
Abstract
Fabrication of magnetic materials with a high level of control down to the nanoscale is a current synthetic challenge. Nature is able to achieve this level of precision under ambient conditions and in aqueous solutions, by using specific biomineralisation proteins to produce highly monodisperse magnetic nanoparticles within the magnetosome organelles of magnetotactic bacteria. This chapter explores the use of such proteins outside the magnetosome, in synthetic magnetite formation reactions, where their ability to control and affect the nanoparticle products in terms of size, morphology and material purity is demonstrated. Understanding how these proteins function to achieve their activity is of particular interest, and we bring together the current literature to assess the roles of sequence and self-assembly in this process. In addition to the magnetosome-derived proteins, researchers are expanding the biological toolkit of available magnetic material mineralising proteins by using and adapting others. We investigate a number of these proteins including ferritin, heat shock protein cages and even small peptides. These can be used without modification, or they can be engineered to contain artificial binding sequences, selected via processes such as phage display. Developing new mineralising sequences allows proteins to be used with materials which are not naturally occurring, such as the platinum alloys of cobalt and iron, which have industrially desirable magnetic characteristics. The proteins/peptides covered in this chapter have the potential to aid future production of precise magnetic nanoparticles for diverse applications in the biomedical and data storage fields.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
- Magnetic Nanoparticles (MNPs)
- Biomineralization Proteins
- CagA Protein
- Magnetotactic Bacteria (MTB)
- Magnetosome Membrane
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Nanoscale inorganic materials are becoming increasingly important as the use of nanotechnology grows. More specifically, magnetic nanoparticles (MNPs) have wide-ranging uses from targeted drug delivery (Muthana et al. 2015) to ultrahigh-density data storage (Terris and Thomson 2005). Magnetite MNPs are particularly useful for biomedical applications owing to their high magnetic saturation and low toxicity, while platinum alloy MNPs such as CoPt and FePt in their L10 phase are ideal for data storage owing to their very high magnetoanisotropy and their ability to retain this at very small particle sizes. However, the reliable production of highly specific monodispersed MNP for such applications is a considerable challenge. There is therefore a critical need to develop new synthetic routes to precisely tailored MNPs.
Natural organisms carefully control the production of a vast range of inorganic minerals through biomineralisation, which is detailed in the previous chapters of this book. MNPs are biomineralised naturally by magnetotactic bacteria (MTB) that take up soluble iron ions from solution and crystallise magnetite MNPs in liposomes within their cells (called magnetosomes). Magnetosomes’ size and morphology varies greatly between strains, but is exactly adhered to within each strain, demonstrating the precision that biomineralisation proteins have over the process. The mechanism of biomineralisation in MTB is the subject of chapters X and Y. The process is controlled by a unique suite of biomineralisation proteins embedded into the magnetosome membrane. Many of these proteins perform functions that we can easily replicate in a chemical test tube, such as the action of redox proteins and iron pumps. In a chemical synthesis, we can simply add iron ions and change the pH at will. However, some of these proteins are able to control the nucleation, crystallisation and morphology of the growing mineral with a very high specificity that chemists cannot currently replicate under ambient conditions, and these proteins will be the focus of this chapter.
In this chapter we explore the prospect of using nature’s MTB biomineralisation proteins independently of the cell. Here we discuss using such proteins in vitro to mediate and control a simple synthetic, solution phase, chemical formation of MNPs. We then explore moving away from natural MTB proteins to synthetic proteins and peptides for the production of magnetite and also other MNPs such as CoPt & FePt. However, first we must consider the chemical synthesis of MNPs.
There are many different magnetic materials and several synthetic routes for each type, leading to a vast number of possible syntheses to discuss. For clarity and relevance to the desired applications, we will only focus on magnetite and CoPt & FePt. For both of these materials, there is a range of synthetic methods to produce them. As this chapter is concerned with simulating biological processes outside the cell, we will only focus on solution-based synthetic methods performed under conditions close to biological/ambient conditions that will be relevant for protein and peptide addition.
Magnetite (Fe3O4) is strongly ferrimagnetic, retains its single-domain character between approximately 30 and 80 nm (depending on the morphology) but is magnetically soft (low coercivity), with its dipole easily switched in a changing magnetic field. Magnetite is biocompatible, being present naturally in many biological organisms. It can be synthesised with a range of particle sizes and shapes, offering different magnetic characteristics. CoPt on the other hand is strongly ferromagnetic. This alloy has a chemically disordered face-centred cubic structure, also known as the A1 phase, where there is an equal probability of finding either Co or Pt in any given site. The CoPt L10 phase (Laughlin et al. 2005) can be obtained through post synthesis high-temperature (>600 °C) annealing. This is a chemically ordered face-centred tetragonal structure with alternating planes of Co and Pt resulting in a very high magnetocrystalline anisotropy energy. This is due to strong structural coupling of anisotropic atoms, giving a very high coercivity and offering single-domain character in particles down to a few nm (meaning these particles can retain their magnetisation direction in varying fields), making them ideally suited to high-density data storage (Mayes et al. 2003).
Chemical co-precipitation is the simplest method of producing both of these MNP materials by oxidising/precipitating Fe salts out of solution via increasing pH to make magnetite (under N2) or by reducing/precipitating Co and Pt salts out of solution with LiBEt3H to form CoPt (Sun et al. 2003). This method is quick and cheap and produces large yields, but the particle quality, size and morphology are very difficult to control and typically result in a heterogeneous population. Variation in precursors, conditions and processing results in different MNP profiles. For example, with magnetite, varying either the type or method of base addition, the speciation and ratio of the iron ions (ferric/ferrous), the available oxygen in the atmosphere and the temperature can result in different sizes and shapes of particles with different iron oxides of varying crystallinity. For example, a co-precipitation of ferric and ferrous ions at room temperature under N2 with KOH used to raise the pH results in tiny nanoparticles of poor crystallinity over a wide shape and size range (<5 nm up to micrometre scales), whereas the partial oxidation of ferrous hydroxide using KOH under N2 at 90°C will precipitate octahedral magnetite particles approximately 20–80 nm in size. Interestingly, changing the base used can result in needle-shaped FeOOH by-products (Regazzoni et al. 1981). Although these methods allow scope to synthesise a range of materials at various sizes, the overwhelming drawback is that it is synthetically demanding to produce a monodisperse and reproducible product with respect to size and shape distribution. There are a range of high-temperature methods of producing higher quality MNP of both magnetite and CoPt with narrower dispersity; however, they require furnaces with high temperatures and pressures and/or organic solvents and toxic precursors (Sun et al. 2004). Furthermore, these conditions are difficult/dangerous to scale up to industrial manufacture and are financially and environmentally expensive.
In this chapter we investigate how we can improve the simple solution phase precipitation of these MNPs using proteins and peptides as synthesis control agents. Firstly, we consider two applications to understand and contextualise the requirements of MNP, and then we turn to nature to consider how MNPs are produced naturally by magnetotactic bacteria. We assess biomineralisation proteins and how they can be used as additives to control chemical MNP formation and consider how they achieve their function in vitro. We investigate the development of new material binding peptides, discovered via the technique of phage display, and how these are opening up the process of biologically controlled MNP synthesis to new materials and synthetic scenarios. Finally we show how these proteins/peptides and MNP can be coupled on planar surfaces to provide a basis for the next generation of data storage technologies.
2 Applications
The development of reliable, reproducible nanomaterials is essential to the evolution of dependable novel technologies, and nanoscale magnetic materials are no exception. Nanomagnets must respond to an applied field in a known and consistent manner, which requires them to have a uniform size and shape distribution. However, their individual specification with respect to size, shape, coating and degree of homogeneity of the population will vary depending on the final application. The scope of applications for MNP ranges from environmental applications such as magnetic bioremediation and contaminant sensors to biomedical diagnostics and therapies to nanotechnology such as data storage and lab-on-a-chip devices. Due to the extensive range of applications, in this chapter we will only briefly consider biomedical and data storage applications as these are directly relevant to the research described later and will enable the ultimate context and specification to be properly understood. For further, more in-depth reading on MNP applications, the reader is directed to excellent reviews describing the various applications (del Puerto et al. 2003; Lu et al. 2007; Terris et al. 2007).
2.1 Biomedical (Magnetite)
Magnetic nanoparticles have a wide range of applications associated with them – one of the most prominent areas of interest lies within the biomedical field. Biomedical applications of magnetite nanocrystals span the remit of both diagnostic and therapeutic areas. Diagnostics encompass any technique used to identify a marker for a certain disease; this could be a specific antigen or an increased level of a certain element/compound. Identification of biomarkers can indicate the presence or absence of a disease in the body. Research is currently being conducted into therapies that are able to treat disease and illness using magnetic nanoparticles. Examples include hyperthermia-based treatments and novel drug delivery systems. Theranostics involving magnetic nanoparticles may also prove to be important in the development of new diagnostic and therapeutic systems. Theranostics refers to the combination of diagnosis and therapy within a single treatment strategy.
When the nanoparticles are used in vivo, it is important that they are coated in a biocompatible layer to ensure it does not lead to an adverse reaction upon addition to the patient and to aid cellular uptake. Sugar or polymer coatings such as dextran or polyvinyl alcohol (PVA) may be used to increase biocompatibility, to increase colloidal stability and to provide a site for binding between the nanoparticle and the biological complex (Sangregorio et al. 1999). Monodispersity with respect to size and shape is also critical to predict the interaction within the body as different sized particles will be directed to different organs of the body. Furthermore, the MNPs must have good dispersity in solution as large sizes and/or aggregation could lead to capillary blockage and embolism.
2.1.1 Magnetic Separation
Magnetic separation of analytes is one instance of their use. For this example, a biological complex (such as an antibody) is tagged with a magnetic nanoparticle (such as magnetite). Following attachment the biological complex is separated from the remaining solvent or serum using magnetic separation of a fluid phase. Magnetic separation of these compounds allows the concentration of sample to be increased and used for further analysis. The magnetically tagged complex is run through a separating system where a magnetic gradient is present. The magnetic gradient immobilises the magnetic complex while the remaining fluid flows through. One disadvantage of this technique is that it is often limited by slow accumulation rates. Instead areas of high magnetism can be suspended throughout the separation vessel, immobilising the magnetic nanoparticles as they float in solution. Further optimisation of the system involves a quadrupolar set-up, where a magnetic moment is produced radially from the centre of the column (Moore et al. 2001). The labelled magnetic nanoparticles move towards the sides of the column where they become immobilised until the magnetic gradient ceases. This technique has been used to detect and diagnose the existence of parasites in the blood by magnetically tagging red blood cells (Paul et al. 1981).
2.1.2 MRI
Magnetic resonance imaging (MRI) is a useful diagnostic tool that utilises magnetic nanoparticles. The iron oxide particles act as contrast agents, providing increased contrast of specific tissues, organs and tumours compared to the rest of the body. The contrast provided by iron oxide nanoparticles was enhanced using magnetite nanocubes (Lee et al. 2011). MRI uses the magnetic moment of protons within the body to produce an image (e.g. of a tumour). Although the magnetic moment of a proton is extremely small, there are so many protons present that the cumulative effect allows for a detectable signal. The problem, however, is that human bodies are mainly composed of water and so protons are indiscriminately ubiquitous, providing negligible differences in contrast between different tissues. This problem is overcome by the addition of a contrast agent. A magnetic contrast agent reduces the relaxation times T1 and T2. T1 is the spin-lattice relaxation time, accounting for loss of heat to the surrounding lattice. T2 is the spin-spin relaxation time and is the measure of the time it takes for the signal to irreversibly decay; this decay occurs as excited nuclear spins in the xy plane interact with each other. Superparamagnetic iron oxide nanoparticles are commonly used contrast agents (Pankhurst et al. 2003). One example of their use is in MRI imaging of the liver. Preferential uptake of the nanoparticles in the liver is partially due to size, as MNPs with diameters of 37 nm MNPs collected more readily in the liver (Huang et al. 2010). This illustrates just how important size control of the MNPs is.
MRI can also be used to assist therapeutic strategies involving magnetic nanoparticles. The external magnetic field applied by the MRI may be used to localise the magnetic nanoparticles to one specific area of the body by directing the magnetic field gradient (Muthana et al. 2015). If this is possible, unwanted side effects of alternative therapeutics may be overcome due to the idea that the doses required for treatment will be significantly reduced and the drug will not affect the whole body – only the desired area. Problems with this method arise due to the biocompatibility of magnetite nanoparticles. Addition of coatings alters the control exhibited over their size and shape distribution during synthesis. This technique would, in theory, work alongside other biomedical applications such as hyperthermia and drug transport.
