRNA duplex. Afterwards, one strand is usually incorporated into the RNA-induced silencing complex (RISC) whereas the other strand is degraded [67]. Regulation of gene expression is mediated through the canonical base pairing of miRNA seed sequence and the complementary sequence of target mRNAs followed by silencing or degradation of target mRNAs [68]. It has been reported an average miRNA has approximately 100 target sites, indicating that miRNAs are capable of regulating a large fraction of protein-coding genes [69]. Given the importance of miRNAs in the regulation of a wide array of cellular functions, it comes with no surprise that deregulating the function of miRNAs could lead to the development of multiple pathological conditions including cancer. Indeed, dysregulated miRNAs have been documented in various cancer types including chronic lymphocytic leukemia [70], breast cancer [71], lung cancer [72], colorectal cancer [73], prostate cancer [74] and ovarian cancer [75].
7.4.1.3 miRNAs in the Progression from NAFLD to HCC
Recent studies have demonstrated aberrant expressions of miRNAs are involved in the acquisition of NAFLD and subsequent progression to NASH. Deregulations of some of the key regulatory miRNAs have been shown to disturb normal glucose, cholesterol and lipid metabolism leading to intra-hepatic excessive accumulation of triglycerides and fatty acids [76]. It has also been demonstrated miRNAs are frequently dysregulated in different phonotypes of NAFLD, from simple steatosis through NASH to cirrhosis and eventually HCC [77].
One of the very first miRNAs associated with lipid metabolism and homeostasis is miR-122, the most abundant miRNA in adult human liver accounting for 70% of the liver’s total miRNAs [78]. Using murine models, Krutzfeldt et al. documented antagomirs targeting miR-122 resulted in reduction of plasma cholesterol levels coupled with altered expression of several genes involved in hepatic lipid biosynthesis [79]. A further study demonstrated inhibition of miR-122 significantly increased hepatic fatty acid oxidation and decreased the biosynthesis of hepatic fatty acid and cholesteral in vivo [80]. Although specific miRNA signatures responsible for NAFLD progression await further investigations, accumulating evidence has implicated a pivotal role of miR-122 in the process. For instance, mice lacking the gene encoding miR-122a were viable but later developed temporally controlled steatohepatitis, fibrosis and HCC [81]. Reduced expression of miR-122 has also been reported to upregulate modulators of tissue remodeling (including hypoxia-inducible factor 1 and vimentin) contributing to NASH-induced liver fibrosis [82]. A comparison of miR-122 levels in hepatocytes and primary human HCC cells revealed that miR-122 was down-regulated in HCC cells and the tumorigenic properties of cancer cells could be reversed by re-introduction of miR-122 [83]. Consistently, diminished expression of miR-122 has recently been shown to contribute to the acquisition of sorafenib chemoresistance in HCC [84] while miR-122 restoration in human stem-like HCC cells was capable of prompting tumour dormancy via Smad-independent TGF-β pathway [85], implicating that miR-122 might serve as a potential therapeutic target in HCC.
Being one of the firstly identified oncomirs, miR-21 upregulation has been demonstrated in the liver of patients with NAFLD and hepatic miR-21 expression is positively correlated with the severity of NASH [86, 87]. Using different mouse models of NASH, Loyer et al. [88] showed miR-21 was overexpressed in hepatic biliary and inflammatory cells while inhibition of miR-21 diminished liver injury, inflammation and fibrosis via restoration of peroxisome proliferator-activated receptor alpha (PPARα). miR-21 is also involved in the pathogenesis of HCC by suppressing expression of the tumour suppressor gene phosphatase and tensin homolog (PTEN) [89]. A recent study reported that miR-21 acted downstream of the oncogenic signal transducer and activator of transcription 3 (STAT3) mediating the tumorigenic properties of HCC cells, suggesting miR-21 inhibition or suppression might prove to be a novel treatment of HCC [90].
miR-34a represents another key oncomir displaying elevated expression in patients with NAFLD and positive association with the degree of NASH [86]. It has been shown that the miR-34a/SIRT1/p53 pro-apoptotic pathway played a significant role in human NAFLD development which could be suppressed by the inhibitor ursodeoxycholic acid (UDAC) [91]. Administration of pifithrin-α p-nitro (PFT), a p53 inhibitor, was capable of attenuating steatosis and liver injury in a mouse model of NAFLD partially attributed to the resulting transcriptional suppression of miR-34a [92]. In contrast, miR-34a functions as a tumour suppressor in HCC. A small molecule modulator of miR-34a, termed Rubone, has recently been demonstrated to dramatically inhibit tumour growth in a mouse xenograft model via restoration of miR-34a expression and functioning [93].
