
Abstract
Neurodegenerative diseases are genetic and/or sporadic disease conditions characterized by progressive nervous system dysfunction involving the atrophy of central or peripheral nervous. The neurodegenerative diseases (NDs) like Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) are responsible for more than 1% deaths and more than 2% disabilities of total world population. These NDs also impart huge socioeconomical burden on families of patients. NDs involve complex etiology with different genetic and environmental factors. The understanding of the etiology may help therapists to develop new effective symptomatic and preventive (genetic) treatments for NDs. The development in Human Genome Project helping to detect the genetic mutations causing HDs and advancement in gene and genome therapy are being implemented to correct these mutations. In this chapter, Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) are discussed in detail for their pathophysiology, etiology, and latest symptomatic and preventive treatment. In preventive treatment, the latest achievements of the gene and genomic therapies are discussed.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Alzheimer’s disease
- Parkinson’s disease
- Huntington’s disease
- Amyotrophic lateral sclerosis
- Genomic treatment
15.1 Introduction
The human brain is the master organ of the body, controlling and regulating all functions of the body. The brain is composed of neurons and glial cells and extends out from the skull as the spinal cord and nerves of the central nervous system (Fig. 15.1a). The axon terminals of the central nervous system connect to the dendrites of neuron cells that service different organs and those of the motor neurons. The nerve tracts of the central nervous system that transmit signals to the muscles and glands are called efferent nerves.

(a) Central and peripheral nervous system. (b) Neuron (Figure adapted from https://en.wikipedia.org/wiki/Neuron#/media/File:Complete_neuron_cell_diagram_en.svg)
The neurons, nerves, and ganglions starting from different organs and connecting to the central nervous system are called peripheral nervous system. The nerves of peripheral nervous system (Fig. 15.1a) are called as afferent nerves that transmit signals from sensory neurons to the central nervous system. The nervous system is functionally differentiated as “autonomic or involuntary” nervous system which works without conscious effort and “somatic or voluntary” nervous system. The autonomic nervous system regulates blood pressure, heart rate, and the breathing rate, while the somatic system operates through the central nervous system consisting of sensory and motor neurons. Thus the neurons and nerve are the basic units controlling all the voluntary and involuntary things happening with our body.
As shown in Fig. 15.1b, neurons are made up of various parts which include the soma (consist of nucleus and extensions called the dendrite tree) and axons (consist of myelin sheath with nucleus and nodes of Ranvier), and axon terminals are fine structures which vary from hundreds to thousands. The axon terminals and dendrites of soma are connected to synapses which are specialized structures where neurotransmitters are released to communicate with target neurons. These neurotransmitters trigger action potential through voltage-dependent sodium channels to start the next wave of electrical impulse. In the brain, glutamate acts as excitatory neurotransmitter, and GABA act as inhibitory neurotransmitter. In motor and sensory neurons, acetylcholine acts as excitatory neurotransmitter, and glycine acts as inhibitory neurotransmitter.
If a nerve is damaged, infected, diseased, autoimmune disrupted, or degenerated, it can lead to various diseases. The causes of damage, infection, immune activation, and degeneration are probably a series of events which include environmental factors, genetic factor, or mixture of damaging genetic and environmental factors. As the age progresses, a person increasingly loses the ability to control this damage, triggering irreversible neurodegeneration or neurodegenerative diseases (NDs).
15.1.1 Neurodegenerative Diseases (NDs)
According to the European Commission Public Health, NDs are defined as genetic and/or sporadic disease conditions characterized by progressive nervous system dysfunction. The atrophy of central or peripheral nervous system is often associated with it. Till date, more than 600 disorders were reported to affect the nervous system and can cause diseases like brain cancer, degenerative nerve diseases, Alzheimer’s disease and other dementias, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis (ALS), encephalitis, epilepsy, genetic brain disorders, hydrocephalus, multiple sclerosis, stroke, prion diseases, and others [1, 2].
15.1.2 Mechanisms for Neuron-Related Diseases
15.1.2.1 Misfolded Protein Aggregates
These aggregates are abnormal clumps of protein which are generated inside motor neurons. They are found in nearly all cases of motor neuron disease and may disrupt the normal working of the motor neurons or put the cell under great strain and can be used as marker for the disease. The most common aggregates found are TDP-43, alpha-synuclein, tau, and beta-amyloid mainly found in Alzheimer’s disease. The diseases like Alzheimer’s disease, Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy are the examples of misfolded protein aggregates [2, 3].
15.1.2.2 Polyglutamine
This disease is caused by genetic mutations leading to formation of multiple CAG nucleotide sequences called polyglutamine (polyQ) tract. CAG is responsible for glutamine amino acid, and its expression results in polyglutamine diseases. The excess glutamine, in the neuron cell, exhibits abnormal properties, such as protein degradation, irregular protein folding, anomalous interactions with other cellular proteins, and altered subcellular localization. Proteasome are the ubiquitin with enzymes which are responsible for degradation of many misfolded proteins like alpha-synuclein and polyQ expansions. If proteasome do not correctly cleave these irregular proteins, it leads to increase in cell toxic protein levels. The diseases like Huntington’s disease and spinocerebellar ataxias are the examples of polyglutamine diseases [4, 5].
15.1.2.3 Cell Transport Disruption
Individual cell has its transport system, which transports nutrients and other chemicals into the cells and throws out the waste toxic products in the form of vesicles or destroys them with the help of antioxidants or enzymatic degradation. Due to changing lifestyle and other environmental factors, antioxidants decrease in the body which leads to the cell transport disruption and buildup of toxic waste in the neuron cell. The diseases like Parkinson’s disease and Huntington’s disease are the examples of cell transport disruption diseases.
15.1.2.4 Glial Cells and Myelin Sheath
The axon of the motor neuron are surrounded and supported by glia cells which provide nutrients to the neuron cells and on outer surface the cells release myelin forming myelin sheath, which is a dielectric fatty white substance. The purpose of a myelin sheath is to increase the speed of impulses propagation through myelinated fiber. Thus, if glial cells or myelin sheath is affected, it leads to hampered nerve signal transfer leading to abnormality. The diseases like multiple sclerosis, chronic inflammatory demyelinating polyneuropathy, acute disseminated encephalomyelitis, Guillain-Barré syndrome, neuromyelitis optica, transverse myelitis, leukodystrophy, and Charcot-Marie-Tooth disease are the examples of glial cell and myelin sheath disorders [6, 7].
15.1.2.5 Mitochondrial Dysfunction
Mitochondria are known as the powerhouse of the cell; if it is damaged, it initiates the intrinsic mitochondrial apoptotic pathway through activation of caspase-9 (cysteine-aspartic acid protease cascade) which releases the cytochrome C from the mitochondrial intermembrane space (IMS). This cytochrome C binds apoptotic protease-activating factor-1 (Apaf-1) and initiates the apoptotic cycle of the cell. The reactive oxygen species (ROS) helps the cytochrome C to bind with Apaf-1 by activating pore-stabilizing proteins (Bcl-2 and Bcl-xL). In large concentration, ROS can cause both apoptosis and necrosis [8,9,10,11]. The diseases like Alzheimer’s, Parkinson’s, Huntington’s, and amyotrophic lateral sclerosis (ALS) are the examples of mitochondrial dysfunction.
15.1.2.6 Programmed Cell Death (PCD)
Programmed cell death (PCD) is an intracellular program-mediated cell death. It is a natural phenomenon used by the cells to commit suicide if cells are infected, diseased, or injured. It involves series of biochemical events which lead to cell death or disruption. Autophagy is a most favored route of self-destruction acquired by any cell if there is presence of aggregate prone proteins in the cell. It is categorized as macro autophagy and chaperone-mediated autophagy (CMA). Both the pathways involve use of lysosome for the destruction of misfolded proteins. The macroautophagy mechanism is involved in macromolecule recycling during starvation, and chaperone-mediated autophagy (CMA) mechanism is involved in macromolecule and other toxic by-product degradations. If any of these cycles are blocked, it leads to accumulation of misfolded proteins and toxic substances leading to cell death. Other PCD like apoptosis involving intrinsic and extrinsic pathways initiated by internal or external cell factors to trigger biochemical events leading to cell death or destruction is also responsible for many nerve diseases [1, 12,13,14]. The diseases like Parkinson’s disease, amyotrophic lateral sclerosis, Alzheimer’s disease, and Huntington’s disease are the examples of programmed cell death mechanism.
15.1.3 Classification of Neurodegenerative Diseases
The neurodegenerative diseases are classified into two main categories:
-
1.