2.1.3 Hyperthermia
Therapeutic devices aid the treatment of a disease. Hyperthermia is one example of a possible therapeutic application of magnetite nanoparticles. Hyperthermia utilises an alternating magnetic field, which causes flipping of the electron spins within the magnetite nanoparticles. The flipping of the electron spins causes heating of the particles (Pankhurst et al. 2003). If the particles are very small, the alternating field results in the whole MNP flipping, generating heat through friction with the environment. Artificially induced hyperthermia has arisen as a possible therapy due to the ability to heat and kill tumour cells while not leading to the death of the surrounding healthy cells. Cancer cells have been found to be more susceptible to these heating effects when compared to healthy cells (Moroz et al. 2001; van der Zee 2002). Magnetic nanoparticles show promise for this application as they can be localised at the site of the diseased tissue (Hatch and Stelter 2001). The technique involves the targeting of magnetic nanoparticles to the desired tissue, followed by the application of an alternating magnetic field. The therapeutic threshold for the destruction of diseased tissue is 42 °C for 30 min. When applying this technology to patients, it is important that the alternating magnetic field is not so strong that it causes undesired effects such as arrhythmia; however, the field must be strong enough to sufficiently heat the tumour. For magnetite to be used for this application, it must first be mixed with a carrier fluid, and the size of the particle must be uniform and on the nanoscale. A monodisperse nanoparticle suspension is essential for uniform heating of the tissue.
In summary, there are various biomedical strategies that utilise magnetic nanoparticles, including (but not limited to) MRI and hyperthermia. MNPs appear to be useful in diagnostic, therapeutic and emerging theranostic applications. It is important to emphasise that the optimisation of the physical parameters of the magnetic nanocrystals is critical for their effective use in these applications.
2.2 Nanotechnology: Bit-Patterned Media Data Storage (CoPt/FePt)
The ideological goal of nanotechnology is to design and synthesise precise nano-components and assemble them accurately into increasingly complex hierarchies and designs to form intricate nanoscale devices. If this was achievable, devices such as sensors, energy accumulators and motors could be vastly miniaturised and interconnected. Data storage is an ideal model nanodevice to fulfil this aim.
2.2.1 Traditional Magnetic Storage
These devices consist of a thin ferromagnetic granular film, typically a Co-based alloy, deposited onto a glass or silicon surface by high-temperature sputtering (Terris and Thomson 2005). Data is recorded and read from these devices by a flying read/write head. To write, the information is transformed into a magnetic signal through an electric current, and this varying magnetic field in the write head magnetically orientates the grains contained within the magnetic film. This forms magnetic patterns in a binary code (0’s and 1’s) that can be read as bits of information by the read head operating in the reverse process. Since IBM introduced the first commercial magnetic hard disc in 1956, increased miniaturisation has driven the storage capacity ever higher, with today’s devices having capacities in excess of 500 Gbit in−2 (more than 20,000,000 times the capacity of IBM’s original device) (Terris and Thomson 2005). Although there are a number of semiconductor-based storage technologies such as dynamic random access memory (DRAM), magnetic devices are the most established technology (Nickolls and Dally 2010) and are significantly cheaper to produce (Terris and Thomson 2005). However, scaling the components of magnetic storage devices to ever smaller dimensions does have its limitations, and current devices are approaching their physical limits. Reducing the grain size of the granular recording media leads to domination of thermal fluctuations which induce the loss of magnetic orientation and thus the information, rendering them unsuitable for data storage as the information cannot be retained (Charap et al. 1997).
2.2.2 Bit-Patterned Media (BPM)
BPM is a new generation of storage media that promises to increase storage densities. It requires the formation of an array of individual magnetic islands with nanoscale dimensions onto which a bit of information is written (Terris and Thomson 2005; Terris et al. 2007). BPM has the ability to form devices with Tbit in−2 storage capacities (Terris et al. 2007). However, there are a number of challenges to overcome before bit-patterned media is used for ultrahigh-density data storage, including the development of a reliable and cost-effective method for the production of a recording medium containing uniform and stable magnetic islands on the nanoscale (Terris et al. 2007).
To become commercially viable, the technique used to manufacture bit-patterned storage media must be cost effective and able to be scaled up for mass production. There are three elements to this that need to be fabricated with a very tight degree of tolerance: (1) the MNP material, (2) the particle dimensions and (3) the pattern dimension.
-
1.
The magnetic material is required to have a high magnetisation to ensure a signal can be clearly read, but not so high as to reorientate its neighbouring MNPs. Similarly, the MNP must be magnetically “hard”, i.e. have a high-coercive force which resists reorientation when a field is reversed. This is important to ensure the information is retained. However, the force should not be so great (like a permanent magnet) as to resist information being written onto the MNP. To get the highest density of data stored in a given area, the MNPs must be as small as possible to reduce the size of each bit but not so small as to become superparamagnetic. The “magnetocrystalline anisotropy” of a material is how easily the material is to magnetise in one direction (easy axis) when compared to another (hard axis) and is a key factor in dictating its coercivity. The L10 phase of CoPt and FePt has received a lot of interest due to being two of the few magnetic materials that has high enough magnetocrystalline anisotropy to remain magnetically stable at volumes of a few nanometres in diameter (Weller et al. 2000). L10 phase 4 nm FePt particles and 18 nm CoPt have shown room temperature coercivity of 7.5 kOe (Sun et al. 2003) and 4.4 kOe (Zhang et al. 2011), respectively; however, the L10 ordered phase is difficult to achieve. Ferromagnetic CoPt alloys formed from a solid solution usually have a disordered face-centred cubic (fcc) structure, also known as the A1 phase (Aboaf et al. 1983). CoPt alloys that have a 50:50 atomic ratio are known to have distinct ferromagnetic properties and a high magnetocrystalline anisotropy energy but only after high-temperature annealing (>600 °C) (Aboaf et al. 1983) where the fcc disordered structure transforms into a face-centred tetragonal (fct) ordered L10 phase. In the A1 phase the probability that each site is occupied by either a Co or Pt atom is the same, whereas in the L10 phase two faces are occupied by one type of atom while the other face is occupied by a different atom giving it a multilayer structure that consists of alternating planes of Co and Pt which promotes the strong magnetic anisotropy and high coercivity (Laughlin et al. 2005). The high magnetocrystalline anisotropy energy of the L10 phase means that the magnetic properties remain stable at dimensions of a few nanometres, which makes L10 CoPt or FePt ideally suited for bit-patterned media (Weller et al. 2000). However, there are problems with current methods to generate the L10 phase due to the current requirement of a high-temperature annealing step, which leads to agglomeration and sintering of the particles (Wang et al. 2014). Interestingly, however, peptide and protein-mediated synthesis of L10 CoPt and FePt at ambient temperature (see Sect. 5.4) have demonstrated the potential of a bottom-up biotemplated route to L10 Pt-alloy MNPs for data storage.
-
2.
Controlling the dimensions of the MNPs arrayed on the surface is paramount to obtain the highest density storage. Ideally MNPs of 5.5 nm in diameter should be spaced 15–20 nm apart (Richter et al. 2006). However, controlling the crystallisation of this size of MNP accurately is a considerable challenge. Within this chapter we will present new possibilities of how proteins and peptide could control the size of MNP and further to this could improve this control when combined with biotemplating within a biological compartment such as a protein cage of the tailored size (see Sect. 5.4).
-
3.
The patterning dimensions can be altered depending upon the technique used. There are many techniques with the capability to produce nanoscale magnetic features (Martın et al. 2003). Top-down methods such as optical lithography, although widely used in the semiconductor industry, are not currently able to achieve patterning resolutions suitable for the fabrication of bit-patterned media (Wollhofen et al. 2013). Electron-beam lithography is widely used for the production of sub-100 nm patterns but is a high-cost serial writing process that is unlikely to ever be scaled up for mass production (Driskill-Smith 2004). Low cost, versatile, soft lithographic printing methods could be financially viable but suffer from issues with reproducibility and scaling down to the nanometre length scale. The most promising top-down lithographic route looks to be interferometric lithography which is discussed in more detail in Sect. 5.5.
3 Mms6, MmsF and MamC for Solution Formation of Magnetite
3.1 Effect of Proteins In Vivo
As described in the previous chapter, the magnetosomes found in magnetotactic bacteria are protein-rich lipid vesicle organelles. The magnetosome membrane is home to a variety of different proteins which are responsible for controlling all the aspects of the magnetic nanocrystal formation, from influx of iron ions to nucleation of the iron oxide mineral and its morphological regulation. Many of these proteins reside in the lipid bilayer which surrounds the magnetosome and as such have transmembrane-spanning (TMS) regions in their sequence. Proteins such as Mms6 which interact with the magnetite nanocrystals directly must do so through the residues which are exposed to the interior of the magnetosome. Depending on the topology of the proteins within the membrane and the number of TMS segments that they have, these interacting regions could be N- or C-terminal domains or solvent-exposed loops connecting TMS helices (Fig. 5.1c). Mms6 and Mms7 both contain a single TMS region (Arakaki et al. 2003), and their C-terminal domain is displayed to the magnetosome interior (Fig. 5.1c). Between different strains of MTB, there are differences in naming nomenclature. Mms13, for instance, from Magnetospirillum magneticum AMB-1, is designated MamC in many other strains of MTB. Studies of Mms13/MamC indicate this protein adopts a two TMS structure with both N- and C-termini exposed to the cytoplasm (Yoshino and Matsunaga 2006), meaning the loop connecting TMS helices 1 and 2 is presented to the magnetosome lumen. MmsF from M. magneticum AMB-1 is a triple TMS protein (Murat et al. 2012; Rawlings et al. 2014). Topological studies indicate that the N-terminal domain is located in the cytoplasm, meaning that the loop regions between TMS helices 1 and 2 must be located inside the magnetosome interior. An odd number of TMS segments can only be accomplished if the protein termini are on different faces of the membrane. Therefore the C-terminal domain of MmsF will be located in the magnetosome interior.

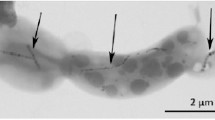
Effect of magnetosome-derived biomineralisation proteins upon magnetite nanoparticle synthesis. (a) Basic reaction set-up for magnetite nanoparticle synthesis. (b) Transmission electron microscopy images showing the effects on nanoparticle products with and without the presence of different proteins. Proteins are Mms6 (Amemiya et al. 2007) and MmsF (Rawlings et al. 2014) from M. magneticum AMB-1 and MamC (Valverde-Tercedor et al. 2015) from Magnetococcus marinus MC-1. c The topology, number of transmembrane-spanning regions and potential magnetite interaction sequences of key morphology affecting proteins in the magnetosome membrane are shown. Mms6/7/F from M. magneticum AMB-1 and MamC from Magnetococcus marinus MC-1 (Images are reproduced with permissions from the references cited)
Mms6, Mms7 and Mms13 were all found to be tightly adhered to the magnetite nanoparticle in vivo (Arakaki et al. 2003). Having a direct binding interaction in this way suggests that these proteins may have a role in controlling the nanoparticle nucleation and/or growth. Several gene knockout and mutational studies have highlighted the importance of the genes encoding these proteins, along with MmsF, for the correct formation of the magnetite nanocrystal (Murat et al. 2012; Tanaka et al. 2010; Yamagishi et al. 2016). This makes these proteins prime targets for in vitro functional analysis.
3.2 Effect of Proteins In Vitro
The proteins present in the magnetosome membrane which are responsible for controlling and affecting the morphology of the resulting nanoparticle are also capable of exerting a similar effect when added to synthetic magnetite formation reactions (Amemiya et al. 2007; Arakaki et al. 2003, 2010; Galloway et al. 2011; Kashyap et al. 2014; Prozorov et al. 2007; Rawlings et al. 2014; Valverde-Tercedor et al. 2015; Wang et al. 2012).
The iron oxide magnetite (Fe3O4) comprises both ferric (Fe3+) and ferrous (Fe2+) iron in the stoichiometric ratio of 2:1. Many different methods exist for the synthesis of magnetite, but most of them require the presence of mixed valence iron and a high pH achieved via the addition of base (Laurent et al. 2008). Incorporating purified magnetosome-derived proteins into these types of reactions allows their effect on particle size, shape and material purity to be assessed in comparison to particles produced in their absence. The exact nature of the biological magnetite precipitation process occurring within the magnetosome has not yet been clearly resolved. However, it is likely that the current synthetic methods for studying Mms6 activity in vitro will share some similarities to the processes which take place within the magnetosome (Table 5.1).
