
Abstract
Human T-cell leukemia virus type 1 (HTLV-1) is the first retrovirus discovered to cause adult T-cell leukemia (ATL), a highly aggressive blood cancer. HTLV-1 research in the past 35 years has been most revealing in the mechanisms of viral oncogenesis. HTLV-1 establishes a lifelong persistent infection in CD4+ T lymphocytes. The infection outcome is governed by host immunity. ATL develops in 2–5% of infected individuals 30–50 years after initial exposure. HTLV-1 encodes two oncoproteins Tax and HBZ, which are required for initiation of cellular transformation and maintenance of cell proliferation, respectively. HTLV-1 oncogenesis is driven by a clonal selection and expansion process during which both host and viral factors cooperate to impair genome stability, immune surveillance, and other mechanisms of tumor suppression. A better understanding of HTLV-1 biology and leukemogenesis will reveal new strategies and modalities for ATL prevention and treatment.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
9.1 Introduction
Human T-cell leukemia virus type 1 (HTLV-1) was discovered in 1980 as the first human retrovirus and the etiological agent of adult T-cell leukemia (ATL) [1, 2]. Since then HTLV-1 research has laid the foundation of viral oncology and human retrovirology [3]. Animal oncogenic retroviruses such as Rous sarcoma virus are replication defective, and they carry viral oncogenes that are originally derived from cellular proto-oncogenes of their host. Unlike these animal retroviruses, HTLV-1 is replication-competent and does not carry any cellular oncogenes. The viral oncoproteins encoded by HTLV-1 are unique, have no cellular counterparts, and are not homologous to any cellular proteins. HTLV-1 has been under intense investigations since HTLV-1 infection causes significant morbidity and mortality in endemic areas. In addition, it also serves as an excellent model for the study of viral oncogenesis. HTLV-1 research has contributed substantially to our understanding of oncogenic viruses and oncogenesis in general.
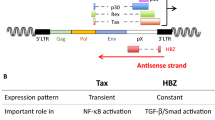
HTLV-1 is a complex deltaretrovirus that harbors additional regulatory genes in addition to gag, pol, pro, and env genes flanked by long terminal repeats (LTR) as found in simple retroviruses (Fig. 9.1). The gag gene encodes the major component of the viral capsid. The pol and pro region provides the reverse transcriptase, protease, and integrase. Interestingly, gag and pol are produced by ribosome frameshift from a single transcript. The env gene codes for a glycoprotein that mediates viral entry. The pX region between env and the 3′-LTR encodes Tax, Rex, as well as other accessory proteins p12, p13, p21, and p30 derived from alternatively spliced transcripts. Tax is a viral transactivator that potently activates transcription from the LTR. Rex mediates nuclear export of viral RNA. The additional accessory proteins are dispensable for viral replication and transformation in vitro but are required for viral propagation and persistence in vivo. In particular, p30 counteracts Toll-like receptor signaling and cooperates with c-Myc to promote cellular transformation [4, 5]. p12 and its cleavage product p8 mediate T-cell activation, immune evasion, and cell-to-cell transmission [6, 7]. Distinct to other retroviruses, HTLV-1 also expresses an antisense transcript encoding the helix-basic loop zipper protein HBZ [8, 9]. Tax and HBZ are two viral oncoproteins that cooperate to drive HTLV-1 leukemogenesis. A full discussion of this will be provided below in part 4 of this review.

HTLV-1 genome organization. The structural genes gag, pro, pol, and env as well as the regulatory genes tax, rex, p21, p12, p13, p30, and HBZ are shown
HTLV-1 has a relative known as HTLV-2. Although they are similar in genome organization and tissue tropism, there is one important difference in pathogenesis: human infection with HTLV-2 is not associated with any malignancy. HTLV-2 and its proteins are therefore commonly used as controls in the study of HTLV-1 oncogenesis. More recently, two new HTLV viruses named HTLV-3 and HTLV-4 have been isolated from Cameroonian hunters of nonhuman primates [10, 11]. Primate counterparts of all four HTLVs have also been identified, and these four pairs of viruses, together with bovine leukemia virus (BLV) and another orphan primate retrovirus, constitute the genus of deltaretroviruses [11, 12]. Whereas infection with HTLV-1 and BLV is associated with leukemia, but HTLV-2 infection is not, it remains to be seen whether HTLV-3 and HTLV-4 are also leukemogenic.