Diseases caused by non-motor neuron degeneration in the cerebral cortex leading to cognitive disturbance like dementia (e.g., Alzheimer disease, Pick disease)
-
2.
Diseases caused by motor neuron degeneration in various parts of the brain and spinal cord, leading to movement disorders which are subcategorized on the basis of the part of the CNS affected as:
-
(a)
Motor neuron weakness and degeneration (e.g., amyotrophic lateral sclerosis (ALS), spinal muscular atrophy)
-
(b)
Motor neuron degeneration at the spinal cord and cerebellum junction (e.g., Friedreich ataxia, ataxia-telangiectasia)
-
(c)
Motor neuron degeneration at substantia nigra and basal ganglia (e.g., Parkinson’s disease, progressive supranuclear palsy)
-
(d)
Motor neuron degeneration at basal ganglia (e.g., Huntington’s disease)
-
(e)
Motor neuron degeneration at multiple areas of CNS leading to complex ND conditions
-
(a)
15.1.4 Severity and Economical Burden of Neurodegenerative Disorders
According to WHO, the diseases like epilepsy, Alzheimer’s and other dementias, Parkinson’s disease, multiple sclerosis, and migraine are categorized under neuropsychiatric neurological disorders [15].
The economic burden due to nerve diseases and brain disorders are very large. These costs include the cost of treatment and the cost due to lost productivity of patients and their family members. In addition to economic burden, the emotional, practical, and financial burden on family members exacerbates the problem. As per the study conducted by the WHO, the World Bank, and the Harvard School of Public Health since 1993, dementias are responsible for the greatest burden with Alzheimer’s disease taking 60–70% share of all dementias and 12% of all neurodegenerative disease cases throughout the world and affecting 0.75% of total world population. Parkinson’s disease (PD) is the second most common neurodegenerative disorder followed by AD, taking 1.8% share of all neurodegenerative disease cases throughout the world and affecting 0.11% of total world population. Similarly multiple sclerosis, migraine, and epilepsy contribute 1.6, 8.3, and 7.9%, respectively, of all neurodegenerative disease cases throughout the world and affect 0.10, 0.52, and 0.50% of total world population, respectively, adding huge economic burden to world economy [16,17,18,19,20,21,22,23].
15.1.5 Current Treatments for Neurodegenerative Diseases
The drug therapy for neurodegenerative disorders involve use of anti-inflammatory drugs, antioxidants, steroids (estrogen), neurotransmitters specifically levo-3,4-dihydroxyphenylalanine (L-dopa) in Parkinson’s disease, GABAergic for Huntington’s disease, and acetylcholine and cholinesterase inhibitors for Alzheimer’s disease.
The functional neurosurgery approaches like lesions, stimulation, and transplants toward the treatment of neurodegenerative disorders are also in use. The lesions and stimulation offer only symptomatic alleviation of the disease, but the surgical knowledge is nowadays being used for transplantation. The transplant being tried includes fetal tissue transplants and cellular transplants which can release neuron protecting growth factors and enzymes. Researchers are also trying to transplant embryonic stem cells and pluripotent stem cells in attempt to create therapies for neurodegenerative diseases with limited success.
Gene therapy approaches to treat neurodegenerative diseases (NDs) which might be inherited or acquired offer many advantages over conventional therapies. It is particularly striking for ND due to BBB’s restricted bioavailability of conventional drugs. The most effective gene delivery can be achieved through lentiviral vectors which offer postmitotic long-term expression of the genes with high titer levels without immunological complications. This therapy showed exciting results in Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, and spinal muscular atrophy [24,25,26].
15.1.5.1 Limitations for Neurodegenerative Diseases Treatments
All the major neurodegenerative diseases (NDs) have their origin in CNS, and the majority of treatments need to concentrate toward the brain and spinal cord. However CNS is strongly protected by several barriers which inhibit the entry of any foreign material. Thus the entry of the drug molecules to the site of action in the CNS is restricted, and it modifies the disease symptoms to a short period of time and does not show any inhibitory effect on the progression of the ND. The main barrier for the drug delivery in the treatment of ND is blood-brain barrier (BBB), which affects the drug release kinetics and many times leads to peripheral side effects. Furthermore, the death of the neurons in any type of NDs is generally caused by multiple factors as mentioned above, thus adding difficulties in ND treatment and management.
15.1.5.2 Way Out for Neurodegenerative Disease Treatments
The major hurdle for ND treatment is BBB, as it limits the drug molecules to reach the target sites in the brain. A vast attention was acquired by nanoparticles as they have a capacity to pass through BBB and carry the drug molecules with it to deliver at target site. More efforts are being made to improve the efficiency of the NP to improve their capacity to carry different drug molecules to treat the symptoms of ND effectively. The other hurdle is to stop the recurrence of the symptoms, which need to be tackled at genetic level. For this the gene therapy approach was tried, as the mechanisms of ND were better understood; it was confirmed that it has more complex biochemical mechanisms involving more than one gene. Thus gene therapy was not very successful in treating the NDs. So the way-out researchers are looking at its genomic treatment along with NP-based symptomatic treatment as cotherapy.
15.1.5.3 Genomic Treatment
Genomic therapy is the branch of medicine which utilizes the maps of human DNA, called the genome, to understand and correlate the biochemistry and pathogenesis of diseases to patients’ genes. It is different from gene therapy, as gene therapy involves modification of just one pair of gene on chromosome resulting in modification of disease condition. This is not much useful in NDs as they involve multiple genetic mutations, while genomic treatment uses many pieces of genetic information to modify disease condition. Genomic treatment is based on three main mechanisms—diagnosing disease, preventing disease, and treating disease. Thus genomic treatment refines diagnoses, prevents adverse drug effects, manages epidemics, develops new therapies, and can individualize the treatments [27,28,29].
The mutations in genes cause diseases by modulating the cell function. Recent successful genomic advances helped to predict few changed phenotype from mutated genotype in single cells [30], but we are still far from being able to relate each mutation to its synthesized molecule and, ultimately, phenotypic outcome in humans.
Detecting and understanding the genetic mutation(s) is necessary for subsequent therapeutic and preventive actions. Thus the major challenge in genome-based medicine is segregating the disease-related mutated and non-mutated genes from a large number of genes and correlating them with the disease. Once the mutations and their phenotypic expressions are identified, the planning and process of designing targeted treatment start. Though this process is slow, little progress has been made to modify the actionable mutations to modify the disease conditions [31]. Genomic treatment is being extensively explored for infectious disease, cardiovascular diseases, cancer, and NDs. In this chapter, the recent trends in genomic treatment of the major four neurodegenerative diseases, namely, Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS), are discussed.
15.2 Alzheimer’s Disease
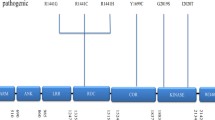
Alzheimer’s disease (AD) is the most common non-motor neurodegenerative disease. It is also the most common type of dementia, involving decline in cognition, specifically in memory, language, and thinking, due to gradual death of brain cells [32]. In the progressed AD, the brain tissue shrinks compared to normal brain, hampering all the cognitive functions, and specifically, the hippocampus and cortex shrinks, damaging the areas involving thinking, planning, remembering, and forming of new memories (Fig. 15.2). It was also observed that the ventricles (fluid-filled spaces within the brain) grow larger. It affects mainly the people aged 65 and older.

Areas in the brain and spinal cord marked for pathogenesis of different neurodegenerative disorders
15.2.1 Global Burden of AD
AD is taking 12% share of all neurodegenerative disease cases throughout the world and affecting 0.75% of total world population. In the USA only, the collective direct and indirect cost of PD, including treatment and lost job income due to work inability, was estimated to be nearly $214 billion per year. According to WHO, the projected deaths attributable to AD as percentage of total deaths for 2015 and 2030 are 0.81 and 0.92%, respectively (Table 15.1).
15.2.2 Pathophysiology
The autopsy samples of AD patients show the presence of plaques and tangles in the brain tissue. The plaques are depositions of the protein called beta-amyloid between the dying cells in the brain and also termed as “amyloid plaques (AP).” The tangles occur between the neurons in the brain and happen due to disintegrated tau protein [33]. In early stages of AD, AP accumulation was observed followed by tau protein tangles and degradation of neurons leading to progressive loss of cognition as the disease progresses [34]. There were different theories proposed based on the AP and tau protein mechanisms of degradation of the nerve cells, and tau protein tangle theory was proved more appropriate for dementia observed in AD (Fig. 15.3a and b) [35].