In an attempt to identify the pattern of altered gene expression at various time points in a high fat diet-induced NAFLD mouse model, Hur et al. [94] found reduced levels of miR-451 in palmitate-exposed HepG2 cells and mouse liver tissue. In vitro analysis further showed miR-451 negatively regulated palmitate-induced interleukin-8 (IL-8) and tumour necrosis factor alpha (TNFα) production supporting a role of miR-451 in preventing the progression from simple steatosis to severely advanced liver disease [94]. Concomitantly, miR-451 has also been documented to function as a potential suppressor of tumour angiogenesis in HCC by targeting IL-6R-STAT3-VEGF signaling, thereby implicating a promising therapeutic role of miR-451 in HCC [95]. miR-221/222, which has been intensively studied in the carcinogenesis of breast cancer, has recently been shown to be overexpressed in human liver in a fibrosis progression-dependent manner [96]. miR-221/222 was further established to promote liver tumorigenesis in a mouse transgenic model [97]. Taken together, studies aiming at elucidating the roles of various miRNAs in the progression from NAFLD to HCC are emerging and further investigations are highly anticipated for detailed insights.
7.4.2 lncRNAs
7.4.2.1 Definition and Functions
Long non-coding RNAs (lncRNAs) are another class of non-protein coding transcripts longer than 200 nucleotides in length that can be further divided into three subtypes: (1) antisense lncRNAs overlapping known protein-coding regions; (2) intronic lncRNAs overlapping transcripts and (3) long intergenic RNAs encoded in the intergenic space between protein-coding areas [98]. The majority of lncRNAs display high specificity with respect to cell subtype, tissue and developmental stage. Although lncRNAs are implicated in the fine-tuning of a wide array of biological processes related to liver homeostasis and cancer including cell proliferation, differentiation and migration, in-depth mechanisms by which the majority of lncRNAs mediate their actions remain largely unknown.
LncRNAs are responsible for the regulation of basal transcription machinery, mRNA processing and stability, protein translation and signal transduction [64]. One of the best-characterized lncRNAs functions in X chromosome inactivation in which the 17 kb transcript Xist recruits suppressive epigenetic factors to guarantee repression of gene expression and proper gene dosage in females [99]. Since then, research interest has been focusing on the emerging roles of lncRNAs in carcinogenesis with few reports mentioning the potential functions of lncRNAs in NAFLD. Until recently, Chen et al. [100] demonstrated the lncRNA steroid receptor RNA activator (SRA) promoted hepatic steatosis in mouse model via repressing the expression of adipose triglyceride lipase. In contrast, quite a number of studies have documented the roles of various lncRNAs in the development of HCC.
Highly upregulated in liver cancer (HULC), a 500 nt transcript discovered by cDNA microarray sequencing, is overexpressed 33-fold in HCC [101]. Using a transient silencing approach, the authors further reported HULC knockdown altered the expression of several genes related to the proliferation of HCC. HULC might also serve as a novel biomarker since it could be detected in the peripheral blood of HCC patients [101]. HOX transcript antisense intergenic RNA (HOTAIR) is a 2158 nt lncRNA displaying overexpression in HCC that is predictive of tumour recurrence in liver transplant patients [102]. Transient knockdown of HOTAIR has been shown to suppress tumorigenesis through inhibition of tumour cell growth and induction of cell cycle arrest [103]. Another recently identified lncRNA MALAT1 acts as a proto-oncogene via Wnt pathway activation and induction of the oncogenic splicing factor SRSF1 [104]. By and large, future studies using next generation sequencing will certainly shed light on the roles of more lncRNAs in hepatocarcinogenesis.