ATL is a heterogeneous disease with four clinical subtypes: acute, lymphoma, chronic, and smoldering. Acute, lymphoma, and unfavorable chronic subtypes are known as aggressive ATL with large tumor burden, blood and lymph node involvement, and hypercalcemia. Favorable chronic and smoldering subtypes are indolent ATL characterized by rash and minimal blood involvement [13]. Smoldering ATL is considered to be an early phase of the disease, which progresses subsequently to acute ATL. Prognosis of aggressive ATL is very poor, with an average survival rate of only a few months.
The mechanisms of HTLV-1 oncogenesis have recently been reviewed [3, 13,14,15,16]. In this chapter, we will revisit the topic with an emphasis on new thoughts and findings. We will start with an overview of epidemiology, followed by a brief summary of experimental models in HTLV-1 research. The interplay of the two HTLV-1 oncoproteins and the mechanisms of HTLV-1 oncogenesis will then be discussed in detail. Finally, we will highlight the treatment options and the new approaches to anti-HTLV-1 therapy.
9.2 Epidemiology
Based on sequence and epidemiological analysis, the primate counterparts of HTLVs are well established in their natural hosts. Several lines of evidence including phylogenetic clustering and geographical coincidence support zoonotic transmission of these primate viruses to humans, plausibly through petting and butchering. The process by which HTLVs establish as a human pathogen through adaptive mutations is similar to that demonstrated for human immunodeficiency viruses. The detection of HTLV-3 and HTLV-4 in African hunters of primates has lent further support to this notion [10, 11]. HTLV-1 and HTLV-2 have obviously acquired the ability to transmit from human to human readily. The identification of HTLV-3 and HTLV-4 has provided a golden opportunity to study their human-to-human transmissibility, pathogenicity, and degree of adaptation. Interestingly, each HTLV has its own primate counterpart. Phylogenetic analysis supports at least four independent introductions of virus into human population. Each of this involves a different species of primate virus.
It is estimated that about 20 million people are infected with HTLV-1 worldwide. About one million of them are in Japan, and the adjusted overall prevalence nationwide is approximately 1% [17]. However, the distribution of HTLV-1 carriers within the country is uneven and highly focal. As such, carrier rate in Japanese women in the age of >50 in endemic areas can be as high as 40%, but these areas are surrounded by areas of low to middle prevalence. HTLV-1 is also highly prevalent in African people resided in the Caribbean islands and tropical Africa; Mongoloid people in South America, Central America, and the Middle East; as well as Melanesian people in northern Oceania [18]. Particularly, some Aboriginal Australians have been found to have the highest prevalence (>50%) of HTLV-1. Interestingly, the study of HTLV-1 prevalence in different populations might even provide useful anthropological information concerning their origin, migration routes, and genetic history.
The prevalence of HTLV-1 in healthy blood donors in China is very low, ranging from 0.01 to 0.08% [19, 20]. The rate could be 0.5% or higher in some professional blood donors and drug addicts. Only some of the Chinese individuals who are seropositive for HTLV-1 have been found to have close contact with Japanese people [21]. Interestingly, prevalence rates of HTLV-1 in healthy blood donors in two cities named Ningde and Putian in the coastal Fujian Province are 0.40% and 0.14%, respectively. These significantly higher rates are indicative of some foci of HTLV-1 carriers [19]. The seropositive rate of HTLV-1 of 0.74% in the ethnic group of Xinjiang Uyghurs is also high. A very small number of sporadic tropical spastic paraparesis (TSP) and ATL cases associated with HTLV-1 infection have also been diagnosed in China in recent years. Several sporadic cases of ATL have been found in Hong Kong, where a more advanced disease surveillance system is in place [22]. Some of these victims in Hong Kong had unsafe sex in Southern Japan. The carrier rate in Hong Kong is estimated to be 0.0041%. In Taiwan, the seropositive rate for HTLV-1 was found to range from to 0.058 to 0.48% [23, 24]. Interestingly, the rates in Aborigines and Hakka Taiwanese are higher than in other ethnic groups. Blood donor screening for HTLV-1 has been implemented in Taiwan since 1996. It will be of some interest to determine whether the Taiwanese HTLV-1 strains are closer to those found in Japan or Fujian, which is geographically closer to Taiwan.