(a) Normal and (b) Alzheimer’s brain TS observed in Alzheimer’s disease; (c) normal and (d) Parkinson’s pallor (depigmentation) of the substantia nigra observed in Parkinson’s disease; (e) normal and (f) Parkinson’s pallor (depigmentation) of the locus coeruleus observed in Parkinson’s disease; (g) normal and (h) Huntington brain TS observed in Huntington’s disease
15.2.3 Etiology
The causes of AD are still not clear, but there are several hypotheses put forth by different investigators ranging from environmental factors to genetic risk factors triggering the pathophysiologic cascade which, over the decades, leads to AD:
-
1.
Genetic causes—Genetic mutations in amyloid precursor protein (APP) gene (chromosome 21), presenilin-1 (PS1) gene (chromosome 14), and presenilin-2 (PS2) gene (chromosome 1) cause early-onset AD (Table 15.2). These faulty genes start producing beta-plated amyloid protein with more sticky 42 amino acid residues instead of normal nonsticky 40 amino acid residues initiating the AP deposition on the nerves [36].
-
2.
Insulin resistance—Researcher observed that there is relation between decreased cerebral glucose metabolic rate due to insulin resistance and onset of AD. Thus, researchers are trying to develop a correlation between extent of insulin resistance and onset of AD as a marker for early detection of AD [37].
-
3.
Infection—It was known that amyloid protein has antimicrobial property. Hence when there is an infection specifically with spirochetes sp., Treponema, and Borrelia burgdorferi resulting in chronic inflammation and neuronal destruction, it was related to the release of amyloid protein. This observation suggests the direct relation between infection and amyloid protein release which might lead to AD if infection becomes chronic [38].
-
4.
Head trauma—Researchers reported that a traumatic brain injury triggers the release of amyloid precursor protein (APP) from axons in extracellular space, and this APP starts depositing amyloid protein on the neurons in the form of amyloid plaques [39].
15.2.4 Symptoms of Alzheimer’s Disease
AD is characterized by shrinking of hippocampus and cortex area of brain tissue. Hippocampus is associated with developing new memories, and thus loss of neurons at this region leads to loss of recent memories (short-term memory loss) which is the earliest and most prominent symptom of AD. This situation worsens over the period of time affecting remote or old memories. As the disease progresses, parietal and temporal lobe starts losing function leading to language dysfunction mainly affecting word search. Thus communication ability of the patient becomes compromised. Posterior cerebral dysfunction causes difficulty in performing simple practiced functions like brushing teeth and using remote control [40]. Patients experience behavioral problems like depression and sleep disturbance. In later stages, verbal and physical aggression, psychomotor agitation, inappropriate sexual behavior, and psychotic symptoms are observed in patients. In advanced stages, patients develop motor signs such as gait disturbance, tremor, and urinary incontinence [41, 42].
15.2.5 Treatment
15.2.5.1 Symptomatic Therapy
There is no cure for Alzheimer’s disease, but some FDA-approved medications can dramatically improve the symptomatic conditions. The first class of symptomatic drug therapy reported to reduce AD symptoms is anticholinesterase inhibitors (AchEI) like donepezil. Other classes are memory booster drugs like galantamine. The parasympathomimetic or cholinergic agent, like rivastigmine, is also used in the treatment of AD. Recently NMDA (N-methyl-D-aspartate) glutamate receptor antagonist drugs, like memantine, are also reported to be used for symptomatic treatment of AD [43].
15.2.5.2 Preventive Therapy
Currently, there are many research-based and clinical trials going on to reduce, delay, or prevent symptoms of AD before they start appearing. One clinical trial ongoing (by Dominantly Inherited Alzheimer Network) on the high-risk genetic AD patients to avoid or delay the AD symptoms includes the use of specific antibodies against beta-amyloid protein which are supposed to neutralize the mutated beta-amyloid and reduce or delay the appearing AD symptoms. Similarly, a clinical trial, known as A4 trial, on the patients aged between 65 and 85 without AD symptoms is being carried out with antibodies against beta-amyloid to see the effect toward avoiding or delaying the appearance of AD symptoms due to age [44].
Tuszynski and coworkers at the University of California performed a clinical trial in 2001 in which they isolated the long-living skin cells from patients and infected them with viral vector containing nerve growth factor (NGF) gene. These cells then started secreting nerve growth factor and acted as mini-bio-pumps of NGF. These NFG-secreting cells were then transferred into patient’s basal forebrain, and patients were studied for 10 years. The studies revealed that patient’s brain started supporting new nerve fibers and the neuron cell size also enhanced. The same group in 2009 reported a gene therapy using brain-derived neurotrophic factor (BDNF)-expressing gene transferred via viral vector to cortical neurons of hippocampus in rat models [45]. They observed that the expression of BDNF gene reverses synapse loss, improves cell signaling, and restores learning and memory (Table 15.2).
In another study, researchers at St. Jude Children’s Hospital developed a gene therapy involving the enzyme neuraminidase 1 (NEU1) gene and tested it in mice. NEU1 is responsible for carrying recycling of unneeded proteins in cells. Researchers observed that the AP buildup on neurons declined dramatically after a few weeks of gene therapy designed to boost NEU1 activity [46]. The effect of low levels of progranulin protein in the brain which increased the formation of amyloid-beta plaques on the neurons leads to memory impairment and triggers the immune response. These researchers then developed the gene therapy in mouse using lentivirus-mediated progranulin (PGRN) gene (GRN) transferred in brain cells of frontotemporal lobe of mouse. The overexpression of this gene lowered amyloid plaque load in AD mice brain [47]. Additionally, CREB-regulated transcription coactivator-1 protein expressed by Crtc 1 gene (released by brain cells mainly at hippocampus region and involved in long-term memory development) is blocked from expression in patients with Alzheimer’s. These researchers then developed an adeno-associated viral-mediated gene therapy for the delivery of Crtc 1 gene in brain cells at hippocampus region of mouse brain. They observed that the gene expression in the hippocampus efficiently reverses Alzheimer-induced spatial learning and memory deficits [48].
15.3 Parkinson’s Disease
Parkinson’s disease (PD) is the second most common neurodegenerative disease followed by Alzheimer’s disease, affecting more than ten million people worldwide. It is a chronic, progressive neurological disorder affecting mainly dopaminergic neurons in substantia nigra area of the brain and development of Lewy bodies (a pathologic hallmark) in dopaminergic neurons (Fig. 15.2). These neurons in substantia nigra are responsible for producing dopamine which is a major neurotransmitter that controls movement and coordination. With the progression of PD, the amount of dopamine produced in substantia nigra goes on decreasing, leading to decreased control of body movements. Primary motor signs of PD are tremor in the hands, arms, legs, jaw, and face, rigidity of limbs and trunk, slowness of movement, and postural instability due to impaired balance and coordination. PD may be diagnosed in some patients in their early 40s and is most common in patients over 60s. It affects men and women equally, and at present there is no known cure for Parkinson’s disease [49,50,51,52,53,54,55].
15.3.1 Global Burden of PD
PD is taking 1.8% share of all neurodegenerative disease cases throughout the world and affecting 0.11% of total world population. In the USA only, the collective direct and indirect cost of PD, including treatment and lost job income due to work inability, was estimated to be nearly $25 billion per year. According to WHO, the projected deaths attributable to PD as percentage of total deaths for 2015 and 2030 are 0.20 and 0.23%, respectively (Table 15.1).
15.3.2 Pathophysiology
As shown in Fig. 15.3, the substantia nigra pars compacta and the pontine locus coeruleus of the brain are affected by typical abnormalities including depigmentation, neuronal loss, and gliosis (Fig. 15.3d and 15.3f). PD is also characterized by Lewy body in a pigmented neuron in substantia nigra. The PD symptoms are prominently observed when 60–70% of the neurons in the substantia nigra pars compacta are degenerated, thus making the treatment more difficult [56, 57].
15.3.3 Etiology
Genetic Mutations
The first and most understood cause of PD is the neuronal deaths at substantia nigra region of the brain which is mainly due to the presence of Lewy bodies in those nerves. These Lewy bodies are composed of defectively aggregated, insoluble alpha-synuclein also known as the non-Abeta component of amyloid plaques [58]. Although the normal function of synuclein remains poorly understood, it was studied for its presence at the nerve terminal and in membrane remodeling. When it is related to PD in Lewy bodies, in contrast to its helical conformation on membranes, synuclein adopts a β-sheet structure in aggregates [59]. Thus, it is a clear mechanism of misfolded protein aggregates as explained in the introduction, and it is clearly the outcome of some genetic mutations that code this protein.