7.5 RNA Methylation
7.5.1 Introduction
The central dogma of molecular biology coined in the 50’s explains the flow of genetic information in living organisms from DNA to RNA and RNA to protein. As such, messenger RNA (mRNA) represents a bridging link faithfully translating the secrets of life encoded in DNA sequences into functional proteins. However, cellular protein levels are not necessarily associated with mRNA levels, suggesting post-transcriptional mRNA modifications are crucial in the regulation of gene expression [105]. In fact, more than 100 different types of chemical modifications have so far been identified in cellular RNA, including mRNA, ribosomal RNA (rRNA), transfer RNA (tRNA), snRNA and lncRNA [106]. The most prevalent modification among all is adenosine methylation, also known as m6A or N6-methyladenosine.
Analysis of nucleic acid modifications and the corresponding effects on epigenetic status has been garnering heated research intention. As mentioned in previous sections, much efforts and interests have been focusing on changes in the chemistry of DNA and the actions of histone proteins as well as their subsequent modifications. It was not until the 1970s with the discovery of m6A in a broad spectrum of eukaryotes-ranging from yeast, Drosophila, viruses to mammals-that investigators had realized and added a whole new RNA dimension to the field of epigenetics [107, 108]. Owing to a series of hindrances including the lack of knowledge of m6A demethylating enzymes, the short life-span of most RNAs, the resulting idea that m6A modifications are unalterable, coupled with technical limitations in detecting m6A-containing mRNAs, however, the pace of RNA epigenetic research had slowed down [109]. In 2011, a new surge of interest was sparked by the discovery that the fat mass and obesity associated protein (FTO) was capable of demethylating RNA, implicating m6A RNA modifications are highly dynamic subject to finely orchestrated regulations [110]. Elucidation of the methylated transcriptome in mammals was further achieved by technical breakthroughs such as m6A RNA immunoprecipitation followed by high-throughput sequencing (MeRIP-Seq) [111, 112]. Since then studies aiming at deciphering novel functions of the m6A modification and more members of the methylation/demethylation machinery have been on the rise.
7.5.2 m6A Modification and Regulation
Following two independent studies unequivocally demonstrating that m6A is a widespread phenomenon in mRNA, further investigations revealed m6A residues are enriched in 5′ untranslated regions (5’ UTRs), around stop codons and in 3’ UTRs adjacent to stop codons in mammalian mRNAs [111,112,113]. Bioinformatic analysis of MeRIP-Seq data identified the recognition sequence for m6A methylation as RRACH (where R = G/A and H = A/C/U). Occurrence of the consensus motif has been estimated at 1 in 2000 bases in human and almost 90% of all m6A peaks contain at least one of the motif variants [111, 112]. The dynamic regulation of m6A methylation is mediated by adenosine methyltransferases (“writers”) and demethylases (“erasers”).
Methyltransferase like 3 (METTL3) is established as the S-adenosyl-L-methionine (SAM)-binding component of a multiprotein methyltransferase complex responsible for catalyzing m6A mRNA methylation [114, 115]. The catalytic function of METTL3 was then confirmed by in vitro studies demonstrating METTL3 knockdown diminished m6A peaks in mRNAs from various cell lines [112, 113]. Intriguingly, localization of METTL3 in both cytoplasm and nucleus has been reported, implying m6A mRNA methylation could occur in both cytoplasmic and nuclear compartments [116]. As a close homologue to METTL3, METTL14 has also been shown to mediate methylation reactions and a complex formed by METTL3 and METTL14 possesses much more efficient activity than separated components [117]. As mentioned previously, the discovery of FTO as the first m6A mRNA demethylase ignited the conception of m6A as reversible modification and resurged research interest in RNA methylation. Functional investigations documented silencing of FTO increased m6A peaks while ectopic expression reduced m6A peaks [110]. AlkB Homolog 5 (ALKBH5), another member of the mRNA demethylase family, was later identified by in vitro and in vivo analyses [118].
7.5.3 Role of m6A Methylation in Disease
Given that m6A modifications have been demonstrated in many housekeeping genes influencing a wide array of biological processes including transcription splicing, nuclear RNA export, translation, energy production and cell differentiation, it comes with no surprise that dysregulation of the modification inevitably contributes to obesity, brain development abnormality and other pathological conditions [119,120,121]. In the field of hepatic diseases, FTO was found to be overexpressed in the livers of NASH patients. In vitro studies showed FTO knockdown was protective against palmitate-induced oxidative stress, mitochondrial dysfunction, ER stress and apoptosis, suggesting inhibition of FTO might serve as a treatment option for NASH [122].