Although HTLV-1 can be divided into six genotypes A to F, sequence variations among genotypes are minor and not as significant as in HIV-1. The sequence divergence among HTLV-1 genotypes is much less than that among different HTLV viruses. Genotype A is predominant. The genotype of HTLV-1 in ATL patients and healthy carriers is not found to be different. Neither is there evidence in support of the influence of genotype on pathogenicity or infection outcome.
HTLV-1 is an infection vertically transmitted from mother to child through breastfeeding. The risk of infection acquired through this route can be as high as 30%. Blood transfusion and unsafe sex are two other routes by which HTLV-1 is transmitted, but sexual transmission is not as efficient as in the case of HIV-1. More than 90% of HTLV-1-infected people remain healthy throughout their lifetime. ATL develops in 2–5% of infected individuals after a prolonged latent period of 30–50 years, during which they remain asymptomatic. Once developed, ATL is highly aggressive and fatal, with very limited and unsatisfactory treatment options [25]. A smaller subset of infected people suffers from TSP or HTLV-1-associated myelopathy, a chronic debilitating neurological disease of the spinal cord. It is not common that TSP and ATL develop sequentially in the same individual. During the long process of ATL development, multiple viral, host, and environmental factors are involved. Notably, high HTLV-1 proviral load is the single major risk factor for ATL development in HTLV-1 carriers. Other reported risk factors include advanced age, family history of ATL, male sex, and HTLV-1 infection early in life [26]. Most ATL cases are associated with breastfeeding. The cumulative risks of developing ATL among HTLV-1 carriers are approximately 6% for males and 2% for females. The high predictive value of proviral load suggests that anti-HTLV-1 therapy might be beneficial in the prevention of ATL.
CD4+ T lymphocytes are the primary and preferential target cells of HTLV-1 in vivo, although other cells such as CD8+ T lymphocytes, monocytes, and dendritic cells (DCs) can also be infected. It remains to be clarified whether HTLV-1 might first infect DCs, which pass on the virus to T cells [27]. One recent report has implicated HTLV-1-transformed CD45RA+ T memory stem cells with stemlike properties as the ATL-initiating cells [28]. These cells could serve as the viral reservoir and a barrier for viral eradication by antivirals. Cell-free transmission of HTLV-1 is highly inefficient except for DCs. All major routes of HTLV-1 transmission including breastfeeding, blood transfusion, and sexual intercourse involve the transfer of infected cells residing in the breast milk, blood, and semen. Cell-to-cell transmission of HTLV-1 is achieved through the virological synapse, which involves the interaction between ICAM-1 on infected cells and LFA-1 on target cells and polarization of the microtubule-organizing center induced by Tax protein [29, 30]. Interestingly, virions at the virological synapse are stored as biofilm-like extracellular assemblies [31]. In addition to cell-to-cell contact, HTLV-1 can also be passed on to daughter cells via mitosis. Thus, the HTLV-1 proviral load in vivo might be determined by both mitotic spread and cell-to-cell transmission. In this connection, it will be of great interest to see how the interaction between Tax and mitotic regulators such as MAD1 [32] might influence both processes. In addition, the HTLV-1 proviral load is also affected by host immune response and particularly by cytotoxic T lymphocyte (CTL) response against viral proteins such as Tax and HBZ [33]. Thus, a better understanding of anti-HTLV-1 CTL response might reveal new strategies for prevention of ATL.
9.3 Experimental Models in HTLV-1 Research
Mechanistic studies of HTLV-1 pathogenesis have largely been conducted in transfected cells in which the HTLV-1 protein of interest is overexpressed. Although these cells provide a good model for the study of HTLV-1 proteins, there are several concerns about their relevance to HTLV-1 infection and biology. First, the expression level of the HTLV-1 protein of interest in transfected cells might be much higher than in infected cells. Second, constitutive expression of the HTLV-1 protein of interest could exhaust or sequester its partners and effectors leading to a squelching effect. Regulated or inducible expression is desired. For example, JPX9 cells, in which Tax expression can be induced by Cd2+ [34], have proved useful in HTLV-1 research. Third, target cells of HTLV-1 are difficult to transfect. Surrogate models such as HeLa and HEK293 cells with high transfection efficiency are helpful, but they are significantly different from CD4+ T cells in many ways. The concern might be addressed by the use of new transfection reagents tailor-made for T cells. Finally, different HTLV-1 proteins interact with each other to fulfill some functions cooperatively or antagonistically. This might not be reconstituted in transfected cells. Tens if not hundreds of cellular binding partners of HTLV-1 proteins such as Tax and HBZ have been identified. Not all of them have been validated in infected cells. Interpretation of these interaction results should be cautious, bearing in mind the limitations of the transfection system. Whenever possible, T-cell lines such as Jurkat and CEMT4 as well as peripheral blood mononuclear cells (PBMCs) infected with HTLV-1 should be used to verify findings obtained from transfected cells.
