
Abstract
Integrative structure modeling is an emerging method for structural determination of protein-protein complexes that are challenging for conventional structural techniques. Here, we provide a practical protocol for implementing our integrated iSPOT platform by integrating three different biophysical techniques: small-angle X-ray scattering (SAXS), hydroxyl radical footprinting, and computational docking simulations. Specifically, individual techniques are described from experimental and/or computational perspectives, and complementary structural information from these different techniques are integrated for accurate characterization of the structures of large protein-protein complexes.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- iSPOT
- Integrative structural biology
- SAXS
- Hydroxyl radical footprinting
- Computational docking simulation
- Structural mass spectrometry
- Protein-protein interaction
14.1 Introduction
Macromolecular interactions provide the molecular underpinning for virtually every biological process. Despite decades of effort, however, structure determination of protein-protein complexes is still a daunting task for conventional techniques due to size, stability, and/or complexity of protein complexes of interest. To advance the ability to characterize these complexes, we have recently established a multi-technique iSPOT platform by integrating small-angle X-ray scattering (SAXS), hydroxyl radical footprinting and computational docking simulations (Huang et al. 2016). iSPOT leverages the widespread availability of individual protein or domain structures and in particular enables the structure determination of complexes in the range of 50–200 kDa that are often challenging for nuclear magnetic resonance (too big) or electron microscopy (too small). Driven by its potential as an emerging technique towards large-scale applications, this iSPOT platform is described here to facilitate broad adoption.
The iSPOT platform overcomes the limitation of individual techniques and succeeds in combining multiple sources of structural information from different techniques that are complementary to each other. For example, computational docking benefits from its combination with experimental scattering/footprinting data, while molecular shape information from SAXS is complemented by solvent accessibility of specific protein sites probed by hydroxyl radical footprinting.
14.2 Implementation of the Integrated iSPOT Platform
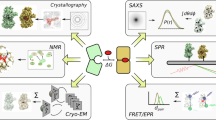
The entire iSPOT platform has three major sources of structural information for each protein-protein complex of interest: (1) molecular shape and structural arrangement from small-angle X-ray scattering (SAXS), (2) solvent accessibility of specific sites probed by hydroxyl radical footprinting, and (3) model prediction by computational protein-protein docking. Figure 14.1 outlines a schematic demonstrating the integration of three different, complementary biophysical techniques in the iSPOT platform.

Multi-technique iSPOT platform for integrated structural modeling of protein-protein complexes. iSPOT represents the integration of small-angle X-ray scattering (SAXS), hydroxyl radical footprinting, and computational docking simulation (Huang et al. 2016). iSPOT also takes advantage of available cystal structures of individual protein components within the complex
It is worth noting that while the integration of all three techniques is emphasized here, a combination of any two approaches can be utilized to generate structure ensembles for a specific question of interest, while the remaining data are used for a validation purpose if available. For this consideration, we describe each component of this iSPOT platform, followed by the integration of all three.
Figure 14.2 provides an overview of the iSPOT workflow. It is arbitrarily divided into four components: (a) computational protein-protein docking for generating structural candidates (or “poses”), (b) parallel SAXS and footprinting data acquisition, (c) candidate scoring against experimental data, and (d) selection and optimization of ensemble structures. A proof-of-principle demonstration of this iSPOT platform has been shown in an earlier publication on several protein-protein complexes with their crystal structures known (Huang et al. 2016). By using the atomic structures of individual proteins (not the complex), iSPOT is able to accurately predict the structures of a large protein-protein complex (TGFβ-FKBP12) and a multidomain nuclear receptor homodimer (HNF-4α), by using simulated SAXS and footprinting data of each complex.

The iSPOT workfolow. It consists of four compoments: (a) computational protein-protein docking, (b) experimental SAXS and footprinting data acquisition, (c) scoring and selection, and (d) structural model optimization
14.2.1 Computational Protein-Protein Docking
Computational studies of protein-protein interaction have been a long-term focus of research (Janin et al. 2003). Quite a few algorithms are now available for docking two proteins into a bound complex. As such, computationally docked conformations or “poses” can be evaluated and compared against experimental data (discussed later). Specifically, rigid-body and flexible docking are described below, as well as post-docking clustering analysis.