The year of 2016 witnessed several inspiring studies documenting the involvement of m6A mRNA modifications in cancer. A hypoxic tumour microenvironment has been reported to stimulate breast cancer stem cell phenotype by increasing NANOG mRNA and protein expression via induction of HIF and ALKBH5 [123]. In lung adenocarcinoma, METTL3 promoted the growth, survival and invasion of cancer cells [124]. Recently, Ma et al. [125] demonstrated a pivotal role of METTL14 in the progression of HCC. Down-regulation of METTL14 accounted for reduced m6A modifications in HCC, acted as an adverse prognostic factor for disease-free survival and was significantly correlated with tumour metastasis in vitro and in vivo. The authors further showed METTL14 depletion reduced expression of the tumour suppressor miR-126 by modulating binding of the microprocessor protein DGCR8 to pri-miR-126 in an m6A-dependent manner [125]. Taken together, while detailed regulations and mechanisms of DNA epigenetics and histone modifications have been thoroughly studied and are already being targeted in various cancer therapies, the emerging RNA epigenetics may represent the next avenue for investigation in the pursuit for novel prognostic and treatment options.
7.6 Concluding Remarks and Future Perspectives
This chapter highlights some of the key epigenetic modulations implicated in the development of NAFLD-HCC. Further in-depth studies would undoubtedly reveal a more comprehensive picture of the role of epigenetics in the development of pathological conditions. On the basis of currently wealthy knowledge of DNA epigenetics, the rapidly growing field of RNA epigenetics will certainly drive forward a new avenue of research direction shedding light on the advancement of better diagnostics, prognostics and therapeutics in the coming era of precision medicine.
References
Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313:2263–73.
Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis. 2010;28:155–61.
Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic fatty liver disease: pathogenesis and disease spectrum. Annu Rev Pathol. 2016;11:451–96.
Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99–S112.
Sun C, Fan JG, Qiao L. Potential epigenetic mechanism in non-alcoholic fatty liver disease. Int J Mol Sci. 2015;16:5161–79.
Berlanga A, Guiu-Jurado E, Porras JA, Auguet T. Molecular pathways in non-alcoholic fatty liver disease. Clin Exp Gastroenterol. 2014;7:221–39.
Lee JH, Friso S, Choi SW. Epigenetic mechanisms underlying the link between non-alcoholic fatty liver diseases and nutrition. Forum Nutr. 2014;6:3303–25.
Anstee QM, Day CP. The genetics of NAFLD. Nat Rev Gastroenterol Hepatol. 2013;10:645–55.
Zilberman D, Henikoff S. Genome-wide analysis of DNA methylation patterns. Development. 2007;134:3959–65.
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5.
Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–3.
Park LK, Friso S, Choi SW. Nutritional influences on epigenetics and age-related disease. Proc Nutr Soc. 2012;71:75–83.
da Silva RP, Kelly KB, Al Rajabi A, Jacobs RL. Novel insights on interactions between folate and lipid metabolism. Biofactors. 2014;40:277–83.
Jacobs RL, Lingrell S, Zhao Y, Francis GA, Vance DE. Hepatic CTP:phosphocholine cytidylyltransferase-alpha is a critical predictor of plasma high density lipoprotein and very low density lipoprotein. J Biol Chem. 2008;283:2147–55.
Martinez-Chantar ML, Corrales FJ, Martinez-Cruz LA, Garcia-Trevijano ER, Huang ZZ, Chen L, et al. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1A. FASEB J. 2002;16:1292–4.
Tryndyak VP, Han T, Muskhelishvili L, Fuscoe JC, Ross SA, Beland FA, et al. Coupling global methylation and gene expression profiles reveal key pathophysiological events in liver injury induced by a methyl-deficient diet. Mol Nutr Food Res. 2011;55:411–8.