Various types of cultured cells including T cells, B cells, DCs, monocytes, endothelial cells, and fibroblasts can be infected in vitro with HTLV-1 through coculture with HTLV-1-infected cells such as MT2 and C8166. These freshly infected cells serve as a good model for acute infection. In contrast, other ATL or derivative cell lines are chronically infected with and transformed by HTLV-1. For example, MT4 and HUT102 cells were derived from ATL patients. MT2 cells were established through coculture of normal cord leukocytes with ATL cells. C8166 cells were obtained by fusion of normal cord leukocytes with ATL cells. Whereas these several lines constitutively express Tax, other lines in which Tax expression has faded but HBZ expression remains robust include ED and TL-Om1 [35, 36]. These cells representative of different phases of infection are widely used in HTLV-1 research. In addition to infection through coculture, infectious molecular clones of HTLV-1 are also available [37]. These clones can be transfected into any cells and spawn HTLV-1 infection in susceptible cells. They greatly facilitate genetic analysis of HTLV-1.
BLV and HTLV-1 share many features in common. Both are transmitted through body fluids requiring cell-to-cell contact. Both are leukemogenic in only a fraction of infected hosts after a prolonged latent period. Thus, BLV serves as a good and relevant model for HTLV-1 research [38]. Particularly, promising results on the prevention of BLV-associated diseases through competitive infection with an attenuated BLV provirus provide useful information as to how proviral load can be reduced with this strategy [39]. BLV can infect several ruminant species with highest prevalence in dairy cattle. Infection of small ruminants such as sheep with BLV has therefore been developed as a productive animal model for both BLV and HTLV-1 research. Sheep can be easily infected and the disease outcome can be observed sooner [40]. By the same reasoning, infection of monkeys such as Japanese macaques with STLV-1 provides useful information about HTLV-1 pathogenesis [41].
Various types of tumor develop in Tax- or HBZ-transgenic mice, but in most cases these are neither leukemia nor lymphoma [42, 43]. Directing the expression of Tax or HBZ more specifically to particular tissues and cells such as thymus and leukocytes is a technical challenge that has only been met partially. Tissue-specific promoters such as those of CD4, CD3ε, Ig, Lck, and granzyme B have been used with some success in the generation of more relevant disease outcomes. Although these transgenic mice are not perfect models for HTLV-1 infection or ATL development, they provide convincing evidence for the oncogenicity of Tax and HBZ proteins, reveal different facets of HTLV-1 oncogenesis, and also serve as platforms for the development of new therapy for ATL. Complementary to transgenic mice, infection of immunocompetent rabbits with HTLV-1 provides another model [38]. However, no disease or symptom related to ATL or TSP can be recapitulated in rabbits.
Many features of ATL can be reproduced in immunocompromised mice engrafted with ATL cells or ATL-derived cell lines [44]. These mouse xenograft models have been used to study HTLV-1 leukemogenesis and to develop anti-HTLV-1 therapy. The immunocompromised mice that have been developed include SCID, NOD-SCID, NSG, NOG, and BRG mice [45]. SCID mice contain a nonsense mutation in the protein kinase required for VDJ recombination of T- and B-cell receptors, leading to a severe combined immunodeficiency (SCID). In NOD-SCID mice, the SCID mutation has been introduced into the nonobese diabetic (NOD) genetic background. This further compromises innate immunity by blocking the function of complements, DCs, and macrophages. Similar to X-linked SCID in human, deficiency in the interleukin-2 receptor common subunit γ (IL2R-γC) in mice results in a complete loss of T, B, and NK cells. In NSG and NOG mice, this mutation in IL2R-γC has been introduced into the NOD/SCID background. Likewise, BRG mice are deficient for IL2R-γC and the recombinase-activating gene 2 (Rag2).