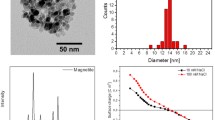
The first recorded in vitro activity of a magnetosome-derived protein was with purified Mms6. In that example, addition of Mms6 to a simple room temperature co-precipitation (RTCP) reaction (20 μg/ml Mms6) resulted in nanoparticle products, of the majority magnetite, with a narrow size distribution and cuboidal morphology which is similar to the particle morphology found in Mms6 containing magnetosomes. Similar results to this have been achieved with reduced amounts of Mms6 (Amemiya et al. 2007) (Fig. 5.1c), alternative ratios of ferrous to ferric iron in the starting reagents (Galloway et al. 2011), as well as in POFH reactions (Amemiya et al. 2007; Galloway et al. 2011), with Mms6 immobilised on planar surfaces (Bird et al. 2016b), and in pluronic gels (Prozorov et al. 2007; Wang et al. 2012). Collected together these reports indicate that the presence of Mms6 in solution typically regulates the size of the nanoparticles produced (30–20 nm) while simultaneously narrowing their size distribution, seemingly irrespective of the protein-free particle size population or reaction conditions. When used in pluronic gels, the protein increases the size of the MNP to around 30 nm, a significant increase compared to protein-free control particles (approximately 10 nm) (Prozorov et al. 2007; Wang et al. 2012). In contrast, when Mms6 is immobilised on surfaces and used in a POFH reaction, the particles produced are approximately 90 nm and much larger than the particles made under protein-free conditions (Bird et al. 2016b; Galloway et al. 2012a, b). These experiments show that using Mms6 in different forms (i.e. with fusion tags, as peptides, etc.) and in different types of experiments (in solution, in gels, on surfaces) a range of effects can be generated, potentially allowing MNP to be tailored to the particular size requirements of different precision applications.
A number of other proteins from the magnetosome have been tested using this kind of in vitro activity assay namely; MmsF, MamF, and MmxF from Magnetospirillum magneticum AMB-1 (Rawlings et al. 2014), and MamC from Magnetococcus marinus MC-1 (Valverde-Tercedor et al. 2015). Although these proteins are normally present within the magnetosome membrane, they are able to be produced in a water-soluble form (Arakaki et al. 2003; Rawlings 2016; Rawlings et al. 2014; Valverde-Tercedor et al. 2015). All of these proteins, including Mms6, show a high degree of self-assembly which results in water-soluble micelle-like structures (Amemiya et al. 2007; Feng et al. 2013; Kashyap et al. 2014; Rawlings et al. 2014; Wang et al. 2012). In these structures the hydrophobic regions of the proteins are believed to be buried within the core of the structure, and the hydrophilic, magnetite interaction regions are exposed to the surrounding aqueous environment. When purified these proteins display secondary structure, suggesting that rather than an amorphous aggregate these proteins actually adopt a specific folding architecture.
When purified, MmsF was added to synthetic RTCP reactions in amounts consistent with previous Mms6 studies. The nanoparticle products consisted of a high degree of magnetite with very little alternative iron oxide. In contrast, the addition of MamF or MmxF (both with highly similar primary and secondary structure to MmsF) shifted the balance of the nanoparticle products in favour of alternative iron oxides and away from the desired magnetite species (Rawlings et al. 2014). This suggests that potentially small residue changes can have significant effects over the type of reaction products which form.
MamC (Mms13), and a truncated form of MamC with a single transmembrane region missing, was also found to produce effects on magnetite nanoparticle synthesis (Valverde-Tercedor et al. 2015). This protein, like Mms6, has the ability to bind ferric ions in solution. When introduced into synthetic magnetite precipitation experiments performed under anaerobic “free drift” conditions over 30 days, the resulting MNP had an increased average size of 30–40 nm compared to the 20–30 nm typically obtained under control conditions. Magnetic measurements of the products confirmed the size increase.
3.3 Understanding the In Vitro Activity
It is evident that Mms6, MmsF and MamC are able to control, to varying degrees, the formation of magnetite MNPs when added to a chemical precipitation. It is clear that an understanding of how these proteins function is now required. The majority of the work has been conducted on Mms6 (which is the subject of a recent review (Staniland and Rawlings 2016)) as this was the first protein to be isolated and used for in vitro mediated synthesis and is also the easier (of Mms5, Mms6, Mms7 and Mms13 initially identified) to purify and work with (Arakaki et al. 2003). However, more recently, MmsF has been shown to be much easier to purify, and thus we predict more analysis will be conducted on this protein in the coming years (Rawlings et al. 2014).
To date Mms7 has yet to be purified and assessed for activity in vitro. Interestingly all four proteins, Mms6, Mms7, MmsF and MamC, have two distinct commonalities, which alludes to their function. Firstly, they all self-assemble, forming micelles or proteinosomes when purified in aqueous solution. This self-assembly is most likely due to the favourable packing of the membrane-spanning regions of the membrane proteins MamC (Valverde-Tercedor et al. 2015) and MmsF (Rawlings et al. 2014). It is believed that both the amphiphilic nature and a specific assembly sequence are responsible for organised self-assembly in the single transmembrane protein Mms6. The second common feature is the presence of multiple acidic amino acids that have been shown to bind to iron ions in the cases of Mms6 and MamC (Arakaki et al. 2003; Feng et al. 2013; Valverde-Tercedor et al. 2015; Wang et al. 2012). Here we can consider both features of self-assembly and iron ion by evaluating the work performed on Mms6 predominately and extrapolating these findings to inform suggestions about the functions of all four proteins.
3.3.1 Self-Assembly
3.3.1.1 Single Transmembrane Region Proteins (Mms6 and Mms7)
The amphiphilic nature of the single transmembrane proteins Mms6 and Mms7 implies they will form micelles in aqueous solution with the C-termini exposed on the surface shielding the hydrophobic N-termini within the core (Fig. 5.1c). This structure was first quantitatively investigated for Mms6 by Wang et al. who found that the micelle was between 200 and 400 kDa made up of 20–40 proteins (Wang et al. 2012). DLS measurements found Mms6 micelles were 10.2 ± 3 nm across (Wang et al. 2012), in agreement with later small-angle X-ray scattering (SAXS) experiments, which found the data fit well to a hydrophobic core of 3.9 ± 0.4 nm radius and hydrophilic corona radius of 1.1 ± 0.2 nm (at pH 3) (Zhang et al. 2015) equating to approximately 200 kDa, in agreement with the initial finding. Mms6 micelles at pH 7.5 have a slightly smaller and tighter dispersity than those at pH 3, as shown by DLS (Wang et al. 2012) and SAXS (Zhang et al. 2015), showing that varying the pH across the pI value of the protein (pI = 4.1) alters the protonation state of the acidic groups displayed on the surface, thereby affecting the structure of the micelle assembly. Interestingly, the SAXS experiment found that addition of iron caused the micelles to form higher-order structures such as discs of micelles, presumably through iron cross-linking (Zhang et al. 2015). Most recently, larger proteinous assemblies of Mms6 (approximately 10× larger) have been visualised in situ in a fluid cell TEM (Kashyap et al. 2014). It is significant that Mms6 is able to control magnetite MNP formation in vitro as in vivo, which leads to the inference that there must be self-assembly in the membrane environment in vivo similar to the aggregation seen in vitro, to retain this characteristic. We have thus suggested that Mms6 (and, due to their similarities, Mms7 also) is not monomeric in the magnetosome membrane but self-assembles to form protein rafts on the interior of the membrane, displaying a charged C-terminal surface akin to the surface of in vitro micelles (albeit of the opposite curvature). We tested this theory by enabling Mms6 to self-assemble on a surface, mimicking the membrane environment. The biomimetic Mms6 surface nucleated and controlled magnetite formation, whereas the C-terminal peptide (C20Mms6) alone could not control magnetite nanoparticle synthesis (Bird et al. 2016b). C20Mms6 is missing the N-terminal region but is still able to assemble on the surface. Thus we deduce that the nature of self-assembly in Mms6 is more specific than generic hydrophobic interactions. A GLGLGLGLG repeating amino acid sequence is notably present and conserved in both of the single transmembrane Mms protein Mms6 and Mms7 (but absent from C20Mms6). Such repeating glycine-leucine motifs are common in self-assembling scaffold proteins such as silk fibroin (Zhou et al. 2001). The large then small repeating side-chain motif lends itself to a repeating knob and holes interaction that we propose interlock with adjacent Mms6 (or even Mms7) to form a regularly packed ordered raft and thus regularly space the iron binding C-terminal sites across the magnetosome’s interior membrane surface (or micelle surface in vitro). We suggest this order is critical as iron ions have been shown to bind to Mms6 (see next section), but we propose that binding in an ordered manner on the protein’s surface initiates nucleation of specifically magnetite, and without this the magnetite nucleation ability is lost, as the C20Mms6 on surfaces demonstrate (Bird et al. 2016b). Supporting this hypothesis, a peptide constructed of a C6Mms6-GL repeat peptide shows much better control over particle formation than the C20Mms6, further demonstrating the importance of this region (Arakaki et al. 2010).
3.3.1.2 Multiple Transmembrane-Spanning Region Proteins (MamC and MmsF)
MamC and MmsF have two and three transmembrane-spanning regions (TMS), respectively, and therefore the sequence exposed to the interior of the magnetosome (and thus the forming magnetite crystal) is constrained in a loop configuration (Fig. 5.1c). The presence of multiple TMS dictates a dominant hydrophobicity leading to the propensity to form inclusion bodies which can be refolded to form water-soluble micelle structures similar to Mms6. This was found to be the case for MamC when it was expressed, refolded and purified (Valverde-Tercedor et al. 2015). However, unexpectedly this was not true for MmsF, with three transmembrane regions. MmsF was found to be extremely soluble (Rawlings et al. 2014). MmsF characterisation revealed it to have self-assembled into soluble “proteinosome” structures (Rawlings et al. 2014). Interestingly, MamF and MmxF (both homologues of MmsF) that are not able to control the formation of magnetite formed differently sized and more polydispersed proteinosomes (Rawlings et al. 2014), perhaps suggesting the packing within the self-assembly to again be precise and critical to magnetite formation.
3.3.2 Iron Binding and Proposed Magnetite Nucleation Function
To date iron binding assays have only been performed on Mms6 and MamC. The data obtained for Mms6 clearly shows the iron binding activity is dominated by interactions with the acidic aspartate and glutamate residues. Mms6 has seven acidic residues within the magnetosome lumen-exposed C-terminus, while Mms7 has five, and the loop regions on MamC and MmsF have five and four, respectively (Fig. 5.1). In addition, MamC, Mms7 and MmsF have an acidic and a basic region in their magnetosome-exposed sequences, perhaps indicating a region for iron ion binding (acidic) and a region for magnetite binding (basic). Mms6 binding studies have typically been performed on ferric (Fe3+) ions, the first being demonstrated by Arakaki et al. that reported the discovery and in vitro function of Mms6 in 2003 (Arakaki et al. 2003). This paper reported a competitive radioactive ion binding assay which showed purified recombinant Mms6 binds Fe3+, as well as Ca2+ and Mg2+. Further, more quantitative ferric binding assays have been performed showing both high- and low-affinity ferric iron binding modes at low pH (Wang et al. 2012). Experiments carried out at pH 7.5 with citrate chelators to solubilise the ferric ions showed a ferric ion binding affinity of kd = 10−16 M, while mutants (with scrambled C-termini) showed no significant binding, demonstrating the importance of amino acid sequence in this area (Wang et al. 2012). Although there is a high affinity for ferric ion binding to Mms6 at a pH that mixed valance iron oxides such as magnetite are formed, there is no structural conformational change between the ferric binding and metal free Mms6 peptide, shown in recent 2D NMR studies, suggesting that the highly charged ferric ion could be interacting due to non-specific electrostatic interactions with the acidic residues on Mms6 (Rawlings et al. 2016). The findings from this works points to a need to assess the activity of Mms6 in situ, suggesting that tracking the chemistry quantitatively throughout the magnetite formation process is crucial to understanding the effect of the Mms6 in vitro. Delicate pH titrations were used to assess the often subtle effect of Mms6 during the magnetite precipitation process (Rawlings et al. 2016). There is no significant difference between the protein-free and Mms6-mediated precipitation at low pH (<pH 4) for a range of different ferric/ferrous ratios. This is the stage in the reaction where more insoluble ferric ions precipitate out as a ferric oxide (such as schwertmannite, hematite, or ferrihydrite depending on the conditions). This is in agreement with Wang et al. ferric binding assay which shows ferric ion binding affinities to be considerably less at pH 3 compared to pH 7 (Wang et al. 2012). It is thus clear that binding at low pH is not the main action of Mms6, being negligible when compared to the bulk precipitation. It is only after the ferric oxides have precipitated out of solution and the mixed valance iron oxides start to precipitate that a difference is seen in the Mms6-mediated titrations, showing its marked effect at this stage (Rawlings et al. 2016). This supports the hypothesis that Mms6 is most active at higher pH when the acidic groups are de-protonated and accessible for iron binding.
Interestingly Mms6 has the most marked effect in ferrous-rich ferric/ferrous ion ratios, favouring increased magnetite production (20%) with Mms6 compared to negligible amounts without protein (Rawlings et al. 2016). This suggests Mms6 directs mineralisation towards magnetite synthesis more markedly at ferrous-dominant conditions that are further from the ideal conditions for magnetite formation, effectively “pulling” minerals formed towards magnetite in preference to forming other iron oxides. Mms6 could therefore be acting as a “mineral/ferrous ion buffer” (Rawlings et al. 2016). Furthermore, this study shows that Mms6’s interaction with ferrous ions is of great importance. Binding assays showed a change in structure of the Mms6 peptide (C20Mms6) with and without ferrous ions by 2D NMR (Rawlings et al. 2016). Ferrous iron produced five fold greater side-chain shifts in all the acidic groups of the DEEVE (49th–53rd amino acid) motif compared to ferric iron, signify a stronger, more specific binding of ferrous ions than ferric. Molecular modelling revealed multisite binding, suggesting Mms6 is a multi-dentate ligand for ferrous ions with the most favourable interactions between the glutamate 50 site and the adjacent carbonyl oxygen on the backbone (Rawlings et al. 2016).