14.2.1.1 Rigid-Body Docking
Rigid-docking techniques have been successfully developed over the years (Chen et al. 2003; Dominguez et al. 2003; Gabb et al. 1997; Tovchigrechko and Vakser 2006). These docking algorithms, such as ClusPro (Comeau et al. 2004) and ZDock (Pierce et al. 2014), are computationally robust and efficient. For this reason, it is a good idea to try rigid-body docking as a first diagnostic step, or even use docking results for evaluating with experimental data if the proteins are relatively non-flexible upon binding. Notably, ZDock is particularly easy to use and provides a simple web interface (http://zdock.umassmed.edu), as well as executable files available for download.
14.2.1.2 Flexible Docking by RotPPR-CGMD Molecule Dynamics Simulation
To account for structural flexibility in protein-protein interaction, we have developed a molecular dynamics (MD) based docking method, termed RotPPR-CGMD (described below), which combines an exhaustive generation of initial poses and subsequent coarse-grained molecular dynamics simulations. This RotPPR-CGMD is composed of (a) conformational sampling by RotPPR and (b) coarse-grained (CG) simulation. The former is to make sure that the conformational space is properly and exhaustively searched; the latter is to use a one-bead-per-residue Cα model to simplify the protein representation as we have shown previously (Ravikumar et al. 2012; Yang et al. 2010a). A suite of source codes and executable files for the setup and configurations of RotPPR-CGMD simulations will be made available for this type of RotPPR-CGMD docking simulations.
Specially, the RotPPR sampling, a combination of a pull-push-release (PPR) strategy along the inter-protein translational axis and a rotational pose generator, collectively enables an extensive conformational sampling in the docking space (Huang et al. 2016). The translation-centric PPR sampling is achieved via a harmonic spring between the centers-of-mass of two proteins to facilitate the docking (Ravikumar et al. 2012), while the pose generator provides a set of different initial docking poses to account for all five rotational degrees-of-freedom (as illustrated in Fig. 14.3).

Computational protein-protein flexible docking. Shown are the six degrees of feedom (five rotational and one translational) involved in two-body protein docking that are extensively sampled by RotPPR-CGMD simulations (Modified with permission Huang et al. 2016)
The energy function used in RotPPR-CGMD simulations is a predictive coarse-grained Cα model, where interaction between two proteins is defined by residue-residue interactions whose parameters are tabulated in a previous publication (Huang et al. 2014). It is worth noting that although the structure of each protein is used for the modeling, it does not require structural knowledge of the entire complex (Ravikumar et al. 2012). Because of its coarse-grained nature, this CGMD is expected to significantly enhance the protein-protein docking, compared to atom-level simulations.
14.2.1.3 Structure Clustering
For post-docking data analysis, structure clustering of RotPPR-CGMD simulation data can be achieved on the basis of structural similarity via two specific metrics: fRMSD and oRMSD. The former is a regular RMSD measure of Cα atoms from the entire complex and the latter is an extension of fRMSD by accounting for the difference in relative orientation between two proteins (Huang et al. 2016). The resulting oRMSD clustering improves the structural ambiguity observed in traditional fRMSD clustering since the measure of oRMSD is more sensitive to protein-protein orientations. As a result, oRMSD clustering is able to group similar simulation-generated structures into one cluster or conformation that appear more homogenous than what was based on fRMSD clustering.
Another notable difference is the input parameter needed for clustering. Traditionally, the number of clusters is used as an input, while a RMSD cutoff value is used in the oRMSD clustering here. Overall, the oRMSD clustering is able to outline top structural candidates to explicitly account for the relative orientations between two proteins.
We have recently illustrated that RotPPR-CGMD is capable of searching various docking conformations (Huang et al. 2016), where the docking conformational space has been visited extensively. Thus, the RotPPR-CGMD provides an MD-based docking strategy to account for the structural flexibility for protein-protein docked conformations, ranging from compacted to extended shapes and from assembled to fully disassembled.
14.2.2 Small-Angle X-Ray Scattering (SAXS)
For characterizing protein-protein complexes, small-angle X-ray scattering (SAXS) data are particularly informative with regard to molecule shape of the entire complex and specifically, subcomponent arrangements. Quite a few excellent reviews have already discussed the basic principles and applications of SAXS (Bernado and Blackledge 2010; Blanchet and Svergun 2013; Kikhney and Svergun 2015; Putnam et al. 2007), and hence we describe the current state-of-the-art SAXS data acquisition and SAXS computing methods below.
14.2.2.1 Experimental SAXS Data Collection
While acquisition of reliable SAXS data is non-trivial, experimental procedures have been recently described in detail (Jeffries et al. 2016; Skou et al. 2014), in addition to what has been covered in this book. Here, we point out that it is becoming a standard option for SAXS data acquisition to use an online chromatography-coupled setup, as illustrated in Fig. 14.4. This chromatography-coupled setup is particularly useful for aggregation-prone samples to allow the separation of a target complex from larger aggregates and/or smaller, excess substrates and thus improve sample homogeneity.