Sookoian S, Puri P, Castano GO, Scian R, Mirshahi F, Sanyal AJ, et al. Nonalcoholic steatohepatitis is associated with a state of betaine-insufficiency. Liver Int. 2017;37:611–9.
Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–18.
Moreira RK. Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med. 2007;131:1728–34.
Friedman SL. Cytokines and fibrogenesis. Semin Liver Dis. 1999;19:129–40.
Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–20.
Pogribny IP, Tryndyak VP, Bagnyukova TV, Melnyk S, Montgomery B, Ross SA, et al. Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. J Hepatol. 2009;51:176–86.
Wu Y, Bu F, Yu H, Li W, Huang C, Meng X, et al. Methylation of Septin9 mediated by DNMT3a enhances hepatic stellate cells activation and liver fibrogenesis. Toxicol Appl Pharmacol. 2016;315:35–49.
Pirola CJ, Scian R, Gianotti TF, Dopazo H, Rohr C, Martino JS, et al. Epigenetic modifications in the biology of nonalcoholic fatty liver disease: the role of DNA Hydroxymethylation and TET proteins. Medicine (Baltimore). 2015;94:e1480.
Murphy SK, Yang H, Moylan CA, Pang H, Dellinger A, Abdelmalek MF, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology. 2013;145:1076–87.
Zeybel M, Hardy T, Robinson SM, Fox C, Anstee QM, Ness T, et al. Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin Epigenetics. 2015;7:25.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Villanueva A, Portela A, Sayols S, Battiston C, Hoshida Y, Mendez-Gonzalez J, et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology. 2015;61:1945–56.
Revill K, Wang T, Lachenmayer A, Kojima K, Harrington A, Li J, et al. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology. 2013;145:1424–1435 e1421–1425.
Li H, Yang F, Gao B, Yu Z, Liu X, Xie F, et al. Hepatitis B virus infection in hepatocellular carcinoma tissues upregulates expression of DNA methyltransferases. Int J Clin Exp Med. 2015;8:4175–85.
Thienpont B, Steinbacher J, Zhao H, D'Anna F, Kuchnio A, Ploumakis A, et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature. 2016;537:63–8.
Chen ZJ, Pikaard CS. Epigenetic silencing of RNA polymerase I transcription: a role for DNA methylation and histone modification in nucleolar dominance. Genes Dev. 1997;11:2124–36.
Gallego-Duran R, Romero-Gomez M. Epigenetic mechanisms in non-alcoholic fatty liver disease: an emerging field. World J Hepatol. 2015;7:2497–502.
Granger A, Abdullah I, Huebner F, Stout A, Wang T, Huebner T, et al. Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J. 2008;22:3549–60.
Tian Y, Wong VW, Chan HL, Cheng AS. Epigenetic regulation of hepatocellular carcinoma in non-alcoholic fatty liver disease. Semin Cancer Biol. 2013;23:471–82.
Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol. 2007;8:284–95.
Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114:2363–73.
Bricambert J, Miranda J, Benhamed F, Girard J, Postic C, Dentin R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J Clin Invest. 2010;120:4316–31.
Jia YL, Xu M, Dou CW, Liu ZK, Xue YM, Yao BW, et al. P300/CBP-associated factor (PCAF) inhibits the growth of hepatocellular carcinoma by promoting cell autophagy. Cell Death Dis. 2016;7:e2400.
Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, et al. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science. 2011;331:1315–9.
Bae HJ, Jung KH, Eun JW, Shen Q, Kim HS, Park SJ, et al. MicroRNA-221 governs tumor suppressor HDAC6 to potentiate malignant progression of liver cancer. J Hepatol. 2015;63:408–19.
Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–35.
Herranz D, Serrano M. SIRT1: recent lessons from mouse models. Nat Rev Cancer. 2010;10:819–23.
Herranz D, Munoz-Martin M, Canamero M, Mulero F, Martinez-Pastor B, Fernandez-Capetillo O, et al. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun. 2010;1:3.
Liu L, Liu C, Zhang Q, Shen J, Zhang H, Shan J, et al. SIRT1-mediated transcriptional regulation of SOX2 is important for self-renewal of liver cancer stem cells. Hepatology. 2016;64:814–27.