Cell Transport Disruption
Ubiquitin-proteasome system is the system present in the nerve cell which is designed to break down and throw out the abnormal proteins like insoluble alpha-synuclein aggregates that become impaired in PD.
Mitochondrial Dysfunction
The neuronal degeneration can be caused by abnormal oxidative stress through reactive oxygen species, which generally occurs when there is a mitochondrial dysfunction [60].
Viral Infection
Braak’s hypothesis claims that pathophysiological progression of PD starts with an unknown, possibly viral, pathogen entering the brain through the olfactory route or the swallowing of nasal secretions introduces the pathogen to the gut from where it enters the vagus nerve and the CNS. It was reported that non-motor symptoms, like sleep disorders, loss of sense of smell, hyposmia, and constipation, may indicate the onset of PD several years earlier to actual motor symptoms. The hypothesis was substantiated by pathologic proofs of presence of Lewy bodies in the intestinal structures, vagus nerve, and brain structures [61].
Environmental Factors
After Braak’s hypothesis, scientists explored possible environmental factors, and their research work suggested possible role of environmental stress and aging in the promotion of neurodegeneration. The researchers found that the exposure to environmental toxins (e.g., pesticides) [62] and abusive drugs or the aging stress leads to a chronic low-level inflammation in the brain. This chronic inflammatory condition might initiate the neuron deterioration in the brain leading to PD [49, 63].
15.3.4 Symptoms of Parkinson’s Disease
Parkinson’s disease is a chronic, progressive neurological disorder affecting mainly dopaminergic neurons in substantia nigra area of the brain which is mainly affecting motor and non-motor functions of the body. The signs and symptoms differ from person to person. Early signs are often mild and remain unnoticed. The motor symptoms often begin from one side of body and progressively worsen as both sides get affected. As per Braak’s hypothesis, we can find out the onset of PD several years earlier to actual motor neuron dysfunction by observing the simple symptoms like sleep disorders, loss of sense of smell, hyposmia, and constipation.
In the early stages of motor neuron dysfunction, patient is unable to show facial expression, the speech becomes soft and slurred, and patient does not swing his/her arms while walking. As condition progresses with time, the following symptoms become worse:
Tremors
A prominent motor symptom characterized with shaking of body; it generally begins with terminal limb organs like fingers or hand. An unintentional back and forth rubbing movement between the thumb and forefinger called pill-rolling tremor or tremor of the hand when at rest is the characteristic of Parkinson’s disease.
Slowed Movement (Bradykinesia) and Muscle Stiffness
As disease progresses, reduced and slower movements and muscle stiffness characterize the disease, as it makes simple tasks difficult and time-consuming. The walking steps may become shorter, patient finds it difficult to get out of a chair, and many times patient drags his/her feet as he/she tries to walk.
Impaired Posture and Balance and Loss of Automatic Movements
In the extreme conditions, maintaining body balance and performing unconscious movements like blinking, smiling, or swinging of arms do not take place naturally.
Other neurodegenerative disorders that can mimic the conditions of PD include dementia with Lewy bodies (DLB), multiple system atrophy (MSA), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD).
15.3.5 Treatment
15.3.5.1 Symptomatic Therapy
Parkinson’s disease cannot be cured, but some medications can dramatically improve the symptomatic conditions, sometimes surgery, to regulate certain regions of the brain. The main medication which is currently available and most effective against motor symptoms is levodopa, but the other medications available are monoamine oxidase type B inhibitors [MAOBIs], amantadine, anticholinergics, β-blockers, or dopamine agonists which are mainly used to avoid levodopa-related motor complications. The non-motor symptoms like hallucinations can be treated by clozapine, dementia can be treated with cholinesterase inhibitors, and depression can be treated with antidepressants and pramipexole. These medications are generally used along with the specific PD symptomatic medications. The specific PD medications available in the market for single or combination therapies are Sinemet (carbidopa/levodopa), Sinemet+Comtan (entacapone), Parlodel (bromocriptine), Permax (pergolide), Requip (ropinirole), Mirapex (pramipexole), and Casbar (cabergoline) [64, 65].
15.3.5.2 Preventive Therapy
Recently, scientists from Nanyang Technological University, Singapore, reported that the existing antimalarial drugs could be a potential treatment for PD. Researchers at NTU reported that activating Nurr1 gene releases protein which protects brain’s ability to generate dopamine. These scientists further reported that potent antimalarial drug molecules like chloroquine and amodiaquine can directly bind to Nurr1 gene and activate it to revert or to stabilize the PD conditions (stop progression) [66]. In addition to pharmacological treatment, gene and genomic therapies are also tried and reported to be successful preclinically and clinically in reverting PD condition.
The scientists from the University of Pittsburgh developed a new gene therapy for PD using the viral vector AAV2 which is in clinical trial phase. They proposed that mitochondria and α-synuclein (encoded by SCNA gene—Table 15.2) can interact and damage the neuron, and if we target the α-synuclein synthesis, it might modulate the PD condition. To prove their hypothesis, they used a harmless virus called adeno-associated virus type 2 (AAV2) which was engineered to transport a SCNA gene silencer code [short hairpin RNA (shRNA)] into the neuron and block the production of α-synuclein. The blocked α-synuclein production ultimately results in avoiding aggregation of faulty Lewy bodies in neurons. It also avoids the interaction of α-synuclein with mitochondria and thus avoids the PD progression [67]. Professor Nicholas Mazarakis and his team from Imperial College London developed a new genomic therapy with a strategy to deliver three different genes (not disclosed) which are coded for enzymes that produce dopamine. He used lentivirus vector which is closely related to HIV to incorporate the set of genetic material into the genome of the neuron cells it infects, ensuring a long-lasting effect. The team reported that the treatment corrected the movement defects of the monkeys for more than 3 years, without any adverse effects. The clinical trials with 15 patients also showed promising results in recovering from motor symptoms and stopping the progression of PD [68].
15.4 Huntington’s Disease
Huntington’s disease (HD) is caused by a defective gene which results in the programmed degeneration of neurons (brain cells) (Fig. 15.2), particularly in basal ganglia and the cerebral cortex. It is an incurable, hereditary brain disorder. HD is named after George Huntington, the physician who described it as hereditary chorea in 1872 [69, 70].
15.4.1 Global Burden of HD
HD is taking 0.3% share of all neurodegenerative disease cases throughout the world and affecting 0.018% of total world population. HD and other hyperactivity disorders (ADHD) also called as Huntington’s disease-like syndrome are showing 5.29% prevalence in the global population. In the USA only, the collective direct and indirect cost of HD, including treatment, and lost job income due to work inability, is estimated to be nearly $1.3 billion per year [71, 72].
15.4.2 Pathophysiology
Pathophysiological evidences can be seen microscopically in the infected brain tissue only at grade 2 HD where neuropathy with striatal atrophy and convex caudate nucleus at neostriatum was observed. Thus the extent of gross striatal atrophy, neuronal loss, and gliosis (Fig. 15.3g and h) is generally used for grading the severity of HD pathology (grades 0–4) [73,74,75,76].
The neuropathy with striatal atrophy in HD is caused due to the expansion and expression of a Nterminus cysteineadenosineguanine (CAG)n repeat sequences in Huntington protein synthesizing gene (e.g., ATXN3, SCA-1) (Table 15.2). This Huntington protein is located in the cytoplasm and is associated with the range of organelles, like transport vesicles, mitochondria, microtubules, synaptic vesicles, etc. The expression of Nterminus cysteineadenosineguanine (CAG) repeat sequences in the Huntington protein might destroy the actual function of the protein. In addition to this, there are some evidences suggesting the mutant Huntington accumulation and formation of inclusions in the brain cell nucleus called as “neuronal intranuclear inclusions (NIIs)” which are toxic/pathogenic and cause HD [77,78,79,80].
15.4.3 Etiology
The disease affects movement, behavior, and cognition in later part, but as discussed, there are microscopic and biochemical clues available for the prediction of the HD. There are different mechanisms like excitotoxicity, oxidative stress, impaired energy metabolism, and apoptosis, which cause these microscopic and biochemical changes in neurons [81]:
-
1.
Excitotoxicity—It is the excessive activation of postsynaptic receptors by excitatory amino acids leading to the neurotoxic effects of HD. This mechanism was proved by injecting kainic acid, an agonist of glutamate receptor, and quinolinic acid, an agonist of NmethylDaspartate (NMDA) receptor in rat, and observing the mimicking neuropathology similar to HD. In addition to excitotoxicity, reduced uptake of glutamate by glial cells was also proposed to play a role in the pathogenesis of HD.
-
2.