Experimental SAXS data acquisition. Two setups are routinely used: one with a simple flow-cell (top) and the other coupled with online chromatography (bottom) (Modified with with permission Yang 2014)
14.2.2.2 SAXS Computing Methods
For the interpretation of experimental SAXS data, how to compute the SAXS profile from a given protein conformation, e.g. those generated from above RotPPR-CGMD simulations, is of particular importance because it is essentially the theoretical foundation of most SAXS data analyses.
CRYSOL and Fast-SAXS-pro are representative among currently available SAXS computing methods. Specially, CRYSOL requires the atomic coordinates (Svergun et al. 1995), while Fast-SAXS-pro takes the coordinates of either all atoms or just Cα atoms alone (Ravikumar et al. 2013). Additional differences include the treatment of excess electron density in a hydration layer by explicitly placing dummy water molecules surrounding the biomolecule. Comparison between these two methods is listed in Table 14.1. It should be noted that CRYSOL can be used for next-step optimization for iSPOT-derived atomic-structure ensembles since it provides an additional capability of best-fitting theoretical and experimental SAXS profiles.
Given its ability of handling the coordinates generated from RotPPR-CGMD docking simulations, Fast-SAXS-pro is thus used for SAXS computing to calculate theoretical scattering profiles, resulting from a collection of efforts (Ravikumar et al. 2013; Tong et al. 2016; Yang et al. 2009, 2010b). A web interface for Fast-SAXS-pro computing is available from the website at http://www.theyanglab.org/saxs.html, as well as executable files will be made available upon request.
14.2.3 Hydroxyl Radical Footprinting
Complementary to shape information obtained from SAXS is the solvent accessibility of specific sites probed by hydroxyl radical footprinting (Huang et al. 2015; Kaur et al. 2015; Xu and Chance 2007). The sites probed can be at the peptide level or at the single-residue level. As described below, specific rate constant measurements from footprinting are correlated to the solvent accessibility of probed amino acids, thereby providing structural information at a rather local residue-specific level.
14.2.3.1 Experimental Footprinting Rate Measurement
The rate constant measurements of probed sites each from a different protein region are illustrated in Fig. 14.5. Typically, irradiation of water by X-rays generates hydroxyl radicals (OH•) that react protein residues via covalent modification. These OH•-modified samples are analyzed via proteolysis and the level of modification or “footprinting” is quantified via mass spectrometry (MS). This MS quantification is normally conducted at a single time point of X-ray exposure or repeated at various time points. In the latter, a dose-response curve of footprinting can be determined for each probed site, thereby establishing a footprinting rate k fp to characterize the overall footprinting effect on each individual site.

Site-specific rate measurement from hydroxyl radical (OH•) footprinting. Following a–f, different regions of a protein are covalently modified by OH• generated from X-ray irradiation of water, which is subsequently quantified by mass spectrometry. A dose-response measurement yields a kinetic rate constant for each site probed (Modified with permission Huang et al. 2015)
14.2.3.2 Protection Factor Analysis and Structural Parameters
To use the footprinting rates k fp for structural characterization, we have established a protection factor (PF) analysis method (Huang et al. 2015; Kaur et al. 2015). This PF analysis can be applied at a single-residue or a peptide level. For example, PFs for single residues (or multiple residues within a peptide) are calculated by dividing the intrinsic reactivity k intrinsic of the residue (or the sum of the intrinsic reactivity for all of the residues within the peptide) by the observed rate k fp ,
This simple conversion to PF values provides structural interpretation of footprinting measurements, enabling for the first time a structural comparison between different amino acid types that were previously impossible because footprinting rates alone are not correlated to any known structural properties. A key advantage of this PF analysis is absolute comparison between different sites that are probed simultaneously within an intact protein, as opposed to the previously limited comparison of a singular site crossing different conformational states. Specially, high-PF regions are structurally buried, while low-PF regions are solvent-exposed.
The PF data are correlated with structural features/parameters of protein sites probed. This is typically examined on a case-by-case basis partially due to the extent of footprinting being dependent on the protein sequence composition and its 3D structure. A list of structural parameters that reflect the related solvent accessibility are solvent accessible surface area (SASA), number of structural contacts (NC), and even the simple binary measure of being exposed or buried. These structural parameters are compared with experimental PF values to quantitatively evaluate the agreement between a protein structure candidate and its corresponding experimental footprinting data.