The research performed to date shows Mms6, MmsF and MamC all self-assemble in aqueous solution. Furthermore, iron binding studies of Mms6 show strong specific ferrous and strong but less specific ferric binding above pH 4 at the stage in the reaction that mixed valance iron oxides nucleate, especially magnetite. We see that Mms6 binds both ferrous and ferric ions, but for a crystal of magnetite to grow, increased quantities of iron ions need to be bound, and thus there is a need to have a negatively charged iron ion binding surface that could be achieved through the protein’s self-assembly. Therefore the evidence suggests that Mms6 is a magnetite nucleating protein. This has clearly been visualised by Kashyap et al., showing iron oxide nucleation on the surface of the Mms6 micelles in situ in a fluid TEM experiment (Kashyap et al. 2014). Remarkably, iron ion association with Mms6 can clearly be seen, and as the pH rises, small iron oxide particles visibly form across the micelle surface (Kashyap et al. 2014).
In vitro Mms6 forms into 10–12 nm-sized Mms6 micelles and at pH 5 and above (when the acidic groups are deprotonated) displays negatively charged surfaces for iron binding. Both ferrous and ferric ions bind, through seemingly specific (ferrous) or indiscriminately and abundantly (ferric), presumably in a 1:2 ratio, respectively. For both, the DEEVE motif is the key binding region, concentrating mixed valence iron on the surface to nucleate magnetite. It is interesting to compare this acidic motif to the other proteins. Most comparable is MmsF which has a similar four acidic residues in the form XXXZX (X = E/D, Z = V/R), albeit with the sequence reversed to Mms6. Mms7 has two double acid residue sites, which could be predicted to be iron ion binding sites. Interestingly the acidic residues on the MamC loop are well separated and appear quite different to the Mms6 binding motif, and thus more research on this specific protein to identify binding motifs involved in the iron binding mechanism is required. The spacing of bound ferric and ferrous ions is critical to nucleate magnetite, and we suggest the GL repeat sequence in Mms6 provides this by interlocking packing, precisely spacing each protein. Evidence for this hypothesis is shown in the fact that the C20Mms6 peptide surfaces (with no GL repeat) cannot nucleate magnetite as effectively (Bird et al. 2016b). Furthermore, in this same study, Mms6 and the C20Mms6 peptide only showed negligible interaction with pre-formed magnetite, showing Mms6 has no specific affinity for a magnetite surface and thus must be active in the formation process before the magnetite surface forms (Bird et al. 2016b). Again, it is indicative to see that the GL repeat motif is common to all the single transmembrane proteins (Mms5, Mms6 and Mms7) suggesting a common purpose.
We propose the action of Mms6 and the other Mms proteins (Mms7, Mms13 and MmsF) in vivo is similar to in vitro. However, instead of self-assembling into micelles or proteinosomes, within the magnetosome membrane, they would assemble as protein rafts at the membrane interior. The protein rafts could be composed of purely one type of protein or be a mixture (in the case of Mms5, Mms6 and Mms7 as they have the common GL repeat). This could be very interesting to investigate further. It is clear that Mms6 and Mms13 regulate the size of particles in vitro, while there is clear evidence Mms6 and MmsF control the iron oxide formation to be specifically magnetite. For Mms6 this size regulation is seen across a range of methods (approx. 21 nm), when nucleated on Mms6 micelles (Table 5.1). Interestingly, Mms6 assembled on planar surfaces nucleates particles approximately 90 nm in size, while MNPs within magnetosomes are 40–50 nm. The key difference between all these systems is surface curvature, moving from convex to flat to concave, respectively. We suggest this difference in degree and angle of contact between the assembled protein surface and the mineral (along with nucleation physics) is another important nucleation parameter that could be responsible for the different particle sizes (Bird et al. 2016b).
It is thought that iron is transported into magnetosomes as Fe2+ with subsequent partial oxidation to Fe3+ by specific oxidase enzymes. Interestingly Mms6 is the most influential in ferrous-rich conditions, where magnetite is more chemically challenging to achieve, but is expected to be dominant in magnetosomes. While the pH inside magnetosomes has not been determined, it must be high enough to enable magnetite to precipitate and thus the Mms proteins to be deprotonated and functional. We propose raft assemblies of Mms6 that specifically bind ferrous and ferric ions to specifically nucleate magnetite formation. Whether or not Mms6 (and by inference the other Mms proteins discussed) is a nucleating or shape controlling protein is debated in the literature. We have presented a nucleation argument for Mms6 specifically, supported by the research discussed above. However, in vivo mms6, mms7 and mmsF knockout mutant studies show poorly formed elongated magnetite crystals, supporting morphological controlling activity. We believe morphology and nucleation activities are coupled; if a particle is not nucleated properly, it cannot form to the correct morphology. Equally nucleation from a specific crystal plane will guide the final morphology.
4 Selected/Engineered Biomineralisation Proteins for Solution Formation of MNP
Naturally occurring proteins and peptides are capable of both interacting with, and affecting the formation of, different inorganic materials at the nanoscale. This makes them ideally placed to play an important role in precision control of nanomaterial fabrication in the future. However, these proteins are not without certain limitations. Firstly, many, particularly magnetosome proteins, are water insoluble (require elaborate refolding strategies) or difficult to produce in large quantities. Secondly, the magnetic materials required for future applications, such as magnetic data storage, require magnetic properties that are not available from nature, so no naturally occurring biomineralisation proteins for such materials can be exploited. Helpfully, there are emerging strategies which allow biomolecules to be developed to overcome these challenges. These include (i) modification of existing biomineralisation proteins to enhance stability, “expressibility” and solubility, (ii) repurposing of existing proteins to new mineralisation scenarios and (iii) development of entirely new peptide sequences (Fig. 5.2).
4.1 Engineering of Magnetosome-Associated Proteins
Many proteins can be modified and adapted to make them more amenable to certain characterisation or applications, and the biomineralisation proteins from magnetotactic bacteria are no exception. Mms6 has been modified with the addition of a single cysteine residue at the N-terminal region (Bird et al. 2016a, b). The incorporation of this non-native thiol group to the Mms6 protein enables direct immobilisation on a gold substrate for QCMD measurements and nano-patterning techniques (see Sect. 5.5.1). It is also possible to produce Mms6 as a C-terminal fusion to the highly soluble maltose binding protein (MBP). By engineering a linker sequence between the two proteins that is cleavable by tobacco etch virus protease, Mms6 can be easily released from the fusion after expression and purification without the need for lengthy refolding strategies (Galloway et al. 2012b).
Another example is where the maltose binding protein (MBP) has been used as a protein scaffold to display peptide loops from magnetotactic bacteria. Nudelman et al. have demonstrated this by fusing the magnetite binding loop of the biomineralisation protein MamC to the C-terminus of MBP (Fig. 5.2a & bi). Two constructs were produced: one short peptide consisting of 17 amino acids (R-61 to G77) and one longer peptide containing 21 amino acid residues (L57-G77). These constructs were made to analyse the relationship between the structure of the MamC loop and its binding capability to magnetite nanoparticles. For this investigation the constructs were added to in vitro magnetite synthesis reactions. The use of the MBP display scaffold aided the discovery of two important amino acid residues for MamC binding to magnetite: Glu66 and Asp70 (Nudelman et al. 2016).
4.2 Peptide and Protein Biopanning Against MNP
4.2.1 Principles of Biopanning
Biopanning is a way of finding novel biomolecules (usually proteins or peptides) which interact with specific substrates (Jijakli et al. 2016; Smith 1985). In this approach, a diverse library of random peptides or proteins are created which have variable amino acid sequences. This library is engineered so that an individual sequence is expressed as a fusion to an endogenous protein exposed to the outer surface of either a cell (cell display) or bacteriophage (phage display). Using a cell or phage in this way is the crux of the biopanning technique, as both the protein and the DNA, which encodes it, are physically linked together. This pool of proteins, each displayed on a cell or virus, is then applied to the desired substrate material. Some of the library members will contain a sequence which has a natural binding affinity to the material and the others will not. By washing the substrate to remove the non-binding sequences, the positive binders are effectively purified from the pool and can be selectively eluted using, for instance, pH switching of the surrounding buffer. The cells or viruses collected in this way can then be amplified, and the sequences of the binding proteins determined via sequencing of the encoding DNA. In each successive round of binding, washing, elution and sequence amplification, the positive binders which show strong affinity to the desired substrate are selectively enriched. This results in a population of high-affinity binders to carry forward for further analysis (Fig. 5.2c).
Biopanning in this way has uncovered a number of sequences which are able to interact with a range of specific materials for either simple binding or for directed material synthesis.
4.2.2 Magnetite Interacting Sequences
One such example of a system used for both magnetite binding and synthesis is the Adhiron protein (Fig. 5.2bii). This is a monomeric protein scaffold that is stable against a large pH range and high temperatures. This protein scaffold expresses well in E. coli, and the X-ray crystal structure has been resolved to 1.75 Å (Tiede et al. 2014). The protein contains up to two loop regions. These loop regions were altered so that they contained nine randomised amino acids which enabled a phage display libraries to be constructed (Tiede et al. 2014). The Adhiron library, containing the randomised loops, was screened against magnetite nanoparticle and, more specifically, magnetite cubes (Rawlings et al. 2015). This library was used to determine whether any peptide sequences preferentially bound to the specific surface of the iron oxide. These hits were named magnetite interacting Adhirons (MIAs) (Fig. 5.2bii). The library produced was found to be enriched with basic, lysine residues (Rawlings et al. 2015). Adhiron proteins containing both a single loop and two loops showed molecular recognition towards magnetite. Furthermore, these proteins appeared to encourage the formation of cubic nanoparticles in room temperature co-precipitation (RTCP) reactions – indicating the protein holds the ability to exhibit a degree of shape control on the forming magnetite nanoparticles (Rawlings et al. 2015). It is expected that the Adhiron has a major role during the growth of the crystal. It is likely that the MIA interacts with the magnetite crystal early, during the crystal growth phase. This interaction may inhibit further growth at this face, therefore promoting growth of the other crystal faces, resulting in a cubic shape. Artificial scaffolds, such as this one, have a number of benefits over naturally occurring proteins (e.g. proteins that have been repurposed). The artificial scaffolds tend to have increased solubility, expression levels and yields of production, especially when compared to proteins such as transmembrane proteins.
In a different approach, short peptides in a phage display library were screened for binding to magnetite (Baumgartner et al. 2014). For this application, phage display was not used for the direct assessment of peptide sequences, but it was used to identify similar sequences in MTB that were likely to have magnetite biomineralisation capabilities (Baumgartner et al. 2014). This removes the need for extensive mutagenesis experiments and subsequent in vivo analysis.
4.2.3 CoPt/FePt Interacting Sequences
Biopanning carried out by Reiss et al. in 2004 identified four peptide sequences that show specificity for L10 FePt. These peptide sequences are HNKHLPSTQPLA, SVSVGMKPSPRP, VISNHRESSRPL and KSLSRHDHIHHH (Reiss et al. 2004). Similarly, an L10 CoPt specific peptide was identified by phage display by Klem et al in 2005 with the sequence KTHEIHSPLLHK (Klem et al. 2005). All these peptide sequences contain a large number of basic amine residues, such as lysine, which is an excellent ligand to bind to Pt. Lysine is believed to be essential in the mechanism for binding of high mobility group domain proteins to the Pt/DNA complex that is formed in cisplatin-based therapies (Mikata et al. 2001). Therefore it is likely lysine will bind to platinum during the synthesis of FePt and CoPt NPs. For FePt HNKHLPSTQPLA was identified through biopanning against FePt NPs and a thin film of L10 phase FePt; therefore this was the only peptide used in subsequent experiments. In that study FePt NPs were produced by mixing FeCl2, H2PtCl6 and NaBH4 in a 1:1:1 molar ratio under ambient conditions, with 1 ml of engineered phage present. NPs with a diameter of 4 nm were produced, and selected area diffraction patterns showed rings corresponding to (001), (110), (111) and (200). The presence of the (001) and (110) planes indicates that a proportion of the NPs are in the L10 phase (Reiss et al. 2004).