Oxidative stress—It is caused by the presence of free radicals like highly reactive oxygen derivatives (ROS) in large amounts. The mechanism was proved by the observation that quinolinic acid-induced striatal damage can be reduced by administration of antioxidants. Oxidative stress can occur as a consequence of mitochondrial breakdown or excitotoxicity and can trigger apoptosis. The probable mechanism of the oxidative stress proposes reduction in the activity of the respiratory chain complexes II and III of mitochondria of neurons which show the increased lactate levels in the basal ganglia and occipital cortex of patients with HD. This mechanism was proved by Revilla and coworkers by injecting 3nitroproprionic acid (3NP) which is an inhibitor of succinate dehydrogenase or complex II of the respiratory chain. This experiment caused dose-dependent ATP depletion, increased lactate concentration in neurons, and neuronal loss in the striatum in rats [81].
-
3.
Apoptosis—It is called as programmed cell death and is the natural phenomenon which the body experiences from embryogenesis where this phenomenon is used to remove supernumerary neurons as part of natural development. Naturally the neurons are protected from apoptosis by CREB-binding protein (CBP), which is a major mediator of survival signals in mature neurons. One theory suggests that expanded polyglutamine repeats interact with other proteins containing short polyglutamine tracts and interference with transcription and expression of CREB-binding protein (CBP) leading to HD [80].
15.4.4 Symptoms of Huntington’s Disease
Huntington’s disease is an autosomal genetic disorder, it requires only one copy of the defective gene to develop the disorder, and there are 50–50% chances of developing HD in children of HD parents. The disease causes the programmed breakdown (degeneration) of neurons in the brain. The HD signs and symptoms generally appear in patients at the age 30s or 40s, but the onset of disease may be earlier (in some cases it was observed in 20s).
This degeneration of neurons in basal ganglia and the cerebral cortex causes uncontrolled movements, loss of thinking (cognitive) ability, and psychiatric disorders. Broadly, the patient gradually loses his/her abilities to walk, talk, think, and reason, and they become totally dependent on other people for their care. Thus, HD has a major negative physical, emotional, and socioeconomic impact on patients and their families’ lives.
15.4.5 Treatment
15.4.5.1 Symptomatic Therapy
HD is not curable but some symptoms of the disease can be treated with medications. Symptoms like depression, obsessive-compulsive behaviors (OCBs), agitation, and irritability can be treated by selective serotonin reuptake inhibitor type of antidepressants. Irritability and impulsive behavior can be treated with anticonvulsants like valproic acid or carbamazepine. Anxiety can be treated with anxiolytic drugs, and delusions and behavioral outbursts can be treated by antipsychotic drugs. Chorea can be treated with dopamine-blocking agents like tetrabenazine. Cognitive disorders are now being treated with new drugs, namely, memantine [82], rivastigmine [83], and donepezil [82,83,84].
15.4.5.2 Preventive Therapy
The molecular and biochemical understanding of the disease suggested the following causes for HD: changes in protein homeostasis, mitochondrial dysfunction, excessive or abnormal neurotransmitter input, and hampered axonal trafficking in brain neuron cells.
Based on the same understanding, new compounds and strategies were designed as HD preventive treatment which include histone deacetylase inhibitors (e.g., phenylbutyrate); neuroprotective compounds (e.g., lithium); antioxidants, mitochondrial enhancers, and energy substrates (e.g., coenzyme Q and creatine); antiapoptotic compounds (e.g., minocycline); transglutaminase inhibitors; chemicals that inhibit protein aggregation or support protein folding; molecules that enhance clearance of mutant protein; molecules that inhibit the kynurenine 3monooxygenase pathway; cell or gene replacement therapy; and RNA silencing agents that “knock down” disease gene expression [85].
Different genetic mutations lead to faulty protein expression (Table 15.2) and ultimately lead to the disease. HD is mainly caused by faulty production of Huntington protein in brain neurons. These faulty proteins contain (CAG)n repeat sequences because of mutant gene transcribing faulty mRNA. Prof. Stephanie Liou developed an antisense gene therapy which is a gene silencing technique, where he inserted short single-stranded pieces of chemically modified nucleotides, known as oligonucleotides (oligos), into the cells. These oligos contain complimentary sequences to that of faulty mRNA and either physically block translation, or they stimulate RNAse H enzyme to degrade the mRNA complex and thus stop synthesis of faulty Huntington protein.
Similar to gene silencing technique, RNA interference (RNAi) technique is also being developed which utilizes large strand of complimentary RNA molecule which binds to faulty mRNA of Huntington protein and activates “dicer” enzyme which cuts this complex into smaller fragments and thus avoids translation of the faulty Huntington protein. Researchers found that inserting the large complimentary RNA fragment is difficult, so they came up with the new idea of small interfering RNA (siRNA), which is a double-stranded RNA fragment with complimentary sequence to the mRNA for Huntington protein. These siRNA are easy to insert in cells, and after entering the cells, they separate, and the complimentary piece attaches to mRNA strand initiating the mRNA degradation by “dicer” enzyme, thus avoiding translation of Huntington protein.
Researchers also found that the caspase enzyme is essential for the specific cleavages of Huntington protein. This enzyme cleaves the protein at 3rd and 6th position from N-terminal. If this enzyme is allowed to cleave the mutated Huntington protein selectively at caspase3 site, but not caspase6 site, it resulted in protection of neuron from neuronal dysfunction and neurodegeneration. These observations suggest that preventing caspase6 cleavage of Huntington protein may be studied for therapeutic interest [86].
15.5 Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, Charcot disease, or motor neuron disease (MND), is a rapidly progressive motor neuron disease which attacks and kills the neurons controlling voluntary muscles such as the diaphragm, face, arms, and legs. The motor neurons in the brain, brain stem, and spinal cord maintain and control the vital communication between voluntary muscles of the body. In ALS the motor neurons transmitting signals from the brain (upper motor neurons) to the spinal cord (lower motor neurons) degenerate or die ultimately failing to send the messages to muscles and ultimately showing no voluntary muscular movement. As there are no movements, muscles gradually weaken and undergo atrophy which is characterized by fine twitches (fasciculations). After 3–5 years from the onset of symptoms, the brain completely loses the ability to control voluntary movement of the body. When voluntary muscles in the diaphragm and chest wall fail, patients lose the ability to breathe without external support and eventually die.
15.5.1 Global Burden of ALS
ALS is taking 0.072% share of all neurodegenerative disease cases throughout the world and affecting 0.002% of total world population. In the USA only, the collective direct and indirect cost of ALS, including treatment and lost job income from work inability, is estimated to be nearly $1.03 billion per year [87].
15.5.2 Pathophysiology
The brain stem and upper spinal cord neurons undergo axonal loss with secondary myelin pallor and gliosis which extend throughout the spinal cord. It was also observed that deeper layers of the gray matter which is underlying subcortical white matter undergo astrocytic gliosis. The presence of CD68 (lysosomal marker) suggest that the glial response at the cortical and spinal tracts are due to microglia activation and presence of active macrophages. It was also observed that ventral roots become thin due to the huge loss of myelinated fibers from motor nerves leading to denervation atrophy (Fig. 15.3g) [88].
15.5.3 Etiology
The exact mechanism of the neuron degradation in ALS is not clear, but researchers believe that there are multiple factors individually or in combination affecting the motor neurons or the cells that support them.
-
1.
Aggregates—As reported earlier, if the mRNA translation is faulty, it leads to generation of faulty protein. TDP-43 is a protein which regulates the expression of the mRNA in neuron. In ALS this TDP-43 protein itself expressed faulty forming aggregates and thus started the cascade of faulty protein expressions which ultimately disrupt the normal working of the motor neurons and destroy it.
-
2.
Cell transport disruption—The normal cell transportation gets rid of metabolic and other toxic substances by packaging them in microvesicles and throwing them out of cells. Antioxidants help the cells in this reaction. This normal function is disrupted in the ALS, and toxic waste buildup in cells will destroy the normal functioning and ultimately complete neuron cell.
-
3.
Glial cells—Glial cells are the nurse cells of the neuron fibers. They supply nutrients and support and excrete lipid layer around it, which help neuron cells to function normally and transfer electrical impulses efficiently. If these glial cells are hampered, neurons will not receive nutrition and support, and lipid layer will be destroyed, resulting in complete loss of nerve conduction and normal function of neuron.
-
4.
Glutamate—If the neuron cells become sensitive (immunosensitive) to the neurotransmitter (glutamate), immune system will destroy the neurons and a complete loss of motor neuron activity will be observed.
-
5.