Humanization of immunocompromised mice by reconstituting their immune system through engraftment of human hematopoietic stem cells has not only opened the door for detailed analysis of human immunity but also provided a powerful new tool for the study of human pathogens including HTLV-1 [46]. The mice are engrafted with CD34+ hematopoietic stem cells from human peripheral and cord blood. Because all CD4+ T lymphocytes in these mice are derived from the engrafted human cells, they are excellent models for lymphotropic viruses such as HTLV-1 [47, 48]. These models have already been used successfully to study HTLV-1 infection and oncogenesis [49, 50]. For example, CD4+ T-cell lymphoma was shown to develop in NOD-SCID mice engrafted with human CD34+ cells infected with HTLV-1 [50]. Although the original paper reporting this finding was later retracted by the editors and it remains to be determined whether HTLV-1 infects CD34+ cells, which subsequently differentiated into CD4+ cells, humanized mice still hold great promises to advance HTLV-1 and ATL research with a biologically relevant model. One challenge in this area is to develop a good model for persistent HTLV-1 infection in humanized mice.
9.4 HTLV-1 Oncoproteins
HTLV-1 encodes two major oncoproteins Tax and HBZ. In this part we will first describe existing findings on Tax and HBZ essentially in chronological order. Then we will summarize how they exert their impacts on the hallmarks of cancer. Finally we will discuss their differential roles in HTLV-1 leukemogenesis.
Tax is a 40-kDa transactivator protein serving as the master regulator of HTLV-1 proviral expression from the LTR. To activate HTLV-1 transcription, Tax forms a homodimer to engage CREB and DNA of three cAMP-response element-like 21-bp repeats in the LTR [51,52,53,54]. Tax has a minimal transactivation domain [55]. Optimal activity of Tax specifically requires the core TATAA promoter of HTLV-1, CREB, and the 21-bp repeats [56]. Transcriptional coactivators including p300/CREB-binding protein (CBP) and CREB-regulating transcriptional coactivators (CRTCs) are then attracted by Tax [52, 57, 58]. Tax also recruits other regulators and protein modification enzymes to modulate this process [59]. For example, p21-activated kinases are recruited to activate LTR-dependent transcription [60], whereas LKB1 and salt-inducible kinases [61], protein deacetylases SIRT1 [62], as well as T cell-specific transcription factors TCF1 and LEF1 [63] are recruited to medicate negative regulation of proviral transcription.
In addition to CREB, NF-κB is another major cellular transcription factor activated by Tax [16, 64]. Tax interacts with NF-κB regulators such as IκB kinase regulatory subunit IKK-γ [65,66,67], ubiquitin-editing enzyme A20 [68, 69], ubiquitin-binding adaptor protein TAX1BP1 [70, 71], E2 ubiquitin-conjugating enzyme UBC13 [72], and E3 ubiquitin ligase RNF8 [73] to modulate NF-κB activation through the ubiquitin-proteasome pathway. Notably, Tax is a powerful modulator of K48-linked, K63-linked, and linear ubiquitination of key adaptors of NF-κB signaling such as IKK-γ and TAB2 [73, 74]. Furthermore, Tax can also interact with and stimulate SRF [75, 76] and c-Jun [73, 77] transcription factors resulting in the activation of transcription from serum response elements and AP-1-binding sites.
It is generally accepted that Tax is required for the initiation of HTLV-1-mediated malignant transformation. Expression of Tax alone can sufficiently transform murine fibroblasts [78], immortalize T lymphocytes [79], and induce tumor formation in nude mice and transgenic mice [42, 43, 80]. Through CREB and NF-κB, Tax activates a wide variety of cellular genes that contribute to transformation. Activation of both CREB and NF-κB signaling is required for full-blown transformation induced by Tax [81, 82].
Tax is a multifunctional protein that activates transcription and transformation primarily through protein-protein interaction [59]. Tax is known to interact with a subset of PDZ domain-containing proteins. For example, Tax interacts with TIP1 [83], PDLIM2 [84], and MAGI1 [85] that contain PDZ domains. Another group of Tax-binding proteins contains the coiled-coil motifs that mediate their interaction with Tax [86]. Proteins in this group include mitotic checkpoint protein MAD1 [32], transcriptional repressor GPS2 [87], regulatory subunit IKK-γ of IκB kinase [65,66,67], centrosomal and ciliary protein TAX1BP2 [88], transcriptional coactivators CRTC1/CRTC2/CRTC3 [57, 58], as well as ubiquitin-binding adaptor protein TAX1BP1 [70, 71]. Tax-binding proteins in both groups are the effectors of Tax in transcriptional regulation and transformation.