Li Y, Xu S, Li J, Zheng L, Feng M, Wang X, et al. SIRT1 facilitates hepatocellular carcinoma metastasis by promoting PGC-1alpha-mediated mitochondrial biogenesis. Oncotarget. 2016;7:29255–74.
Chen J, Chan AW, To KF, Chen W, Zhang Z, Ren J, et al. SIRT2 overexpression in hepatocellular carcinoma mediates epithelial to mesenchymal transition by protein kinase B/glycogen synthase kinase-3beta/beta-catenin signaling. Hepatology. 2013;57:2287–98.
Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;17:41–52.
Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. 2011;44:177–90.
Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, et al. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell. 2003;12:1591–8.
Fei Q, Shang K, Zhang J, Chuai S, Kong D, Zhou T, et al. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat Commun. 2015;6:8651.
Ueda K, Hayaishi O. ADP-ribosylation. Annu Rev Biochem. 1985;54:73–100.
Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–9.
Corda D, Di Girolamo M. Functional aspects of protein mono-ADP-ribosylation. EMBO J. 2003;22:1953–8.
Nomura F, Yaguchi M, Togawa A, Miyazaki M, Isobe K, Miyake M, et al. Enhancement of poly-adenosine diphosphate-ribosylation in human hepatocellular carcinoma. J Gastroenterol Hepatol. 2000;15:529–35.
Lampiasi N, Umezawa K, Montalto G, Poly CM. (ADP-ribose) polymerase inhibition synergizes with the NF-kappaB inhibitor DHMEQ to kill hepatocellular carcinoma cells. Biochim Biophys Acta. 2014;1843:2662–73.
Verger A, Perdomo J, Crossley M. Modification with SUMO. A role in transcriptional regulation. EMBO Rep. 2003;4:137–42.
Hay RT. SUMO: a history of modification. Mol Cell. 2005;18:1–12.
Muller S, Hoege C, Pyrowolakis G, Jentsch S. SUMO, ubiquitin’s mysterious cousin. Nat Rev Mol Cell Biol. 2001;2:202–10.
Yeh ET. SUMOylation and De-SUMOylation: wrestling with life’s processes. J Biol Chem. 2009;284:8223–7.
Shiio Y, Eisenman RN. Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci U S A. 2003;100:13225–30.
Jin ZL, Pei H, Xu YH, Yu J, Deng T. The SUMO-specific protease SENP5 controls DNA damage response and promotes tumorigenesis in hepatocellular carcinoma. Eur Rev Med Pharmacol Sci. 2016;20:3566–73.
Liu J, Tao X, Zhang J, Wang P, Sha M, Ma Y, et al. Small ubiquitin-related modifier 1 is involved in hepatocellular carcinoma progression via mediating p65 nuclear translocation. Oncotarget. 2016;7:22206–18.
Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66.
Chaudhuri K, Chatterjee R. MicroRNA detection and target prediction: integration of computational and experimental approaches. DNA Cell Biol. 2007;26:321–37.
Alvarez-Garcia I, Miska EA. MicroRNA functions in animal development and human disease. Development. 2005;132:4653–62.
Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol. 2006;13:1097–101.
Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol. 2010;11:252–63.
Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA-target recognition. PLoS Biol. 2005;3:e85.
Mraz M, Pospisilova S. MicroRNAs in chronic lymphocytic leukemia: from causality to associations and back. Expert Rev Hematol. 2012;5:579–81.
Wang F, Zheng Z, Guo J, Ding X. Correlation and quantitation of microRNA aberrant expression in tissues and sera from patients with breast tumor. Gynecol Oncol. 2010;119:586–93.
Sotiropoulou G, Pampalakis G, Lianidou E, Mourelatos Z. Emerging roles of microRNAs as molecular switches in the integrated circuit of the cancer cell. RNA. 2009;15:1443–61.
Yang L, Belaguli N, Berger DH. MicroRNA and colorectal cancer. World J Surg. 2009;33:638–46.
Walter BA, Valera VA, Pinto PA, Merino MJ. Comprehensive microRNA profiling of prostate cancer. J Cancer. 2013;4:350–7.
Iorio MV, Visone R, Di Leva G, Donati V, Petrocca F, Casalini P, et al. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007;67:8699–707.
Rottiers V, Naar AM. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol. 2012;13:239–50.