Mitochondria—They are the energy house of the cells and contain SOD1 protein in intermembrane space, matrix, and outer membrane. If there is a presence of abnormal misfolded SOD1 protein, it can cause mitochondrial dysfunction with damaged mitochondrial membrane, and it leads to motor neuron diseases.
15.5.4 Symptoms of Amyotrophic Lateral Sclerosis Disease
The early symptoms like cramps, stiff muscles, muscle weakness affecting arms and legs, slurred speech, and difficulty in chewing and swallowing were very difficult to distinguish from non-ALS progressing patients. As the disease progresses, patients find it difficult to perform simple tasks such as buttoning a shirt, writing, or turning a key in a lock. Many patients experience tripping or stumbling while walking or running. There is muscle weakness, and atrophy spreads through other parts of the body which is indicative of lower motor neuron degeneration. A characteristic abnormal reflex action, Babinski’s sign (the large toe extends upward as the sole of the foot is stimulated in a certain way), is an indication of upper motor neuron damage. Thus, to confirm the diagnosis of ALS, patients must show signs and symptoms of both upper and lower motor neuron damages which cannot be confused with other disease symptoms. In the later part of disease, severe conditions were observed where patients find it difficult to stand, walk, use their hands and arms, and get in or out of bed on their own. There is a risk of choking due to the difficulty in chewing and swallowing. As ALS is mainly a motor neuron disease, the cognitive abilities of the patients remain relatively intact and as patients are aware of their progressive loss of function, they become anxious and depressed. In end stages of the disease, patients feel difficulty in breathing as the muscles of the respiratory system weaken and ultimately lose the ability to breathe on their own [89].
15.5.5 Treatment
15.5.5.1 Symptomatic Therapy
There is no cure for ALS to date, but a symptomatic treatment for glutamate-related motor neuron degeneration is available in the market. FDA approved riluzole (Rilutek) in 1995 which decreases the release of glutamate and thus reduces damage to motor neurons. This drug does not reverse the damage already done to motor neurons but slows down the damage further and helps the patients with swallowing difficulty. It also helps the patients to delay the ventilation support as the disease progresses. Scientists are trying to develop the combination therapy to further slow down the progression of ALS. Other symptoms like fatigue, muscle cramps, excess saliva and phlegm, pain, depression, sleep disturbances, and constipation can be treated with the help of available medications. Physical therapy, speech therapy, nutrition support, ventilators and Diaphragm Pacing System, respirators, home care, and hospice nurses can help patients to some extent, but it does not help in ALS progression.
15.5.5.2 Preventive Therapy
The understanding of molecular mechanisms of the ALS helps the scientist to develop the preventive therapy for ALS. Scientists are focusing on the molecular mechanisms of RNA molecules and recycling of proteins, impaired energy metabolism, hyperactivation of motor neurons, and degradation of glial cells. Scientists are working on developing gene/genome therapy to correct single or multiple mutations in the gene responsible for ALS. One of the important and most understood mutations responsible for ALS is antioxidant enzyme Cu/Zn superoxide dismutase 1 (SOD1). Recently researchers found TDP-43 and FUS protein aggregates in the spinal cord which are responsible for ALS, and the respective mutations were observed in chromosome 9 (C9orf72). Scientists used adeno-associated virus serotype 6 (AAV6) to deliver small hairpin RNAs (shRNAs) to knockdown mutant superoxide dismutase 1 (mSOD1) in mouse models, and the treatment showed neuroprotection and halted muscle atrophy in mouse [90]. Scientists also tested neuron protection action of expressed PRDX3 or NRF2 genes by transferring these genes to NSC34 cells lines (ALS tissue culture model expressing the human SOD1G93A mutation) using lentiviral vectors. The cell lines showed the overexpression of PRDX3 or NRF2 with 40 and 50% decrease in endogenous oxidation stress levels [91]. Some researchers used antisense oligonucleotides (ASOs) to bind to the antisense (GGCCCC) direction C9orf72 RNA foci which specifically reduced the expression C9orf72 and showed significant neuron protection action. In the same study, researchers also observed that siRNAs inserted in cells with viral vectors fail to reduce nuclear RNA foci but showed marked reduction in C9orf72 RNAs which is well tolerated by mice brain with no symptoms of ALS [92].
Conclusion
This chapter summarizes the pathophysiology, etiology, and latest symptomatic (drug) and preventive (gene and genomic) therapies for Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis. The etiology of the HDs suggested that there are multiple genes responsible for these conditions. The recent studies suggested that genomic treatment (multiple gene treatment) can reverse or at least stop further neuron degeneration and effectively provide the cure for different HDs. These treatments are site-specific with no or little side effects, suggesting genes can be ultimately be used as medicine. Thus it can be concluded that gene and genome therapy can provide effective treatment for HDs.
References
Bredesen DE, Rao RV, Mehlen P (2006) Cell death in the nervous system. Nature 443(7113):796–802
Thompson LM (2008) Neurodegeneration: a question of balance. Nature 452(7188):707–708
Rubinsztein DC (2008) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443(7113):780–786
Marsh JL, Lukacsovich T, Thompson LM (2009) Animal models of Polyglutamine diseases and therapeutic approaches. J Biol Chem 284(12):7431–7435
Zoghbi HY, Orr HT (2009) Pathogenic mechanisms of a Polyglutamine-mediated neurodegenerative disease, Spinocerebellar ataxia type 1. J Biol Chem 284(12):7425–7429
Coleman MP, Freeman MF (2010) Wallerian degeneration, WldS and Nmnat. Ann Rev Neurosci 33:245–267
De Vos KJ, Grierson AJ, Ackerley S, Miller CC (2008) Role of axonal transport in neurodegenerative diseases. Ann Rev Neurosci 31:151–173
DiMauro S, Schon EA (2008) Mitochondrial disorders in the nervous system. Ann Rev Neurosci 31:91–123
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443(7113):787–795
Martindale JL, Holbrook NJ (2002) Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 192(1):1–15
Tafani M, Karpinich NO, Hurster KA, Pastorino JG, Schneider T, Russo MA, Farber JL (2002) Cytochrome C release upon Fas receptor activation depends on translocation of full-length bid and the induction of the mitochondrial permeability transition. J Biol Chem 277(12):10073–10082
Engelberg-Kulka H, Amitai S, Kolodkin-Gal I, Hazan R (2006) Bacterial programmed cell death and multicellular behavior in bacteria. PLoS Genet 2(10):e135
Kimichi A, Kroemer G, Zalckvar E, Chiara MM (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8:741–752
Vila M, Przedbroski S (2003) Targeting programmed cell death in neurodegenerative diseases. Nat Rev Neurosci 4:1–11
Neurological disorders: public health challenges (2006) Report by World Health Organization, Geneva, Switzerland. http://www.who.int/mental_health/neurology/neurological_disorders_report_web.pdf
Alzheimer’s association (2015) Alzheimer’s disease facts and figures. Alzheimer’s Dementia 11(3):332–384
Huse DM, Schulman K, Orsini L, Castelli-Haley J, Kennedy S, Lenhart G (2005) Burden of illness in Parkinson’s disease. Mov Disord 20(11):1449–1454
Kandale VV, Mujawar SN, Welasly PJ, Nimbalkar JM (2013) Development of integrated database of neurodegenerative diseases (IDND). Rev Res 2(9):1–5
Kowal SL, Dall TM, Chakrabarti R, Storm MV, Jain A (2013) The current and projected economic burden of Parkinson’s disease in the United States. Mov Disord 28(3):311–318
Mathers CD, Loncar D (2005) Updated projections of global mortality and burden of disease, 2002–2030: data sources, methods and results. World Health Organization, Geneva. (Evidence and Information for Policy Working Paper). http://www.who.int/healthinfo/statistics/bod_projections2030_paper.pdf
Mathers CD, Salomon JA, Ezzati M, Begg S, Lopez AD (2006) Sensitivity and uncertainty analyses for burden of disease and risk factor estimates. In: Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJL (eds) Global burden of disease and risk factors. The World Bank, Oxford University Press, Washington, pp 399–426