Magnetic measurements performed at 5 and 300 K showed coercivities of 1350 and 200 Oe, respectively (Reiss et al. 2004). The coercivity measured at room temperature is lower than that expected for a sample containing L10 NPs. This could be because the particles produced in the reaction are close to the SPM limit of L10 FePt or the NPs have less L10 character than predicted. As a control experiment, FePt particles were synthesised in the presence of a peptide that is not specific to FePt (SPPRNYYSSMSS) (Reiss et al. 2004). From the SAED of these particles, only one band can be seen, and this relates to the (111) plane of FePt; this plane is not indicative of L10 and therefore suggests that very little chemical ordering occurs when a non-specific peptide is used (Reiss et al. 2004).
4.3 Repurposing Protein Cages for MNP Synthesis
Protein cages can be used as a biotemplate in the synthesis of magnetic nanoparticles. Due to the multiple subunit nature of protein cages, modification of a single subunit will result in modification of the fully assembled cage. There are three surfaces that can be modified in a protein cage: the internal and external surfaces and the interface between the subunits. Modification of a protein cage can result in the formation of a biological template that has a high affinity for a specific material.
4.3.1 Unmodified Protein Cages
4.3.1.1 Ferritin
Ferritin is a naturally occurring, cage protein used for the storage and biomineralisation of inorganic minerals. The mammalian ferritin precursor, apoferritin, consists of a 24-subunit assembly with a four-helix bundle forming a dodecameric cage. The interior compartment of the protein is between 7 and 8 nm in diameter and can act as a nanoreactor, promoting the synthesis of monodisperse nanomaterials in the protein core. These nanomaterials can be used for a wide range of applications (Jutz et al. 2015). The outer shell of ferritin can also be utilised. It can be functionalised chemically or genetically. This functionalisation enables the ferritin protein shell to act as a multivalent scaffold. For example, functionalisation of the protein shell can render ferritin particles soluble in organic solvents, which can be advantageous for many applications. This has been shown by Wong et al. (1999) and Sengonul et al. (2007). Functionalisation of the outer surface of ferritin, combined with a controlled inorganic core, gives technologically useful, biocompatible complexes. The core acts as a template for the formation of monodisperse nanocrystals, with inorganic material becoming incorporated into the core through the internal channels or through reversible disassembly and reassembly of the cage. Nucleation can occur on the surface of the ferritin internal cavity; this occurrence depends on the metal ion binding capability of the exposed charged amino acids on the protein. Alternatively inorganic cores can form by directed mineralisation within the ferritin cage; this is thought to occur by forming favourable electrostatic interactions with the metal to induce crystal nuclei formation. The first mineral cores to be synthesised within ferritin were iron sulphide and magnesium oxide (Meldrum et al. 1991). Later studies showed the formation of nanoparticles which possess properties that make them useful for various applications, such as magnetic nanoparticles (i.e. magnetite). The magnetic properties of the MNP core can be altered without changing the size of the crystal. This is because the size is tightly controlled by the size of the channels and the rigidity of the protein template. Doping with other transition metal elements such as cobalt and manganese can increase the magnetic hardness of the complex. The development of clinically applicable systems was enhanced further upon the synthesis of gadolinium (III) oxide hydroxide by incubating a gadolinium nitrate solution with ferritin (Sanchez et al. 2009).
As mentioned previously, phage display libraries have been designed to identify peptide sequences that bind strongly and specifically to certain inorganic surfaces. These sequences can be used to link inorganic particles to biomolecules fused to the peptide. These peptides often exhibit stronger, more specific binding when attached to a rigid scaffold. In phage display, the phage can occupy this role; however, once the peptide has been released from the phage, it often requires attachment to another display protein – such as ferritin. A specific binding sequence for titanium was inserted into ferritin (via a DNA cassette encoding the peptide) between the first and second codons of the L-chain gene. The horse L-chain that was used consists of 174 amino acids, arranged into four long helices. The adapted ferritin cages still maintain the capability to biomineralise iron but are also capable of selectively binding other inorganic material, such as titanium, with a higher affinity than the titanium-binding peptide in the absence of the ferritin scaffold (Sano et al. 2005).
4.3.1.2 MjHsp
A well-studied example of a protein cage is the small heat shock protein cage from Methanococcus jannaschii (MjHsp). MjHsp assembles into an empty 24-subunit cage with exterior and interior diameters of 12 nm and 6 nm, respectively. Large 3 nm pores at the threefold axes allow exchange between the interior of the cage and the bulk solution (Kim et al. 1998). The protein cage is stable up to 70 °C and in a pH range of 5–11. MjHsp has been used to encapsulate ferrihydrite through air oxidation of Fe(II) in the presence of the protein cage; this is analogous to the ferritin reaction (Flenniken et al. 2003). These particles were imaged using TEM, and an average diameter of 9.0 nm was observed; the presence of iron was confirmed by electron energy loss spectroscopy (EELS). As a control Fe(II) was oxidised in the absence of MjHsp, resulting in a rapid precipitation of bulk ferric oxide due to unconstrained particle growth. Electron diffraction data suggested that the bulk solution was consistent with lepidocrocite and the particles synthesised in the presence of MjHsp were consistent with ferrihydrite (Flenniken et al. 2003), suggesting that the MjHsp protein cage acts as a constrained reaction vessel for the mineralisation of Fe(O)OH.
4.3.1.3 PepA
PepA is protein shell from Streptococcus pneumoniae that has a hollow tetrahedral structure with a 6 nm interior cavity and a 12 nm exterior diameter. The cavity at the centre has four wide channels at the faces of the tetrahedron and four narrow channels at the edges (San et al. 2013). The wide and narrow channels have a diameter of 4 nm and 1 nm, respectively. PepA has been used to mediate the growth of CoPt NPs inside the protein cavity under ambient reaction conditions (San et al. 2013). The mechanism of how the protein shell mediates NP growth is unclear (Douglas et al. 2002). However, it has been suggested that it occurs via a complementary electrostatic mechanism. In this proposed method, it is thought that Co2+ and Pt2+ diffuse in and accumulate in PepA through the channels. The positively charged precursor metals interact with the negatively charged interior of the cage resulting in the formation of nucleating intermediates. CoPt NPs then grow by further deposition at the NP seeds (Douglas et al. 2002).
CoPt NPs synthesised using this method have a size range of 1.1–2.8 nm. The 1.1 nm particles exhibit superparamagnetic behaviour, while NPs greater than 2.1 nm exhibit ferromagnetic behaviour but only at 5 K. XRD data shows diffraction peaks at (111), (200), (220) and (311) all of which are typical for CoPt. Coercivities of 775 and 800 Oe were observed for particles of 2.1 nm and 2.8 nm, respectively, and a general trend of decreased magnetisation was observed as particle size decreased (San et al. 2013).
4.3.1.4 Dps
Dps is a ferritin-like protein from the bacteria Listeria innocua that forms a 12-subunit cage structure with 3:2 tetrahedral symmetry (Bozzi et al. 1997). The Dps protein has some motifs that are structurally similar to ferritin and also contain a ferroxidase centre. The cage has an exterior and interior diameter of 9 and 6 nm, respectively. Ferrimagnetic Fe3O4 nanoparticles have been synthesised by the controlled oxidation of iron at an elevated temperature (65 °C) and pH (8.5). The samples were characterised via TEM, and a narrow size distribution of 4.5–6.0 nm was identified with an average of 500 Fe atoms per cage (Allen et al. 2002; Bozzi et al. 1997). It is also possible to synthesise Co3O4 nanoparticles in the Dps protein cage by substitution of Co (II) for Fe(II) (Allen et al. 2003).
4.3.2 Modified Protein Cages
By combining sequences obtained from phage display with protein cage architectures, magnetic nanoparticles can be synthesised inside the protein cavity, resulting in a high level of control (Fig. 5.2biii).
4.3.2.1 MjHsp
MjHsp has external and internal surfaces that can be chemically or genetically modified. The sequence for the L10 phase of CoPt peptide (KTHEIHSPLLHK) was incorporated at the N-terminus of the protein cage, which expressed on the interior (Klem et al. 2005). Reaction of Co(II) and Pt(II) salts under reducing conditions led to the formation of CoPt NPs inside the protein cages. Nanoparticles synthesised in this reaction were analysed by TEM and have a mean diameter of 6.5 nm, which corresponds to the interior diameter of the protein cage (Klem et al. 2005).
Magnetic measurements of MNPs produced in the presence of the modified protein cage showed a coercivity of 150 Oe at room temperature. No hysteresis was observed at room temperature for NPs mineralised in the presence of the wild-type protein cage; instead a superparamagnetic component was identified, and this is consistent with the absence of L10 structure (Klem et al. 2005). High-temperature annealing (650 °C) leads to a coercivity of 650 Oe for NPs mineralised with the modified protein cage. This high-temperature annealing step leads to the alignment of the c-axis and therefore increased magnetism (Klem et al. 2005). However, high-temperature annealing can cause agglomeration of adjacent particles, which increases particle size and size distributions, therefore removing the advantages of the biotemplating process.
4.3.2.2 CPMV
The cowpea mosaic virus (CPMV) is an example of a virus that has been used for the formation of MNPs. CPMV is 28 nm in diameter, and the surface of the virus particle has exposed amino acids suitable for the attachment of proteins (Love et al. 2014). For example, there are exposed amines (lysine), carboxylates (aspartic acid and glutamic acid) and hydroxyl (tyrosine) groups. Exposed functional groups have been used to conjugate surface amines to a peptide that directs specific mineralisation processes. Peptides specific for the mineralisation of FePt (HNKHLPSTQPLA) and CoPt (CNAGDHANC) were bound to CPMV through chemical coupling of exposed surface amine groups (Aljabali et al. 2011a, b). The peptide that has been used in this study for the mineralisation of CoPt is different from that in previous studies (KTHEIHSPLLHK); this is because it is not specific for the L10 phase of CoPt but just to the mineralisation of CoPt (Klem et al. 2005). By attaching these peptides to the surface of CPMV, CoPt- and FePt-coated nanospheres with a diameter of 32 nm could be formed at room temperature (Aljabali et al. 2011b).
5 Patterned Surface Arrays of MNP Using Proteins and Peptides
In the previous section of this chapter, we have clearly identified active peptide regions of both Mms proteins and biopanned selected MIA (magnetite) proteins and explained how they may control nucleation and crystal growth/morphology. These proteins and “easier to produce” mimics have been used extensively and reliably as additives to control magnetite MNP formation in “green” chemical precipitation. Additionally, this has been expanded into the synthesis of other MNP materials such as CoPt using active peptide regions, and these can be displayed on cage protein scaffolds. Furthermore, we have explored the potential of such a route for nanotechnological applications, namely, high-density data storage. In this section we expand on this in vitro methodology to develop more complex architectures, producing MNPs attached to surfaces and the patterning of these surfaces to make defined arrays of MNPs on surfaces (Fig. 5.3). In doing so, we are expanding the function of the protein, giving it a dual purpose. This is because the protein precisely controls the particles’ composition, size and/or morphology while also locating and immobilising the particle on the surface to facilitate patterning.
The first benefit of mineralising MNPs on proteins displayed on surfaces is that this 2D arrangement more closely mimics the 2D native lipid membrane environment of the protein in vivo. This has been used as a model to understand how the protein functions. However, the key motivation behind producing protein-templated MNPs in arrays on surfaces is the potential to develop a “green” biosynthetic method to produce precise and tailored nanodevices, specifically high-density data storage media (see Sect. 5.4.2). To this end, bio-additives to control the formation L10 Pt-alloy MNPs are particularly relevant as ideal information storage nanoparticles. Magnetite and L10 CoPt MNPs biotemplated on surfaces are therefore discussed in this section.
The methodology can be broken down into three distinct components:
-
1.
The type of MNP to be patterned and thus the choice of protein or peptide. In this section we will review the formation of magnetite MNPs on surfaces using Mms6, ferritin cages, CoPt in ferritin cages and the CoPt-binding peptide KTHEIHSPLLHK previously discussed.
-
2.
The method of attachment to the surface. Patterning a self-assembled monolayer (SAM) with contrasting functional groups is a well-established field of surface chemistry. Briefly, a thin film composed of long-chain organic molecules with a head group that bonds directly to the surface and a functional tail self assembles into a monolayer in a dense brush-like configuration. SAMs were first discovered in the 1940s (Bigelow et al. 1946), but the field of surface science using SAMs developed rapidly from the 1980s onwards with the crucial development of thiol-SAMs where a sulphur head group binds to gold surfaces (Nuzzo and Allara 1983) and trichlorosilane-SAM using silane’s affinity to silica (Maoz and Sagiv 1984). SAMs offer tight control over the surface characteristics. For example, a terminal carboxylic group will give a hydrophilic SAM surface, whereas an alkane tail group will give a hydrophobic SAM surface. This level of control and flexibility of SAM surface chemistry, along with the advent of nanoscale analytical instrumentation over this time, leads to a revolution in surface science over the last few decades led by researchers such as Whitesides (Qin et al. 2010). Generally, the physisorption of a hydrophobic protein or fraction of a protein onto a hydrophobic SAM can be used as a non-specific form of patterning, demonstrated in later sections (Arakaki et al. 2009; Martinez et al. 2011). For a more specific attachment, polyethylene-terminated alkanethiol (PEG) SAM molecules are used. These are hydrophobic and create a SAM surface on gold that resists biofouling by the protein. These can be patterned with hydrophilic carboxylic acid-terminated PEG-alkanethiols (PEG-COOH) where the COOH group can be activated to form a peptide bond with a primary amine of the protein to attach it (Lahiri et al. 1999; Rusmini et al. 2007). This creates a surface with distinct regions of protein-attached and protein-repellent areas. However, this multistep process is not trivial, and more simple routes have been explored using the direct attachment of a protein to the substrate by modifying the protein with a cysteine amino acid or “silica affinity peptide” end group to strongly and specifically bind directly to the gold or silica surface, respectively. In this method a PEG SAM surface is patterned by particle attachment to an alternative SAM on the surface in the chosen pattern or removing some PEG leaving exposed a pattern of underlying substrate so the protein can bind directly to it (Bird et al. 2015, 2016a; b; Galloway et al. 2015, 2016).