Celikbilek M, Baskol M, Taheri S, Deniz K, Dogan S, Zararsiz G, et al. Circulating microRNAs in patients with non-alcoholic fatty liver disease. World J Hepatol. 2014;6:613–20.
Lewis AP, Jopling CL. Regulation and biological function of the liver-specific miR-122. Biochem Soc Trans. 2010;38:1553–7.
Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–9.
Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98.
Tsai WC, Hsu SD, Hsu CS, Lai TC, Chen SJ, Shen R, et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest. 2012;122:2884–97.
Csak T, Bala S, Lippai D, Satishchandran A, Catalano D, Kodys K, et al. microRNA-122 regulates hypoxia-inducible factor-1 and vimentin in hepatocytes and correlates with fibrosis in diet-induced steatohepatitis. Liver Int. 2015;35:532–41.
Coulouarn C, Factor VM, Andersen JB, Durkin ME, Thorgeirsson SS. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene. 2009;28:3526–36.
Kishikawa T, Otsuka M, Tan PS, Ohno M, Sun X, Yoshikawa T, et al. Decreased miR122 in hepatocellular carcinoma leads to chemoresistance with increased arginine. Oncotarget. 2015;6:8339–52.
Boix L, Lopez-Oliva JM, Rhodes AC, Bruix J. Restoring mir122 in human stem-like hepatocarcinoma cells, prompts tumor dormancy through smad-independent TGF-beta pathway. Oncotarget. 2016;7:71309.
Cheung O, Puri P, Eicken C, Contos MJ, Mirshahi F, Maher JW, et al. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology. 2008;48:1810–20.
Gori M, Arciello M, Balsano C. MicroRNAs in nonalcoholic fatty liver disease: novel biomarkers and prognostic tools during the transition from steatosis to hepatocarcinoma. Biomed Res Int. 2014;2014:741465.
Loyer X, Paradis V, Henique C, Vion AC, Colnot N, Guerin CL, et al. Liver microRNA-21 is overexpressed in non-alcoholic steatohepatitis and contributes to the disease in experimental models by inhibiting PPARalpha expression. Gut. 2016;65:1882–94.
Vinciguerra M, Sgroi A, Veyrat-Durebex C, Rubbia-Brandt L, Buhler LH, Foti M. Unsaturated fatty acids inhibit the expression of tumor suppressor phosphatase and tensin homolog (PTEN) via microRNA-21 up-regulation in hepatocytes. Hepatology. 2009;49:1176–84.
Zhang N, Duan WD, Leng JJ, Zhou L, Wang X, Xu YZ, et al. STAT3 regulates the migration and invasion of a stemlike subpopulation through microRNA21 and multiple targets in hepatocellular carcinoma. Oncol Rep. 2015;33:1493–8.
Castro RE, Ferreira DM, Afonso MB, Borralho PM, Machado MV, Cortez-Pinto H, et al. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J Hepatol. 2013;58:119–25.
Derdak Z, Villegas KA, Harb R, Wu AM, Sousa A, Wands JR. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J Hepatol. 2013;58:785–91.
Xiao Z, Li CH, Chan SL, Xu F, Feng L, Wang Y, et al. A small-molecule modulator of the tumor-suppressor miR34a inhibits the growth of hepatocellular carcinoma. Cancer Res. 2014;74:6236–47.
Hur W, Lee JH, Kim SW, Kim JH, Bae SH, Kim M, et al. Downregulation of microRNA-451 in non-alcoholic steatohepatitis inhibits fatty acid-induced proinflammatory cytokine production through the AMPK/AKT pathway. Int J Biochem Cell Biol. 2015;64:265–76.
Liu X, Zhang A, Xiang J, Lv Y, Zhang X. miR-451 acts as a suppressor of angiogenesis in hepatocellular carcinoma by targeting the IL-6R-STAT3 pathway. Oncol Rep. 2016;36:1385–92.
Ogawa T, Enomoto M, Fujii H, Sekiya Y, Yoshizato K, Ikeda K, et al. MicroRNA-221/222 upregulation indicates the activation of stellate cells and the progression of liver fibrosis. Gut. 2012;61:1600–9.