HTLV-1 expresses both unspliced and spliced forms of HBZ, with the latter form being more abundant in infected cells [9]. The expression of HBZ appears to be required for HTLV-1 infectivity in vivo [89, 90]. Interestingly, HBZ RNA and protein show differential activity on apoptosis, but both promote cell cycle progression into S phase [87]. HBZ protein was initially identified as a heterodimerization partner of ATF4 [8]. In most cases dimerization of HBZ with ATF4, CREB, c-Jun, and other bZIP transcription factors results in repression of their activity. Thus, HBZ is a negative regulator of proviral transcription and it counteracts the activity of Tax. In addition, HBZ suppresses canonical pathway of NF-κB activation. Collectively, HBZ plays an important role in the proliferation of infected T cells as well as the induction and maintenance of latent infection [3, 16, 89,90,91,92,93].
The hallmarks of cancer include self-sufficiency in growth signal, insensitivity to antigrowth signals, resisting cell death, enabling replicative immortality, evading immune surveillance, genome instability and mutation, as well as tumor-promoting inflammation [94]. Although Tax and HBZ are antagonistic in many scenarios, they cooperate with each other to impinge on the different hallmarks of cancer. Some key examples are summarized below. HBZ activates Wnt signaling to sustain T-cell proliferation [95]. Tax perturbs tumor suppressor function of p53 [96] and Rb [97, 98]. HBZ suppresses apoptosis by targeting FoxO3a that activates proapoptotic genes [99]. Tax suppresses innate antiviral response by preventing TBK1-induced type I interferon production [100]. HBZ induces the expression of immune checkpoint molecule TIGIT to evade T-cell response [101]. Tax impairs DNA damage response [100,101,102,103,104,105], mitotic checkpoint [32], and centrosome duplication [88] leading to genome instability and a mutator phenotype. HBZ activates the transcription of hTERT to elevate telomerase activity [106]. Whereas HBZ enhances transforming growth factor-β signaling leading to overproduction of IFN-γ [43, 107, 108], Tax activates NF-κB to induce various cytokines [64]. Both result in activation of pro-inflammatory response.
Tax and HBZ play different roles in HTLV-1 oncogenesis. Whereas Tax is required for the initiation of oncogenic transformation, HBZ is essential for the induction and maintenance of HTLV-1 persistence and T-cell proliferation. Consistent with this model, Tax is abundantly expressed in the early stage of infection and transformation, but its expression and activity are suppressed through multiple mechanisms. First, promoter hypermethylation occurs in the 5′-LTR, leading to inhibition of Tax gene transcription [109]. Second, deletions and inactivating mutations are commonly found in the 5′-LTR and Tax coding region in the HTLV-1 genome in ATL cells [3]. Third, Tax recruits a group of inhibitors of proviral transcription such as LKB1, SIRT1, TCF1, and LEF1 through a negative feedback loop [61,62,63]. Last but not least, the strong CTL response directed against Tax essentially selects for T cells with low or no expression of Tax [33, 110]. In contrast, HBZ is constitutively expressed in all stages of infection and transformation. The differential activation of 5′- and 3′-LTR in the HTLV-1 genome is governed by chromatin insulator CTCF and the CTCF-binding site in the pX region [111].
The expression of Tax and HBZ in infected individuals is highly dynamic. There exist a large number of HTLV-1+ clones, each of which is characterized by a unique integration site of the HTLV-1 provirus in the host genome [112]. The pattern and level of Tax and HBZ expression could vary from one to another clone. They might even be passed on to the daughter cells through mitosis. In particular, Tax expression is known to be influenced by the distance and transcriptional direction of the provirus relative to the host gene in the closest vicinity. Importantly, the abundance of Tax- and HBZ-expressing cells is also governed by the CTL response. HBZ is less immunogenic than Tax [110], but CTL response targeting HBZ can potently suppress T-cell proliferation and has protective effect [113]. As mentioned above, the CTL response confers a survival advantage to HTLV-1+ clones that do not express Tax [33, 110]. From another perspective, HTLV-1-infected cells express cyclin-dependent kinase inhibitors p21 and p27 to high levels and enter cellular senescence in an NF-κB-dependent manner [110]. Cells in which Tax is abundantly expressed, NF-κB activity is high, and HTLV-1 replication is robust would be eliminated by apoptosis. Only latently infected cells with high HBZ expression and low NF-κB activation would survive [16, 114].