-
3.
The method of patterning the surface. In this section we discuss two patterning methods. The first is the soft lithographic technique of micro-contact printing (μCP)/nanocontact printing (nCP) which is a cheap and versatile stamping method. Here a PDMS elastomer stamp is “inked” with the chosen SAM molecule and printed onto the substrate surface. Once the master stamp is made, the method is easy to use and can accommodate a range of patterned designs. However, as it is a manual process, there are issues with reproducibility and quality of the pattern, limiting the patterning feature size to about 1 μm (below this more complex PDMS materials are required). To pattern reliably below this size, we consider a second patterning method: interferometric lithography (IL). IL uses the interference pattern of a split laser beam (using a Lloyds mirror arrangement) to etch a pattern in the SAM down to 20 nm scale features (Fig. 5.3).
The literature is summarised by these categories in table below (Table 5.2).
5.1 Mms6-Mediated Magnetite Patterned Surfaces
The first report of protein-mediated synthesis of magnetite on a surface was in 2009 where Mms6 was used to direct the location of magnetite MNP synthesis (Arakaki et al. 2009). An octadecyltrimethoxysilane (OTS) SAM molecule was used to form a hydrophobic SAM on a silica surface. A macroscale pattern was formed by simply pipetting Mms6 onto the surface. Mms6 is thought to physisorb to the hydrophobic surface via its N-terminal region, presumably exposing the more hydrophilic iron ion binding region to the mineralisation process. Contact angle measurements showed the hydrophobic surface became hydrophilic after protein addition. Iron oxide was mineralised on the Mms6 areas, but not on the hydrophobic areas of the surface. Mineralisation was not seen on areas covered in a control protein, BSA (Arakaki et al. 2009).
In 2012 this idea was advanced by Galloway et al. who improved this protocol by attaching Mms6 to the surface specifically by the N-terminal amine, ensuring the protein is in the correct orientation (with the C-termini active and exposed) to control magnetite MNP synthesis (Galloway et al. 2012a). This method was the first to demonstrate μm patterning using a μCP method to form 20 μm squares. Here a hydrophobic PEG SAM was patterned on a gold surface, and a 10% PEG-COOH SAM was backfilled into the non-SAM patterned regions. Proteins can be bound to COOH groups via their N-termini through a covalent peptide bond.
Ethyl(dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) form an activated ester on a carboxylic acid surface, which preferentially bind to the N-terminal amine of a protein, when the protein is buffered to ≈1 pH point below its isoelectric point (pI) (Lahiri et al. 1999; Rusmini et al. 2007). This protocol was subjected to extensive optimisation for different mineralisation conditions and analysis (Galloway et al. 2012a, b) and showed that the magnetite MNPs formed on the surface have a very narrow size distribution but are much larger than MNPs formed in an Mms6-mediated in vitro synthesis and bigger than those grown in the native magnetosome. Although this method clearly demonstrates the activity of Mms6 and its ability to pattern MNP on surfaces, the EDC/NHS attachment adds an additional synthetic step. Therefore the attachment method was altered by modifying the Mms6 protein to bind directly to the substrate. A cysteine residue was added to the N-terminal region of Mms6. The sulphur atom of the cysteine binds directly to the gold surface. The SAM patterning protocol was unchanged, but now instead of backfilling with a PEG-COOH SAM to bind the protein, the Cys-Mms6 protein backfills directly onto the exposed gold areas. This was successfully achieved by Bird et al. in 2015, and importantly the patterning was much cleaner (Fig. 5.3f-h) (Bird et al. 2015). Furthermore, this meant that the size of the pattern could be reduced to the μCP limit of 1 μm wide lines that showed excellent definition between the MNPs and the SAM (Bird et al. 2015). Importantly the MNPs formed on these surfaces were smaller than the SAM bound Mms6 surface MNP. At 90 ± 15 nm they are closer in size to native magnetosomes and have tighter population distribution.
The patterning methods so far have gone from the macro- to the microscale, but are not yet on the nanoscale. Therefore, the cysteine-modified Mms6 protocol was advanced in 2016 by Bird et al. with the patterning method of IL (Fig. 5.3a-c) (Bird et al. 2016a). A PEG SAM surface was prepared and 350 nm wide lines were etched to expose the underlying gold, and the Cys-Mms6 protein was deposited into these areas. Mineralisation on these surfaces occurs with high pattern definition and yields well-defined magnetite MNPs of a size and distribution in agreement with the previous study (86 ± 21 nm). The size increase of MNP on surfaces compared to the solution phase and the native magnetosome may be a result of the curvature on the environment the Mms6 protein is displayed in (see Sect. 5.2) (Bird et al. 2016b). Work with Mms6 demonstrates the potential of this methodology to pattern arrays on MNPs on surfaces down to the nanoscale for potential nanodevice fabrication. However, although magnetite has many uses, it is not the ideal material for data storage, and Mms6 restricts the size of the MNPs to dimensions which are too large for many applications.
5.2 Ferritin-Mediated MNP Patterned Surfaces
Ferritin has been successfully electrostatically absorbed on μCP patterned surfaces, reported by Martinez et al. in 2011 (Martinez et al. 2011). Approximately 360 nm wide OTS SAMs were stamped on a silica surface, and this was backfilled with 3-aminopropyltriethoxysilane (APTES). The negatively charged ferritin adsorbed onto the positively charged amino-terminated APTES, giving strips approximately 400 nm wide. Interestingly, by varying the pressure on the stamp, only the edges of the strips can be printed leading to a different pattern of alternating 400 nm and 90 nm ferritin strips. The iron mineral inside the ferritin is ∼7 nm ferrihydrite and thus poorly magnetic. In this study they heat their surfaces to 500 °C in O2 to remove the ferritin protein cage. Further plasma etching in O2 causes the size of the iron oxide particles to decrease to 2 nm. While this demonstrates good patterning of biotemplated iron oxides on surfaces within a ferritin cage, the heating step should perhaps be avoided as the effect this has on the iron oxide core has not been assessed and the magnetism of the particle cores are low. Interestingly, the ferritin cage has been used as a template to mineralise other MNPs (see Sect. 5.3). However, we have only found one example of these immobilised on a surface reported by Mayes et al. in 2003 (Mayes et al. 2003). CoPt was deposited within apoferritin to form 7 nm CoPt particles. These were predominately the semicrystalline fcc disordered phase. They were assembled on a silica substrate surface which was then subjected to high-temperature (500–650 °C) annealing to obtain the L10 phase. Mayes argues that the ferritin not only defines the MNPs size but also provides a shell to prevent sintering and agglomeration during the annealing step. Although the ferritin protein is removed during annealing, and the particles are seen to cluster somewhat after this step. This highlights the clear advantage in being able to remove all high-temperature processes related to fabrication. Most interestingly, the authors performed a contact drag test to assess information recording and reading. Although the surfaces need optimisation, there is a promise to this method as a clear read-back signal is demonstrated.
5.3 Peptide-Mediated CoPt Patterned Surfaces
5.3.1 Dual Affinity Peptide
Previous biopanning experiments carried out by Klem et al. in 2005 identified one key peptide sequence (KTHEIHSPLLHK) that binds to the L10 phase of CoPt (Klem et al. 2005). This CoPt-binding peptide has been used to create a dual affinity peptide (DAP) that is able to bind to a silica surface (Galloway et al. 2015). The DAP contains a peptide sequence known to bind to silica (HPPMNASHPHMH) connected to the CoPt-binding peptide by a flexible linker (GSG) so the DAP has the sequence HPPMNASHPHMH-GSG-KTHEIHSPLLHK (Eteshola et al. 2005; Klem et al. 2005). In this study surfaces were prepared via μCP. A PDMS stamp was inked with 1 mg/ml CoPt-DAP solution, and after 1 min excess solution was removed and the stamp dried. The inking and drying procedure was repeated at least ten times before the stamp was placed into conformal contact with a silicon surface. The surface was then placed into a reaction of Co(II) and Pt(II) salts under reducing conditions, at room temperature. This led to the formation of CoPt NPs attached to a silica surface via the DAP. X-ray diffraction data suggests that the DAP is able to biotemplate the L10 phase of CoPt (Galloway et al. 2015). This is due to the presence of (001) and (100) planes that are expected for CoPt L10 reflections. It is thought that the DAP is able to lower the activation energy for the formation of L10 phase in the aqueous phase. This is because the DAP is more closely packed when on a silica surface and therefore may enhance crystallographic alignment and improve the templating abilities of the peptide. Another possibility is that the peptide may organise particles into the tetragonal phase and aid in the reduction to metallic CoPt when bound to the surface (Galloway et al. 2015).
However, magnetisation data shows that the coercivity is very low and this could be because the biotemplated L10 MNPs are not aligned in the same way as sputtered L10 films. When hysteresis measurements have been carried out for sputtered films that have misaligned grains loops with reduced or no coercivity were observed. Therefore, it is possible that the magnetic orientations of the MNPs are not aligned correctly to achieve maximum coercivity.
5.3.2 Cys-CoPt Peptide
The peptide (KTHEIHSPLLHK) previously identified from biopanning as being specific to the L10 phase of CoPt has been further modified for attachment onto surfaces. The modification involved incorporation of a cysteine amino acid residue connected to the original peptide via a flexible linker (GSG), C-GSG-KTHEIHSPLLHK (Galloway et al. 2016).
In this study IL and μCP were used to pattern gold surfaces allowing nano- and microscale lines to be produced. In IL PEG SAMs were first formed on the surface and then exposed to the interferometer. Regions that were exposed to the interferometer resulted in removal of the SAM to reveal bare gold. For μCP the PEG SAM was applied to the surface using a PDMS stamp, and bare regions on both IL and μCP surfaces were then backfilled with cys-CoPt peptide before mineralisation of CoPt. SEM images of the biotemplated surfaces show that regions functionalised with cys-CoPt were covered in a densely packed thin film of MNPs, whereas PEG SAM regions remained free of any NPs. An average line width of 226 nm and 6.1 μm was observed for lines of CoPt NPs generated by IL (Fig. 5.3d-e) and μCP (Fig. 5.3i,j), respectively (Galloway et al. 2016). Grain size analysis was performed on these SEM images, and NPs mineralised using cys-CoPt had an average diameter of 12 nm (Galloway et al. 2016). A previous study that uses a dual affinity peptide to mineralise NPs had an average diameter of 17 nm (Galloway et al. 2015). Therefore this suggest that immobilisation of the cys-CoPt peptide onto a substrate enhances its ability to control the formation of uniform MNPs. As the cys-CoPt is shorter than the DAP, this could result in closer packing of the peptide onto the surface and enhance templating of smaller more uniform particles. EDX data for the biotemplated surfaces shows that CoPt has been formed in a 1:1 stoichiometric ratio by using the cys-CoPt peptide. This is typical for L10 CoPt. From XRD and SAED data, peaks that correspond to the (001), (101) and (110) planes of L10 CoPt were observed for the biotemplated surfaces. CoPt3 peaks were also present, but values for these were not as close to literature values. Therefore, due to the EDX data, the peaks on the XRD were tentatively assigned as L10 CoPt. No L10 structure was observed for bulk particles, therefore suggesting that immobilisation of the cys-CoPt peptide to a surface shows control over the particles formed (Galloway et al. 2016).
Magnetic data indicated that the nanoparticles formed on the surface were ferromagnetic at room temperature with coercivities of 20–30 Oe. This is lower than expected for L10 CoPt. To achieve high coercivity of the L10 phase, the c-axis needs to be aligned perpendicular to the surface (Galloway et al. 2016). Therefore, as the biotemplated surfaces in this study have a low coercivity, it suggests that they lack this c-axis alignment. Mineralisation was also carried out in the presence of a 0.2 T magnetic field; these surfaces did not show any increase in coercivity and had a lack of long-range ordering.