Callegari E, Elamin BK, Giannone F, Milazzo M, Altavilla G, Fornari F, et al. Liver tumorigenicity promoted by microRNA-221 in a mouse transgenic model. Hepatology. 2012;56:1025–33.
Hardy T, Mann DA. Epigenetics in liver disease: from biology to therapeutics. Gut. 2016;65:1895.
Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–6.
Chen G, Yu D, Nian X, Liu J, Koenig RJ, Xu B, et al. LncRNA SRA promotes hepatic steatosis through repressing the expression of adipose triglyceride lipase (ATGL). Sci Rep. 2016;6:35531.
Panzitt K, Tschernatsch MM, Guelly C, Moustafa T, Stradner M, Strohmaier HM, et al. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology. 2007;132:330–42.
Yang Z, Zhou L, Wu LM, Lai MC, Xie HY, Zhang F, et al. Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann Surg Oncol. 2011;18:1243–50.
Fu WM, Zhu X, Wang WM, Lu YF, Hu BG, Wang H, et al. Hotair mediates hepatocarcinogenesis through suppressing miRNA-218 expression and activating P14 and P16 signaling. J Hepatol. 2015;63:886–95.
Malakar P, Shilo A, Mogilavsky A, Stein I, Pikarsky E, Nevo Y, et al. Long noncoding RNA MALAT1 promotes hepatocellular carcinoma development by SRSF1 up-regulation and mTOR activation. Cancer Res. 2017;77(5):1155–67.
Wu L, Candille SI, Choi Y, Xie D, Jiang L, Li-Pook-Than J, et al. Variation and genetic control of protein abundance in humans. Nature. 2013;499:79–82.
Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, et al. MODOMICS: a database of RNA modification pathways--2013 update. Nucleic Acids Res. 2013;41:D262–7.
Wei CM, Moss B. Methylated nucleotides block 5′-terminus of vaccinia virus messenger RNA. Proc Natl Acad Sci U S A. 1975;72:318–22.
Rottman FM, Desrosiers RC, Friderici K. Nucleotide methylation patterns in eukaryotic mRNA. Prog Nucleic Acid Res Mol Biol. 1976;19:21–38.
Cao G, Li HB, Yin Z, Flavell RA. Recent advances in dynamic m6A RNA modification. Open Biol. 2016;6:160003.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–46.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–6.
Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15:707–19.
Bokar JA, Rath-Shambaugh ME, Ludwiczak R, Narayan P, Rottman F. Characterization and artial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J Biol Chem. 1994;269:17697–704.
Bokar JA, Shambaugh ME, Polayes D, Matera AG, Purification RFM. cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. 1997;3:1233–47.
Chen T, Hao YJ, Zhang Y, Li MM, Wang M, Han W, et al. m(6)A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell. 2015;16:289–301.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–5.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29.
Boissel S, Reish O, Proulx K, Kawagoe-Takaki H, Sedgwick B, Yeo GS, et al. Loss-of-function mutation in the dioxygenase-encoding FTO gene causes severe growth retardation and multiple malformations. Am J Hum Genet. 2009;85:106–11.
Klungland A, Dahl JA. Dynamic RNA modifications in disease. Curr Opin Genet Dev. 2014;26:47–52.
Blanco S, Frye M. Role of RNA methyltransferases in tissue renewal and pathology. Curr Opin Cell Biol. 2014;31:1–7.
Lim A, Zhou J, Sinha RA, Singh BK, Ghosh S, Lim KH, et al. Hepatic FTO expression is increased in NASH and its silencing attenuates palmitic acid-induced lipotoxicity. Biochem Biophys Res Commun. 2016;479:476–81.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113:E2047–56.
Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62:335–45.
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N6 -methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65:529–43.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Zhao, F. (2018). Dysregulated Epigenetic Modifications in the Pathogenesis of NAFLD-HCC. In: Yu, J. (eds) Obesity, Fatty Liver and Liver Cancer. Advances in Experimental Medicine and Biology, vol 1061. Springer, Singapore. https://doi.org/10.1007/978-981-10-8684-7_7
Download citation
DOI: https://doi.org/10.1007/978-981-10-8684-7_7
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-8683-0
Online ISBN: 978-981-10-8684-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)