Murray CJL, Lopez AD (1997) Alternative projections of mortality and disability by cause, 1990–2020: global burden of disease study. Lancet 349:1498–1504
The world health report (2004) Changing history. World Health Organization, Geneva
Azzouz M, Kingsman SM, Mazarakis ND (2004) Lentiviral vectors for treating and modeling human CNS disorders. J Gene Med 6(9):951–962
Nanou A, Azzouz M (2009) Gene therapy for neurodegenerative diseases based on lentiviral vectors. Prog Brain Res 175:187–200
Wong LF, Goodhead L, Prat C, Mitrophanous KA, Kingsman SM, Mazarakis ND (2006) Lentivirus-mediated gene transfer to the central nervous system: therapeutic and research applications. Hum Gene Ther 17(1):1–9
Geller G, Dvoskin R, Thio CL, Duggal P, Lewis MH, Bailey TC, Sutherland A, Salmon DA, Kahn JP (2014) Genomics and infectious disease: a call to identify the ethical, legal and social implications for public health and clinical practice. Genome Med 6(11):106–118
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E et al (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366(10):883–892
Tuteja S, Rader DJ (2012) Genomic medicine in the prevention and treatment of atherosclerotic cardiovascular disease. Pers Med 9(4):395–404
Karr JR, Sanghvi JC, Macklin DN, Gutschow MV, Jacobs JM, Bolival B Jr et al (2012) A whole-cell computational model predicts phenotype from genotype. Cell 150(2):389–401
Dancey JE, Bedard PL, Onetto N, Hudson TJ (2012) The genetic basis for cancer treatment decisions. Cell 148(3):409–420
National Institute of Neurological Disorders and Stroke. Dementia: hope through research. Bethesda, MD (2013) Office of Communications and Public Liaison, National Institute of Neurological Disorders and Stroke, US National Institutes of Health. Published online; version last updated June 26th, 2013, accessed November 1st, 2013
Alzheimer’s Association (2013) The role of plaques and tangles. Published online, accessed https://www.alz.org/braintour/plaques_tangles.asp. Accessed 01 Nov 2013
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Biol 3(9):a006189
Davinelli S, Intrieri M, Russo C, Di Costanzo A, Zella D, Bosco P, Giovanni S (2011) The “Alzheimer’s disease signature”: potential perspectives for novel biomarkers. Immune Ageing 20(8):7–17
Goldman JS, Hahn SE, Catania JW, LaRusse-Eckert S, Butson MB, Rumbaugh M, Strecker MN, Roberts JS, Burke W, Mayeux R, Bird T (2011) Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical Genetics and the National Society of genetic Counselors. Genet Med 13(6):597–605
Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, Craft S (2011) Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch Neurol 68(1):51–57
Miklossy J (2011) Emerging roles of pathogens in Alzheimer disease. Expert Rev Mol Med 13:e30
Magnoni S, Brody DL (2010) New perspectives on amyloid-beta dynamics after acute brain injury: moving between experimental approaches and studies in the human brain. Arch Neurol 67(9):1068–1073
Dubois B, Feldman HH, Jacova C, Dekosky ST, Barberger-Gateau P, Cummings J, Delacourte A, Galasko D, Gauthier S, Jicha G, al e (2007) Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet Neurol 6:734–746
Ballard CG, Gauthier S, Cummings JL, Brodaty H, Grossberg GT, Robert P, Lyketsos CG (2009) Management of agitation and aggression associated with Alzheimer disease. Nat Rev Neurol 5:245–255
Savva GM, Zaccai J, Matthews FE, Davidson JE, McKeith I, Brayne C (2009) Medical Research Council cognitive function and ageing study. Prevalence correlates and course of behavioural and psychological symptoms of dementia in the population. Br J Psychiatry 194:212–219
Galimberti G, Scarpini E (2013) Treatment of Alzheimer’s disease: symptomatic and disease-modifying approaches. Curr Aging Sci 3(1):46–56
Prevention and Risk of Alzheimer’s and Dementia (2015) Alzheimer’s Association, Chicago, IL, USA. http://www.alz.org/research/science/alzheimers_prevention_and_risk.asp
Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH (2009) Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med 15:331–337
Annunziata I, Patterson A, Helton D, Hu H, Moshiach S, Gomero E, Nixon R, d’Azzo R, Lysosomal NEU (2013) Deficiency affects amyloid precursor protein levels and amyloid-β secretion via deregulated lysosomal exocytosis. Nat Commun 4:2734
Minami SS, Min SM, Krabbe G, Wang C, Zhou Y, Asgarov R, Li Y, Martens LH, Elia LP, Ward ME, Mucke L, Farese Jr RV, Gan L (2014) Progranulin protects against amyloid β deposition and toxicity in Alzheimer’s disease mouse models. Nat Med 20:1157–1164
Parra-Damas A, Valero J, Chen M, España J, Martin E, Ferrer I, Rodríguez-Alvarez J, Saura CA (2014) Crtc1 activates a transcriptional program deregulated at early Alzheimer's disease-related stages. J Neurosci 34(17):5776–5787
Chinta S, Lieu C, Demaria M, Laberge R, Campisi J, Anderson J (2013) Environmental stress, ageing, and glial cell senescence: a novel mechanistic link to Parkinson’s disease. J Internal Med 273:429–436
Chou K (2013) Clinical manifestations of Parkinson Disease. Up-to-date. www.uptodate.com. Accessed 22 July 2013
Fritsch T, Smyth K, Wallendal M, Hyde T, Leo G, Geldmacher D (2012) Parkinson disease: research update and clinical management. South Med J 105(12):650–656
Gazewood J, Richards D, Clebak K (2013) Parkinson disease: an update. Am Family Phys 87(4):267–273
MacPhee G, Stewart D (2001) Parkinson’s disease. Rev Clin Gerontol 11:33–49
Parkinson’s Disease Foundation: Statistics on Parkinson’s (2013) Retrieved on 22 July 2013 from http://www.pdf.org/en/parkinson_statistics
Sherer TB, Chowdhury S, Peabody K, Brooks DW (2012) Overcoming obstacles in Parkinson's disease. Mov Disord 27(13):1606–1611
Jankovic J, Hurtig H, Dashe J (2013) Etiology and pathogenesis of Parkinson Disease. Up-to-date. www.uptodate.com. Accessed 22 July 2013
Postuma R, Gagnon J, Montplaisir J (2009) Clinical prediction of Parkinson’s disease: planning for the age of neuroprotection. J Neurol 81(9):1008–1013
Duda JE, Lee VM, Trojanowski JQ (2000) Neuropathology of synuclein aggregates. J Neurosci Res 61(2):121–127
Bendor JT, Logan TP, Edwards RH (2013) The function of α-Synuclein. Neuron 79(6):1044–1066
Beitz JM (2014) Parkinson’s disease: a review. Front Biosci S6:65–74
Hawkes C, Del K, Braak TH (2007) Review: Parkinson's disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol 33:599–614
Brown T, Rumsby P, Capleton A, Rushton L, Levy L (2006) Pesticides and Parkinson's disease: is there a link? Environ Health Persp 14(2):156–164
Ceccatelli S (2013) Mechanisms of neurotoxicity and implications for neurological disorders. J Internal Med 273:426–429
Connolly BS, Lang AE (2014) Pharmacological treatment of Parkinson disease - a review. JAMA 311(16):1670–1683
Olanow CW (2004) The scientific basis for the current treatment of parkinson’s disease. Annu Rev Med 55:41–60
Kim C, Han B, Moon J, Kim D, Shin J, Rajan S, Nguyen QT, Sohn M, Kim W, Han M et al (2015) Nuclear receptor Nurr1 agonists enhance its dual functions and improve behavioral deficits in an animal model of Parkinson’s disease. PNAS 112(28):8756–8761
Zharikov AD, Cannon JR, Tapias V, Bai Q, Horowitz MP, Shah V, Ayadi AE, Hastings TG, Greenamyre JT, Burton EA (2015) shRNA targeting α-synuclein prevents neurodegeneration in a Parkinson’s disease model. J Clin Investig 125(7):2721–2735
Palfi S, Gurruchaga JM, Ralph GS, Lepetit H, Lavisse S, Buttery PC, Watts C, Miskin J, Kelleher M, Deeley S et al (2014) Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson's disease: a dose escalation, open-label, phase 1/2 trial. Lancet 383(9923):1138–1146
Huntington G (1872) On chorea. Med Surg Report 26:320–321
Parikshak NN, Gandal MJ, Geschwind DH (2015) Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat Rev Genetics 16:441–458
Polanczyk G, de Lima MS, Horta BL, Biederman J, Rohde LA (2007) The worldwide prevalence of ADHD: a systematic review and Metaregression analysis. Am J Psychiatry 164(6):942–948
Schneider SA, Bhatia KP (2012) In: Weiner WJ, Tolosa E (eds) Chapter 5 – Huntington’s disease look-alikes in handbook of clinical neurology (hyperkinetic movement disorders), vol 100 (3rd. series). Elsevier, Amsterdam, pp 101–111
Paulson HL, Albin RL (2011a) In: Lo DC, Hughes RE (eds) Chapter 1 Huntington’s disease - clinical features and routes to therapy in neurobiology of Huntington's disease: applications to drug discovery. CRC Press, Boca Raton, pp 1–38