6 Conclusion
This chapter has shown that specific proteins can act as powerful tools in controlling the synthesis of magnetic nanoparticles. Proteins derived from the magnetosome, such as Mms6, MmsF and MamC, display remarkable self-assembly properties when studied in vitro and retain the ability to reduce the heterogeneity of nanoparticle products in synthetic MNP precipitation reactions. Not only can these proteins be used synthetically, but they provide a valuable model system for understanding protein-mediated biomineralisation. They also provide a proof of concept for the successful utilisation of proteins in magnetic material synthesis, and the field has now widened to encompass designed proteins such as the MIA and the re-engineering of naturally occurring proteins such as the heat shock protein MjHsp.
One important element of the magnetosome, which is often overlooked, is the presence of the lipid vesicle. Constraining the synthesis of magnetite within the lumen of the magnetosome offers a level of size control of the magnetite nanoparticle which is hard to achieve any other way. Studying biomineralisation proteins such as Mms6 in a water-soluble micelle may be quite different compared to when the protein is embedded in the magnetosome membrane. An important future direction for this field will be the incorporation of such proteins into vesicle structures to create artificial magnetosomes. This approach would provide two distinct advantages over solution phase synthesis: (i) it offers a close to native environment in which to study the mechanism of each protein, and (ii) it can provide a further level of control over MNP synthesis and may result in particles with a higher degree of homogeneity than can be achieved with the protein alone. Steps towards this goal have already been made, with the successful precipitation of iron oxides within lipid and polymer vesicles (Bain et al. 2015a, b; Bakhshi et al. 2016). Introducing proteins into this type of system could lead to new advances and insights.
In addition to the benefits on particle formation, if used on surfaces, these proteins have a dual role of forming and binding the particles while simultaneously anchoring them to the surface. This has been demonstrated successfully with Mms6 on gold surfaces patterned with both IL and micro-contact printing. A similar approach has been used with CoPt nanoparticles using a dual affinity peptide which has binding interactions with both the CoPt NP and the silica surface. These approaches demonstrate the flexibility available to the protein engineer, as a single protein can be designed to fulfil two key roles.
We are entering a critical era for protein-mediated nanomaterial synthesis. The understanding gleaned from the wealth of previous studies, some of which are highlighted in this chapter, coupled with advances in material synthesis, protein engineering and new patterning technologies, will lead to new innovations in this fast-moving field. A future with cleaner, cheaper and more precise magnetic nanoparticles is moving a step closer to reality.

The link between magnetotactic bacteria, phage display and scaffold proteins. (a) A magnified cartoon of the magnetosomes of magnetic bacteria including the transmembrane MamC biomineralisation protein. (b) Examples of protein scaffold additives used for magnetite synthesis in vitro. Left: MBP-MamC (PDB: 5E7U) (Nudelman et al. 2016). Top right: Adhiron protein scaffold (PDB: 4N6T) (Tiede et al. 2014). Bottom right: apoferritin (PDB: 5ERK) (Pontillo et al. 2016). c The biopanning cycles implemented during phage display; used in the production of MIAs and modified ferritin cages

Schematic outlining the two methods of patterning discussed, micro-contact printing and interferometric lithography. Scanning electron micrograph images show patterns produced using these two methods: (a–c) Mms6 biotemplated MNP arrays patterned by IL (Bird et al. 2016a), (d, e) cys-CoPt peptide biotemplated MNP arrays patterned by IL (Galloway et al. 2016) and (f–h) μCP cobalt-doped magnetite arrays (Bird et al. 2015) and (i, j) μCP patterned arrays of CoPt (Galloway et al. 2016). Scale bars represent: (a) 2 μm, (b) 1 μm, (c) 500 nm, (d) 20 μm, (e) 200 nm, (f) 100 μm, (g) and (h) 25 μm, (i) 20 μm and (j) 2 μm (Images are reproduced with permission from The Royal Society of Chemistry)
References
Aboaf J, Herd S, Klokholm E (1983) Magnetic-properties and structure of cobalt platinum thin-films. IEEE Trans Magn 19:1514–1519. https://doi.org/10.1109/TMAG.1983.1062575
Aljabali AAA, Barclay JE, Cespedes O, Rashid A, Staniland SS, Lomonossoff GP, Evans DJ (2011a) Charge modified cowpea mosaic virus particles for templated mineralization. Adv Funct Mater 21:4137–4142
Aljabali AAA, Shah SN, Evans-Gowing R, Lomonossoff GP, Evans DJ (2011b) Chemically-coupled-peptide-promoted virus nanoparticle templated mineralization. Integr Biol 3:119–125
Allen M, Willits D, Mosolf J, Young M, Douglas T (2002) Protein cage constrained synthesis of ferrimagnetic iron oxide nanoparticles. Adv Funct Mater 14:1562–1565
Allen M, Willits D, Young M, Douglas T (2003) Constrained synthesis of cobalt oxide nanomaterials in the 12-subunit protein cage from Listeria innocua. Inorg Chem 42:6300–6305
Amemiya Y, Arakaki A, Staniland SS, Tanaka T, Matsunaga T (2007) Controlled formation of magnetite crystal by partial oxidation of ferrous hydroxide in the presence of recombinant magnetotactic bacterial protein Mms6. Biomaterials 28:5381–5389. https://doi.org/10.1016/j.biomaterials.2007.07.051
Arakaki A, Webb J, Matsunaga T (2003) A novel protein tightly bound to bacterial magnetic particles in Magnetospirillum magneticum strain AMB-1. J Biol Chem 278:8745–8750. https://doi.org/10.1074/jbc.M211729200
Arakaki A, Masuda F, Matsunaga T (2009) Iron oxide crystal formation on a substrate modified with the Mms6 protein from magnetotactic bacteria. MRS Online Proc Libr Arch 1187:1187–KK1103-1108. (1185 pages. https://doi.org/10.1557/PROC-1187-KK03-08
Arakaki A, Masuda F, Amemiya Y, Tanaka T, Matsunaga T (2010) Control of the morphology and size of magnetite particles with peptides mimicking the Mms6 protein from magnetotactic bacteria. J Colloid Interface Sci 343:65–70. S0021–9797(09)01488-X [pii]. https://doi.org/10.1016/j.jcis.2009.11.043
Bain J, Berry M, Dirks C, Staniland S (2015a) Synthesis of ABA tri-block Co-polymer magnetopolymersomes via electroporation for potential medical application. Polymer 7:2558–2571. https://doi.org/10.3390/polym7121529
Bain J, Ruiz-Perez L, Kennerley A, Muench S, Thompson R, Battaglia G, Staniland S (2015b) In situ formation of magnetopolymersomes via electroporation for MRI. Sci Rep 5. https://doi.org/10.1038/srep14311
Bakhshi PK, Bain J, Gul MO, Stride E, Edirisinghe M, Staniland SS (2016) Manufacturing man-made Magnetosomes: high-throughput in situ synthesis of biomimetic magnetite loaded Nanovesicles. Macromol Biosci. https://doi.org/10.1002/mabi.201600181
Baumgartner J, Carillo MA, Eckes KM, Werner P, Faivre D (2014) Biomimetic magnetite formation: from biocombinatorial approaches to mineralization effects. Langmuir 30:2129–2136. https://doi.org/10.1021/la404290c
Bigelow WC, Pickett DL, Zisman WA (1946) Oleophobic monolayers. J Colloid Sci 1:513–538. https://doi.org/10.1016/0095-8522(46)90059-1
Bird SM, Galloway JM, Rawlings AE, Bramble JP, Staniland SS (2015) Taking a hard line with biotemplating: cobalt-doped magnetite magnetic nanoparticle arrays. Nanoscale 7:7340–7351. https://doi.org/10.1039/C5NR00651A
Bird SM, El-Zubir O, Rawlings AE, Leggett GJ, Staniland SS (2016a) A novel design strategy for nanoparticles on nanopatterns: interferometric lithographic patterning of Mms6 biotemplated magnetic nanoparticles. J Mater Chem C. https://doi.org/10.1039/C5TC03895B
Bird SM, Rawlings AE, Galloway JM, Staniland SS (2016b) Using a biomimetic membrane surface experiment to investigate the activity of the magnetite biomineralisation protein Mms6. RSC Adv 6:7356–7363. https://doi.org/10.1039/C5RA16469A
Bozzi M, Mignogna G, Stefanini S, Barra D, Longhi C, Valenti P, Chiancone E (1997) A novel non-heme Iron-binding ferritin related to the DNA binding proteins of the Dps family in Listeria innocua J Biol Chem 272:3259–3265
Burkhard P, Stetefeld J, Strelkov S (2001) Coiled coils: a highly versatile protein folding motif. Trends Cell Biol 11:82–88. https://doi.org/10.1016/S0962-8924(00)01898-5
Charap SH, Lu P-L, He Y (1997) Thermal stability of recorded information at high densities magnetics. IEEE Trans on 33:978–983
del Puerto MM, Gonzalez-Carreno T, Tartaj P, Veintemillas-Verdaguer S, Serna CJ (2003) The preparation of magnetic nanoparticles for applications in biomedicine. J Phys D Appl Phys 36:R182–R197. https://doi.org/10.1088/0022-3727/36/13/202
Douglas T, Strable E, Willits D, Aitouchen A, Libera M, Young M (2002) Protein engineering of a viral cage for constrained nanomaterials synthesis. Adv Mater 14:415–418
Driskill-Smith AA (2004) Electron-beam and emerging lithography for the magnetic recording industry. In: Microlithography 2004. International Society for Optics and Photonics, pp 16–30
Eteshola E, Brillson LJ, Lee SC (2005) Selection and characteristics of peptides that bind thermally grown silicon dioxide film. Biomol Eng 22:201–204
Feng SR, Wang LJ, Palo P, Liu XP, Mallapragada SK, Nilsen-Hamilton M (2013) Integrated self-assembly of the Mms6 magnetosome protein to form an Iron-responsive structure. Int J Mol Sci 14:14594–14606. https://doi.org/10.3390/ijms140714594
Flenniken ML, Willits DA, Brumfield S, Young MJ, Douglas T (2003) The small heat shock protein cage from Methanococcus jannaschii is a versatile nanoscale platform for genetic and chemical modification ACS. Nano Lett 3:1573–1576
Galloway JM, Arakaki A, Masuda F, Tanaka T, Matsunaga T, Staniland SS (2011) Magnetic bacterial protein Mms6 controls morphology, crystallinity and magnetism of cobalt-doped magnetite nanoparticles in vitro. J Mater Chem 21:15244–15254. https://doi.org/10.1039/C1jm12003d
Galloway JM, Bramble JP, Rawlings AE, Burnell G, Evans SD, Staniland SS (2012a) Biotemplated magnetic nanoparticle arrays. Small 8:204–208. https://doi.org/10.1002/smll.201101627
Galloway JM, Bramble JP, Rawlings AE, Burnell G, Evans SD, Staniland SS (2012b) Nanomagnetic arrays formed with the biomineralization protein Mms6. J Nano Res Sw 17:127–146. https://doi.org/10.4028/www.scientific.net/JNanoR.17.127
Galloway JM, Talbot JE, Critchley K, Miles JJ, Bramble JP (2015) Developing biotemplated data storage: room temperature biomineralization of L10 CoPt magnetic nanoparticles. Adv Funct Mater 25:4590–4600. https://doi.org/10.1002/adfm.201501090
Galloway JM et al (2016) Nano- and micro-patterning biotemplated magnetic CoPt arrays. Nanoscale 8:11738–11747. https://doi.org/10.1039/C6NR03330J
Gurnon DG, Whitaker J, Oakley MG (2003) Design and characterization of a homodimeric antiparallel coiled coil. J Am Chem Soc 125:7518–7519. https://doi.org/10.1021/ja0357590
Hatch GP, Stelter RE (2001) Magnetic design considerations for devices and particles used for biological high-gradient magnetic separation (HGMS) systems. J Magn Magn Mater 225:262–276. https://doi.org/10.1016/S0304-8853(00)01250-6
Huang J, Bu L, Xie J, Chen K, Cheng Z, Li X, Chen X (2010) Effects of nanoparticle size on cellular uptake and liver MRI with polyvinylpyrrolidone-coated iron oxide nanoparticles. ACS Nano 4:7151–7160. https://doi.org/10.1021/nn101643u
Jijakli K et al (2016) The in vitro selection world. Methods 106:3–13. https://doi.org/10.1016/j.ymeth.2016.06.003