9.5 Mechanisms of HTLV-1 Oncogenesis
The long latency period of ATL development indicates that HTLV-1 oncogenesis is a slow and multistage process. Above we describe with examples the impact of Tax and HBZ oncoproteins on the different hallmarks of cancer. The subversion of genome instability, the evasion from immune response, and the induction of pro-inflammatory response are particularly attractive mechanisms that warrant further investigations. These mechanisms are critically important and they contribute to different stages of HTLV-1 oncogenesis. However, we should also bear in mind that ATL does not develop overnight, and it is the collective effect of Tax and HBZ over several decades that ultimately gives rise to ATL. Currently there is no consensus model that could fully explain the process of HTLV-1 oncogenesis.
Although insertional mutagenesis is a widely accepted mechanism for retroviral oncogenesis, how this applies in the case of HTLV-1 remains to be clarified. Integration site analysis in asymptomatic carriers and ATL patients indicates nonrandom insertion of HTLV-1 provirus into the host genome, with a preference for transcriptional start sites and CpG islands. Although no integration hot spots are found in ATL, a strong bias toward certain binding sites for transcription factors such as STAT1 and p53 is seen [115]. Because HTLV-1 contributes a CTCF site with the potential of forming a chromatin loop with another CTCF site in the host genome [111], it might modify host chromatin structure both in the vicinity and over a long distance, leading to aberrant activation of proto-oncogene, which confers a growth and survival advantage in natural selection. This model could explain why ATL development is a rare accident that occurs only in a small subset of HTLV-1-infected subjects. Further investigations are required to elucidate whether and how CTCF-mediated DNA looping might contribute to HTLV-1 oncogenesis.
Analysis of HTLV-1 clonality by deep sequencing reveals the difference in the frequency distribution of HTLV-1-infected T-cell clones in asymptomatic carriers and ATL patients [112]. In an asymptomatic carrier, there are about 10,000 lower-abundance clones. In a TSP patient, the number increases to about 30,000. These clones contribute substantially to the HTLV-1 proviral load, which is the major risk factor for TSP and also ATL. That is to say, the total number of clones but not the degree of oligoclonal expansion is influential in ATL development. Tax expression is more common in the lower-abundance clones than their high-abundance counterparts. Whether ATL arises from these lower-abundance clones is an issue of debate. Evidence in support of this model comes from integration site analysis and comparison with HTLV-2 [112, 116].
ATL cells are aneuploid and exhibit a mutator phenotype. Various genetic mutations have been found to accumulate in ATL cells, many of which are known to affect NF-κB activation [82]. For example, mutations in CARD11, PRKCB, and PLCG1 are thought to be critical in the activation of NF-κB signaling [117]. Plausibly, some of these mutations might serve as the second or third hit to drive full development of ATL. In light of the requirement of CREB signaling in HTLV-1 oncogenesis, it will not be too surprising if some of the genetic mutations might also be found in the future to have an impact on CREB activation. For instance, mutations of E3 ubiquitin ligase FBW7 have been found to affect Notch signaling [118]. FBW7 is a well-characterized tumor suppressor gene. It will be of interest to see whether these mutations might affect other pathways critically involved in tumor suppression. Through its Tax and HBZ oncoproteins, HTLV-1 can also induce alterations in epigenetic regulators, promoter methylation profiles, and microRNA expression patterns [82, 119]. In this regard, it will be of great importance to determine to what extent the epigenetic and genetic alterations in ATL cells would contribute to leukemogenesis. A working model for HTLV-1 oncogenesis that has incorporated some of the points mentioned above is presented in Fig. 9.2.