The intrinsic reactivity data can be from the website at http://www.theyanglab.org/protection.html. This weblet also provides the rate-PF conversion for single-residue footprinting data.
14.2.4 Data Integration by iSPOT
The multi-technique iSPOT platform is a result of these developments made in computational docking, SAXS and footprinting (illustrated in Fig. 14.1). These techniques are different but complementary, so the integration enabled by iSPOT provides a novel approach for structure determination of previously uncharacterized protein-protein complexes. Following the iSPOT workshop described in Fig. 14.2, we here show that each docking pose is used for evaluation against experimental SAXS and footprinting data via two specific scoring functions χ 2 and φ 2 as detailed below.
14.2.4.1 The Goodness of Fit to SAXS Data χ 2
For each docked pose (or conformational cluster), the goodness of fit between the theoretical (I cal) and experimental (I exp) SAXS profiles is scored by a unitless χ 2 (Yang et al. 2010a),
where σ(q) is the uncertainty of logI exp(q) and N is the number of data points in I exp(q). Theoretical SAXS profiles I cal (q) can be calculated from the docking configuration by either Fast-SAXS-pro or CRYSOL as described earlier. Specifically, a lower χ 2 value represents a better fit between theoretical and experimental SAXS data. For example, χ 2 often approaches 1–3 when experimental and theoretical SAXS profiles start to agree well.
14.2.4.2 The Goodness of Fit to Footprinting Data φ 2
For the same docked pose, the goodness of fit between experimental footprinting PFs and structural parameters is scored by another unitless φ 2 (Huang et al. 2016),
where log(PF i ) is the protection factor of each site i probed by footprinting (either at a single-residue or peptide level) (Huang et al. 2015; Kaur et al. 2015), δ i is the uncertainty of logPF i , and N fp is the total number of probed sites. As aforementioned, a list of structural parameters of solvent accessibility SA i include solvent accessible surface area (SASA) and number of neighboring contacts (NC). The scaling constant of c is to offset the linear fitting between SA and logPF. Similar to χ 2, here φ 2 is the difference between experimental footprinting PFs and theoretical solvent accessibility of each docked conformation. For example, a lower φ 2 value indicates a better fit of the candidate toward the target structure.
14.2.4.3 iSPOT Model Selection and Refinement
The best-fit structural models that are selected by iSPOT are among the lowest χ 2 and φ 2 values. This selection is illustrated in Fig. 14.2, where the orthogonal information provided by SAXS (about overall shape) and footprinting (about local solvent accessibility) is able to accurately select the crystal-like ensemble structures of a large complex. By testing on several protein-protein complexes with known structures, we have showed that the iSPOT is able to narrow down the correct target structure of bound complexes such as TGFβ-FKBP12 (Huang et al. 2016).
Refinement of the iSPOT-derived structure models of a protein-protein complex can be achieved by force-field based molecular dynamics (MD) simulations. Based on the atomic coordinates of individual protein components of the complex, a realistic structure of the complex can be constructed for all-atom, explicit-solvent MD simulations, as illustrated in the bottom of Fig. 14.2. As such, iSPOT is able to generate atomic structure ensembles of protein-protein complexes that can be further tested for model validation.
14.3 Summary
Structure determination of protein-protein complexes has been a challenging task. The multi-technique iSPOT platform is therefore a niche method available to structurally characterize such biomolecular complexes that are in the range of 50–200 kDa, although the method will work well for complexes of any size. We should stress that compared to other structural techniques that are quite matured or currently in their prime time, the development and application of iSPOT is still at its infancy. This early-stage technology development thus provides a critical step for future iSPOT applications to many biologically and biomedically important protein complexes.