Jutz G, van Rijn P, Miranda BS, Boker A (2015) Ferritin: A Versatile Building Block for Bionanotechnology. Chem Rev 115:1653–1701. https://doi.org/10.1021/cr400011b
Kashyap S, Woehl TJ, Liu X, Mallapragada SK, Prozorov T (2014) Nucleation of iron oxide nanoparticles mediated by Mms6 protein in situ. ACS Nano 8:9097–9106. https://doi.org/10.1021/nn502551y
Kim KK, Kim R, Kim S-H (1998) Crystal structure of a small heat-shock protein. Nature 394:595–599
Klem MT, Willits D, Solis DJ, Belcher AM, Young M, Douglas T (2005) Bio-inspired synthesis of protein encapsulated CoPt nanoparticles. Adv Funct Mater 15:1489–1494
Lahiri J, Isaacs L, Tien J, Whitesides GM (1999) A strategy for the generation of surfaces presenting ligands for studies of binding based on an active ester as a common reactive intermediate: a surface plasmon resonance study. Anal Chem 71:777–790
Laughlin D, Srinivasan K, Tanase M, Wang L (2005) Crystallographic aspects of L1(0) magnetic materials. Scr Mater 53:383–388. https://doi.org/10.1016/j.scriptamat.2005.04.039
Laurent S, Forge D, Port M, Roch A, Robic C, Vander Elst L, Muller RN (2008) Magnetic iron oxide nanoparticles: synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem Rev 108:2064–2110. https://doi.org/10.1021/cr068445e
Lee N et al (2011) Magnetosome-like ferrimagnetic iron oxide nanocubes for highly sensitive MRI of single cells and transplanted pancreatic islets. Proc Natl Acad Sci U S A 108:2662–2667. https://doi.org/10.1073/pnas.1016409108
Love AJ, Makarov V, Yaminsky I, Kalinina NO, Taliansky ME (2014) The use of tobacco mosaic virus and cowpea mosaic virus for the production of novel metal nanomaterials. Virology 449:133–139
Lu AH, Salabas EL, Schuth F (2007) Magnetic nanoparticles: synthesis, protection, functionalization, and application. Angew Chem Int Ed 46:1222–1244. https://doi.org/10.1002/anie.200602866
Maoz R, Sagiv J (1984) On the formation and structure of self-assembling monolayers. I. A comparative atr-wettability study of Langmuir—Blodgett and adsorbed films on flat substrates and glass microbeads. J Colloid Interface Sci 100:465–496. https://doi.org/10.1016/0021-9797(84)90452-1
Martın J, Nogues J, Liu K, Vicent J, Schuller IK (2003) Ordered magnetic nanostructures: fabrication and properties. J Magn Magn Mater 256:449–501
Martinez RV, Chiesa M, Garcia R (2011) Nanopatterning of ferritin molecules and the controlled size reduction of their magnetic cores. Small 7:2914–2920. https://doi.org/10.1002/smll.201100366
Mayes E et al (2003) Biologically derived nanomagnets in self-organized patterned media. IEEE Trans Magn 39:624–627. https://doi.org/10.1109/tmag.2003.808982
Meldrum FC, Wade VJ, Nimmo DL, Heywood BR, Mann S (1991) Synthesis of inorganic nanophase materials in supramolecular protein cages. Nature 349:684–687
Mikata Y, He Q, Lippard SJ (2001) Laser-induced photo-cross-linking of cisplatin-modified DNA to HMG-domain proteins. Biochemistry 40:7533–7541
Moore LR et al (2001) Progenitor cell isolation with a high- capacity quadrupole magnetic flow sorter. J Magn Magn Mater 225:277–284. https://doi.org/10.1016/S0304-8853(00)01251-8
Moroz P, Jones SK, Gray BN (2001) Status of hyperthermia in the treatment of advanced liver cancer. J Surg Oncol 77:259–269. https://doi.org/10.1002/jso.1106
Murat D et al (2012) The magnetosome membrane protein, MmsF, is a major regulator of magnetite biomineralization in Magnetospirillum magneticum AMB-1. Mol Microbiol 85:684–699. https://doi.org/10.1111/j.1365-2958.2012.08132.x
Muthana M et al (2015) Directing cell therapy to anatomic target sites in vivo with magnetic resonance targeting. Nat Commun:6. https://doi.org/10.1038/ncomms9009
Nickolls J, Dally WJ (2010) The GPU computing era. IEEE Micro 30:56–69
Nudelman H et al (2016) Structure-function studies of the magnetite-biomineralizing magnetosome-associated protein MamC. J Struct Biol 194:244–252. https://doi.org/10.1016/j.jsb.2016.03.001
Nuzzo RG, Allara DL (1983) Adsorption of bifunctional organic disulfides on gold surfaces. J Am Chem Soc 105:4481–4483. https://doi.org/10.1021/ja00351a063
Pankhurst QA, Connolly J, Jones SK, Dobson J (2003) Applications of magnetic nanoparticles in biomedicine. J Phys D Appl Phys 36:R167–R181. https://doi.org/10.1088/0022-3727/36/13/201
Paul F, Melville D, Roath S, Warhurst D (1981) A bench top magnetic separator for malarial parasite concentration magnetics. IEEE Trans on 17:2822–2824. https://doi.org/10.1109/TMAG.1981.1061711
Pontillo N, Pane F, Messori L, Amoresano A, Merlino A (2016) Cisplatin encapsulation within a ferritin nanocage: a high-resolution crystallographic study. Chem Commun 52:4136–4139. https://doi.org/10.1039/c5cc10365g
Prozorov T et al (2007) Protein-mediated synthesis of uniform superparamagnetic magnetite nanocrystals. Adv Funct Mater 17:951–957. https://doi.org/10.1002/adfm.200600448
Qin D, Xia Y, Whitesides GM (2010) Soft lithography for micro- and nanoscale patterning. Nat Protoc 5:491–502
Rawlings A (2016) Membrane proteins: always an insoluble problem? Biochem Soc Trans 44:790–795. https://doi.org/10.1042/BST20160025
Rawlings AE, Bramble JP, Walker R, Bain J, Galloway JM, Staniland SS (2014) Self-assembled MmsF proteinosomes control magnetite nanoparticle formation in vitro. Proc Natl Acad Sci USA 111:19094–19099. https://doi.org/10.1073/pnas.1409256111
Rawlings AE et al (2015) Phage display selected magnetite interacting Adhirons for shape controlled nanoparticle synthesis. Chem Sci. https://doi.org/10.1039/C5SC01472G
Rawlings AE, Bramble JP, Hounslow AM, Williamson MP, Monnington AE, Cooke DJ, Staniland SS (2016) Ferrous iron key to Mms6 magnetite biomineralisation:a mechanistic study to understand magnetite formation using pH titration and NMR. Chem Eur J 22:7885–7894
Regazzoni AE, Urrutia GA, Blesa MA, AJG M (1981) Some observations on the composition and morphology of synthetic magnetites obtained by different routes. J Inorg Nucl Chem 43:1489–1493
Reiss BD, Mao C, Solis DJ, Ryan KS, Thomson T, Belcher AM (2004) Biological routes to metal alloy ferromagnetic nanostructures ACS. Nano Lett 4:1127–1132
Richter HJ et al (2006) Recording on bit-patterned media at densities of 1 Tb/in <img src="/images/tex/413.Gif" alt="^2"> and beyond. IEEE Trans Magn 42:2255–2260. https://doi.org/10.1109/TMAG.2006.878392
Rusmini F, Zhong Z, Feijen J (2007) Protein immobilization strategies for protein biochips. Biomacromolecules 8:1775–1789. https://doi.org/10.1021/bm061197b
San BH, Lee S, Moh SH, Park J-G, Lee JH, Hwang H-Y, Kim KK (2013) Size-controlled synthesis and characterization of CoPt nanoparticles using protein shells. J Mater Chem B t1:1453. https://doi.org/10.1039/c2tb00290f
Sanchez P et al (2009) MRI relaxation properties of water-soluble apoferritin-encapsulated gadolinium oxide-hydroxide nanoparticles. Dalton Trans:800–804. https://doi.org/10.1039/b809645g
Sangregorio C, Wiemann J, Connor CJ, Rosenzweig Z (1999) A new method for the synthesis of magnetoliposomes. J Appl Phys 85:5699–5701
Sano K, Ajima K, Iwahori K, Yudasaka M, Iijima S, Yamashita I, Shiba K (2005) Endowing a ferritin-like cage protein with high affinity and selectivity for certain inorganic materials. Small 1:826–832. https://doi.org/10.1002/smll.200500010
Sengonul M, Ruzicka J, Attygalle AB, Libera M (2007) Surface modification of protein nanocontainers and their self-directing character in polymer blends. Polymer 48:3632–3640. https://doi.org/10.1016/j.polymer.2007.04.017
Smith GP (1985) Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228:1315–1317
Staniland S, Rawlings A (2016) Crystallizing the function of the magnetosome membrane mineralization protein Mms6. Biochem Soc Trans 44:883–890. https://doi.org/10.1042/BST20160057
Sun S et al (2003) Controlled synthesis and assembly of FePt nanoparticles. J Phys Chem B 107:5419–5425
Sun X, Jia Z, Huang Y, Harrell J, Nikles D, Sun K, Wang L (2004) Synthesis and magnetic properties of CoPt nanoparticles. J Appl Phys 95:6747–6749. https://doi.org/10.1063/1.1667441
Tanaka M, Mazuyama E, Arakaki A, Matsunaga T (2010) Mms6 protein regulates crystal morphology during nano-sized magnetite biomineralization in vivo. J Biol Chem. M110.183434 [pii]. https://doi.org/10.1074/jbc.M110.183434
Terris B, Thomson T (2005) Nanofabricated and self-assembled magnetic structures as data storage media. J Phys D Appl Phys 38:R199
Terris B, Thomson T, Hu G (2007) Patterned media for future magnetic data storage. Microsyst Technol 13:189–196
Tiede C et al (2014) Adhiron: a stable and versatile peptide display scaffold for molecular recognition applications. Protein Eng Des Sel 27:145–155. https://doi.org/10.1093/protein/gzu007
Valverde-Tercedor C et al (2015) Size control of in vitro synthesized magnetite crystals by the MamC protein of Magnetococcus marinus strain MC-1. Appl Microbiol Biotechnol 99:5109–5121. https://doi.org/10.1007/s00253-014-6326-y
van der Zee J (2002) Heating the patient: a promising approach? Ann Oncol 13:1173–1184. https://doi.org/10.1093/annonc/mdf280
Wang LJ et al (2012) Self-assembly and biphasic Iron-binding characteristics of Mms6, a bacterial protein that promotes the formation of superparamagnetic magnetite nanoparticles of uniform size and shape. Biomacromolecules 13:98–105. https://doi.org/10.1021/Bm201278u
Wang Y, Zhang X, Liu Y, Jiang Y, Zhang Y, Yang J (2014) Microstructure and magnetic properties of L1(0) CoPt nanoparticles by ag addition. J Sol-Gel Sci Technol 70:528–533. https://doi.org/10.1007/s10971-014-3318-4
Weller D et al (2000) High K-u materials approach to 100 Gbits/in(2). IEEE Trans Magn 36:10–15. https://doi.org/10.1109/20.824418
Wollhofen R, Katzmann J, Hrelescu C, Jacak J, Klar TA (2013) 120 nm resolution and 55 nm structure size in STED-lithography. Opt Express 21:10831–10840
Wong KKW, Colfen H, Whilton NT, Douglas T, Mann S (1999) Synthesis and characterization of hydrophobic ferritin proteins. J Inorg Biochem 76:187–195. https://doi.org/10.1016/s0162-0134(99)00114-2
Yamagishi A, Tanaka M, Lenders J, Thiesbrummel J, Sommerdijk N, Matsunaga T, Arakaki A (2016) Control of magnetite nanocrystal morphology in magnetotactic bacteria by regulation of mms7 gene expression. Sci Rep 6. https://doi.org/10.1038/srep29785
Yoshino T, Matsunaga T (2006) Efficient and stable display of functional proteins on bacterial magnetic particles using Mms13 as a novel anchor molecule. Appl Environ Microbiol 72:465–471. https://doi.org/10.1128/AEM.72.1.465-471.2006
Zhang YJ et al (2011) A novel approach to the synthesis of CoPt magnetic nanoparticles. J Phys D Appl Phys 44:295003
Zhang H et al (2015) Morphological transformations in the magnetite Biomineralizing protein Mms6 in Iron solutions: a small-angle X-ray scattering study. Langmuir 31:2818–2825. https://doi.org/10.1021/la5044377
Zhou C-Z, Confalonieri F, Jacquet M, Perasso R, Li Z-G, Janin J (2001) Silk fibroin: structural implications of a remarkable amino acid sequence proteins: structure. Funct Bioinforma 44:119–122. https://doi.org/10.1002/prot.1078
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

{kind=link}
Cite this chapter
Rawlings, A.E., Jarrald, R.M., Somner, L.A., Staniland, S.S. (2018). Protein and Peptide-Mediated Synthesis of Magnetic Nanoparticles and Arrays for Biomedicine and Future Data Storage. In: Matsunaga, T., Tanaka, T., Kisailus, D. (eds) Biological Magnetic Materials and Applications. Springer, Singapore. https://doi.org/10.1007/978-981-10-8069-2_5
Download citation
DOI: https://doi.org/10.1007/978-981-10-8069-2_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-8068-5
Online ISBN: 978-981-10-8069-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)