Shirasaki DI, Greiner ER, Al-Ramahi I, Gray M, Boontheung P, Geschwind DH, Botas J, Coppola G, Horvath S, Loo JA, Yang XW (2012) Network organization of the Huntington proteomic interactome in mammalian brain. Neuron 75:41–57
Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, Marder K, Penchaszadeh G, Roberts SA, Gayán J et al (2004) Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. PNAS 101(10):3498–3503
Wexler NS, Young AB, Tanzi RE, Travers H, Starosta-Rubinstein S, Penney JB, Snodgrass SR, Shoulson I, Gomez F, Ramos Arroyo MA et al (1987) Homozygotes for Huntington's disease. Nature 326(6109):194–197
Cleret de Langavant L, Fénelon G, Benisty S, Boissé MF, Jacquemot C, AC BL (2013) Awareness of memory deficits in early stage Huntington's disease. PLoS One 8(4):e61676
Ho A, Hocaoglu M (2011) Impact of Huntington's across the entire disease spectrum: the phases and stages of disease from the patient perspective. Clin Genet 80(3):235–239
Loy CT, McCusker EA (2013) Is a motor criterion essential for the diagnosis of clinical huntington disease? PLoS Curr 5. ecurrents.hd.f4c66bd51e8db11f55e1701af937a419. doi:https://doi.org/10.1371/currents.hd.f4c66bd51e8db11f55e1701af937a419
Nucifora FC Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Taka- hashi H, Tsuji S, Troncoso J, Dawson VL et al (2001) Interference by Huntington and atrophin1 with cbpmediated transcription leading to cellular toxicity. Science 291(5512):2423–2428
Revilla FJ, Grutzendler J, Larsh TR (2015) Huntington disease- background, pathophysiology, Etiology. In: Benbadis SR, Talavera F (eds) Medscape reference - drugs, diseases and procedures. Article 1150165
Beister A, Kraus P, Kuhn W, Dose M, Weindl A, Gerlach M (2004) The NmethylD aspartate antagonist Memantine retards progression of Huntington’s disease. J Neural Transm Suppl 68:117–122
de Tommaso M, Di Fruscolo O, Sciruicchio V, Specchio N, Livrea P (2007) Two years’ followup of Rivastigmine treatment in Huntington disease. Clin Neuropharmacol 30(1):43–46
Bonelli RM, Hofmann PA (2007) Systematic review of the treatment studies in Huntington’s disease since 1990. Expert Opin Pharmacother 8(2):141–153
Paulson HL, Albin RL (2011b) Chapter 1 Huntington’s disease - clinical features and routes to therapy. In Huntington’s disease neurobiology of Huntington's disease. CRC Press, Boca Raton, pp 1–35
Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G (2006) Cleavage at the caspase6 site is required for neuronal dysfunction and degeneration due to mutant Huntington. Cell 125(6):1179–1191
Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, White LA (2013) Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 41(2):118–130
Rossi FH, Franco MC, Estevez AG (2013) Chapter 1 - pathophysiology of amyotrophic lateral sclerosis in current advances in amyotrophic lateral sclerosis. Intech publisher, Rijeka, pp 1–34
Amyotrophic Lateral Sclerosis (ALS) - fact sheet (2013) U.S. department of health and human services, public health service, National Institutes of Health, NIH Publication No. 13 916, http://www.ninds.nih.gov/disorders/amyotrophiclateralsclerosis May 2013
Towne C, Setola V, Schneider BL, Aebischer P (2011) Neuroprotection by gene therapy targeting mutant SOD1 in individual pools of motor neurons does not translate into therapeutic benefit in fALS mice. Mol Ther 19(2):274–283
Nanou A, Higginbottom A, Valori CF, Wyles M, Ning K, Shaw P, Azzouz M (2013) Viral delivery of antioxidant genes as a therapeutic strategy in experimental models of amyotrophic lateral sclerosis. Mol Ther 21(8):1486–1496
Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, Qiu J et al (2013) Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. PNAS 110(47):E4530–E4539
Bergeron D, Lapointe C, Bissonnette C, Tremblay G, Motard J, Roucou X (2013) An out-of-frame overlapping reading frame in the ataxin-1 coding sequence encodes a novel ataxin-1 interacting protein. J Biol Chem 288(30):21824–21835
Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M et al (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299:256–259
Brun A (1987) Frontal lobe degeneration of nonAlzheimer type. I. Neuropathology. Arch Gerontol Geriatr 6:193–208
Burchell VS, Nelson DE, Sanchez-Martinez A, Delgado-Camprubi M, Ivatt RM, Pogson JH, Randle SJ, Wray S, Lewis PA, Houlden H et al (2013) The Parkinson’s disease-linked proteins Fbxo7 and parkin interact to mediate mitophagy. Nat Neurosci 16(9):1257–1265
Cardone F, Principe S, Schininà ME, Maras B, Capellari S, Parchi P, Notari S, Di Francesco L, Poleggi A, Galeno R et al (2014) Mutant PrPCJD prevails over wild-type PrPCJD in the brain of V210I and R208H genetic Creutzfeldt-Jakob disease patients. Biochem Biophys Res Commun 454(2):289–294
Chou AH, Chen YL, Hu SH, Chang YM, Wang HL (2014) Polyglutamine-expanded ataxin-3 impairs long-term depression in Purkinje neurons of SCA3 transgenic mouse by inhibiting HAT and impairing histone acetylation. Brain Res 1583:220–229
Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, Halloran MA, Gleeson PA, Blair IP, Soo KY, King AE, Atkin JD (2014) C9orf72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet 23(13):3579–3595
Forabosco P, Ramasamy A, Trabzuni D, Walker R, Smith C, Bras J, Levine A, Hardy J, Pocock JM, Guerreiro R et al (2013) Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol Aging 34:2699–2714
Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L et al (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349:704–706
Ingelsson M, Hyman BT (2002) Disordered proteins in dementia. Ann Med 34:259–271
Kato S, Shaw P, Wood-Allum C, Leigh PN, Shaw C (2003) Amyotrophic lateral sclerosis. In: Dickson D (ed) Neurodegeneration — the molecular pathology of dementia and movement disorders. ISN Neuropath Press, Basel, pp 350–368
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N, Kitada T (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K et al (1995) Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269:973–977
Morris HR, Baker M, Yasojima K, Houlden H, Khan MN, Wood NW, Hardy J, Grossman M, Trojanowski J, Revesz T et al (2002) Analysis of tau haplotypes in Pick’s disease. Neurology 59(3):443–445
Neuenschwander AG, Thai KK, Figueroa KP, Pulst SM (2014) Amyotrophic lateral sclerosis risk for spinocerebellar ataxia type 2 ATXN2 CAG repeat alleles: a meta-analysis. JAMA Neurol 71(12):1529–1534
Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM, Khan N et al (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44:595–600
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R et al (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T et al (1995) Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376:775–778
Savinkova L, Drachkova I, Arshinova T, Ponomarenko P, Ponomarenko M, Kolchanov N (2013) An experimental verification of the predicted effects of promoter TATA-box polymorphisms associated with human diseases on interactions between the TATA boxes and TATA-binding protein. PLoS One 8(2):e54626
Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD (1993) Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in lateonset Alzheimer disease. PNAS 90:9649–9653
Schneider SA, Marshall KE, Xiao J, LeDoux MS (2012) JPH3 repeat expansions cause a progressive akinetic-rigid syndrome with severe dementia and putaminal rim in a five-generation African-American family. Neurogenetics 13(2):133–140
Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K et al (1995) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375:754–760
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. PNAS 90:1977–1981
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG et al (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160
Yapijakis C, Gatzonis S, Youroukos S, Kollia V, Karachristianou S, Anagnostouli M (2014) Juvenile myoclonic epilepsy is not associated with the DRPLA gene in a European population. In Vivo 28(6):1193–1196
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ et al (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

{kind=link}
Cite this chapter
Badhe, R.V., Chejara, D.R., Kumar, P., Choonara, Y.E., Pillay, V. (2018). Neurodegenerative Disease Conditions and Genomic Treatment for Better Health. In: Pathak, Y. (eds) Genomics-Driven Healthcare. Adis, Singapore. https://doi.org/10.1007/978-981-10-7506-3_15
Download citation
DOI: https://doi.org/10.1007/978-981-10-7506-3_15
Published:
Publisher Name: Adis, Singapore
Print ISBN: 978-981-10-7505-6
Online ISBN: 978-981-10-7506-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)