References
Bernado P, Blackledge M (2010) Structural biology: proteins in dynamic equilibrium. Nature 468:1046–1048
Blanchet CE, Svergun DI (2013) Small-angle X-ray scattering on biological macromolecules and nanocomposites in solution. Annu Rev Phys Chem 64:37–54
Chen R, Li L, Weng Z (2003) ZDOCK: an initial-stage protein-docking algorithm. Proteins 52:80–87
Comeau SR, Gatchell DW, Vajda S, Camacho CJ (2004) ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res 32:W96–W99
Dominguez C, Boelens R, Bonvin AM (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc 125:1731–1737
Gabb HA, Jackson RM, Sternberg MJE (1997) Modelling protein docking using shape complementarity, electrostatics and biochemical information. J Mol Biol 272:106–120
Huang W, Ravikumar KM, Yang S (2014) A newfound cancer-activating mutation reshapes the energy landscape of estrogen-binding domain. J Chem Theory Comput 10:2897–2900
Huang W, Ravikumar KM, Chance MR, Yang S (2015) Quantitative mapping of protein structure by hydroxyl radical footprinting mediated structural mass spectrometry: a protection factor analysis. Biophys J 108:1–9
Huang W, Ravikumar KM, Parisien M, Yang S (2016) Theoretical modeling of multiprotein complexes by iSPOT: integration of small-angle X-ray scattering, hydroxyl radical footprinting, and computational docking. J Struct Biol 196:340
Janin J, Henrick K, Moult J, Eyck LT, Sternberg MJ, Vajda S, Vakser I, Wodak SJ, Critical Assessment of, P.I (2003) CAPRI: a critical assessment of predicted interactions. Proteins 52:2–9
Jeffries CM, Graewert MA, Blanchet CE, Langley DB, Whitten AE, Svergun DI (2016) Preparing monodisperse macromolecular samples for successful biological small-angle X-ray and neutron-scattering experiments. Nat Protoc 11:2122–2153
Kaur P, Kiselar J, Yang S, Chance MR (2015) Quantitative protein topography analysis and high-resolution structure prediction using hydroxyl radical labeling and tandem-ion mass spectrometry. Mol Cell Proteomics (in press)
Kikhney AG, Svergun DI (2015) A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett 589:2570–2577
Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z (2014) ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 30:1771–1773
Putnam CD, Hammel M, Hura GL, Tainer JA (2007) X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys 40:191–285
Ravikumar KM, Huang W, Yang S (2012) Coarse-grained simulations of protein-protein association: an energy landscape perspective. Biophys J 103:837–845
Ravikumar KM, Huang W, Yang S (2013) Fast-SAXS-pro: a unified approach to computing SAXS profiles of DNA, RNA, protein, and their complexes. J Chem Phys 138:024112
Skou S, Gillilan RE, Ando N (2014) Synchrotron-based small-angle X-ray scattering of proteins in solution. Nat Protoc 9:1727–1739
Svergun D, Barberato C, Koch MHJ (1995) CRYSOL – a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr 28:768–773
Tong D, Yang S, Lu L (2016) Accurate optimization of amino acid form factors for computing small-angle X-ray scattering intensity of atomistic protein structures. J Appl Crystallogr 49:1148–1161
Tovchigrechko A, Vakser IA (2006) GRAMM-X public web server for protein-protein docking. Nucleic Acids Res 34:W310–W314
Xu G, Chance MR (2007) Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem Rev 107:3514–3543
Yang S (2014) Methods for SAXS-based structure determination of biomolecular complexes. Adv Mater 26:7902–7910
Yang S, Park S, Makowski L, Roux B (2009) A rapid coarse residue-based computational method for x-ray solution scattering characterization of protein folds and multiple conformational states of large protein complexes. Biophys J 96:4449–4463
Yang S, Blachowicz L, Makowski L, Roux B (2010a) Multidomain assembled states of Hck tyrosine kinase in solution. Proc Natl Acad Sci U S A 107:15757–15762
Yang S, Parisien M, Major F, Roux B (2010b) RNA structure determination using SAXS data. J Phys Chem B 114:10039–10048
Acknowledgements
This work was supported by the NIH (R01GM114056 and P30EB009998), the DoD (W81XWH-11-1033), and by the Ministry of Education of Singapore (2014-T2-1-065). Beamtime access was supported via the BioCAT at the Advanced Photo Source and the LiX beamline at the NSLS-II by the DoE (DE-AC02-06CH11357 and KP1605010) and by the NIH (9P41GM103622 and P41GM111244). Additional support was provided by the Ohio Supercomputer Center and the Clinical and Translational Science Collaborative of Cleveland (4UL1TR000439). The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Hsieh, A., Lu, L., Chance, M.R., Yang, S. (2017). A Practical Guide to iSPOT Modeling: An Integrative Structural Biology Platform. In: Chaudhuri, B., Muñoz, I., Qian, S., Urban, V. (eds) Biological Small Angle Scattering: Techniques, Strategies and Tips. Advances in Experimental Medicine and Biology, vol 1009. Springer, Singapore. https://doi.org/10.1007/978-981-10-6038-0_14
Download citation
DOI: https://doi.org/10.1007/978-981-10-6038-0_14
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-6037-3
Online ISBN: 978-981-10-6038-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)