Working model for ATL development. The roles of clonal expansion, genomic instability, and anti-Tax CTL are highlighted. Shown below are the roles of Tax and HBZ on the hallmarks of cancer. At the initial phase, there exists a wide variation in Tax and HBZ expression in HTLV-1-infected T cells. Although some ATL cells might express Tax, abrogation of Tax expression is more common in ATL cells
9.6 Treatment of ATL
Treatment options for ATL are very limited and unsatisfactory [25, 120, 121]. Indolent ATL can be managed by watchful waiting until disease progression. However, this strategy has been found to result in an even poorer long-term outcome. An alternative treatment for indolent ATL uses a combination of zidovudine and interferon-α (AZT/IFN-α). This treatment has been shown to be effective for ATL [122, 123] and has been successfully used in the USA and Europe, but the mechanism of action remains unclear. Whether antiviral or cytotoxic effect is more important needs to be clarified. It is possible that the antiviral effect of AZT relieves viral suppression of IFN signaling, which has immunomodulatory and proapoptotic effect. The AZT/IFN-α therapy is not recommended in Japan pending the final result from an ongoing clinical trial. For patients with aggressive ATL, AZT/IFN-α or intensive chemotherapy is the first-line treatment. Whereas the outcome of AZT/IFN-α treatment is good for leukemia-type ATL [123], chemotherapy is reserved for lymphoma-type ATL. In addition to AZT/IFN-α and chemotherapy, allogeneic hematopoietic stem cell transplantation (allo-HSCT) is a potentially curative option for aggressive ATL. Using unrelated bone marrow and umbilical cord blood as alternative donor source in allo-HSCT has been successful in Japan. Notably, the antitumor effect of allo-HSCT provides the proof of principle for novel immunotherapy of ATL, including immune checkpoint therapy.
An anti-CCR4 monoclonal antibody is a novel targeted therapy for ATL [124]. CCR4 is selectively expressed in regulatory T cells and T helper type 2 cells. It is found in most ATL cells and its expression is induced by HBZ [125]. CCR4 expression is an indicator of poor prognosis [126]. Thus, anti-CCR4 can selectively eliminate ATL cells primarily through antibody-dependent cell-mediated cytotoxicity. Identification of new biomarkers that can be used to select patients who will benefit most from anti-CCR4 antibody might be the next challenge. In addition to anti-CCR4, an antibody against CD25, the α-subunit of IL2R, has also been tested for targeted therapy of ATL [127].
9.7 Concluding Remarks
More than 35 years have passed since the discovery of HTLV-1. Research findings in the field have not only advanced our understanding of HTLV-1 biology and oncogenesis but also provided new strategies and modalities in the management of ATL. A group of international experts in the field has formed a task force under the Global Virus Network and suggested priorities and open questions in HTLV-1 research [128]. Below I would echo their five suggestions as the concluding remarks of this chapter. First, global prevalence of HTLV-1 infection should be reviewed to identify opportunities and means to expand epidemiological studies [17]. A method to reduce mother-to-child transmission by breastfeeding in low-income countries should be developed. Second, biomarkers to predict disease progression should be identified. Searching for driver mutations through deep sequencing [117] should be continued and their clinicopathological significance determined. Third, preventive and therapeutic vaccines should be developed. Fourth, existing drugs should be screened and novel drugs should be developed to improve therapy [25, 120, 121]. Last but not least, basic research should be strengthened. Unraveling mechanisms of viral replication, persistence, and pathogenesis will open insights into novel treatments. This includes studies on HTLV-1 oncoproteins Tax and HBZ that promote viral replication and persistence [14, 16], viral entry, infectious and mitotic cycles of replication, genetic and epigenetic mechanisms that underlie ATL [82], the role of host immunity in the control of HTLV-1 infection [33], as well as HTLV-2, HTLV-3, and HTLV-4 pathogenesis [12]. We are optimistic that better answers to many of these open questions will be obtained in the near future.
References
Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC (1980) Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U S A 77:7415–7419
Yoshida M, Miyoshi I, Hinuma Y (1982) Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc Natl Acad Sci U S A 79:2031–2035
Matsuoka M, Jeang KT (2011) Human T-cell leukemia virus type 1 (HTLV-1) and leukemic transformation: viral infectivity, Tax, HBZ and therapy. Oncogene 30:1379–1389
Fenizia C, Fiocchi M, Jones K, Parks RW, Ceribelli M, Chevalier SA, Edwards D, Ruscetti F, Pise-Masison CA, Franchini G (2014) Human T-cell leukemia/lymphoma virus type 1 p30, but not p12/p8, counteracts toll-like receptor 3 (TLR3) and TLR4 signaling in human monocytes and dendritic cells. J Virol 88:393–402
Romeo MM, Ko B, Kim J, Brady R, Heatley HC, He J, Harrod CK, Barnett B, Ratner L, Lairmore MD, Martinez E, Lüscher B, Robson CN, Henriksson M, Harrod R (2015) Acetylation of the c-MYC oncoprotein is required for cooperation with the HTLV-1 p30(II) accessory protein and the induction of oncogenic cellular transformation by p30(II)/c-MYC. Virology 476:271–288
Pise-Masison CA, de Castro-Amarante MF, Enose-Akahata Y, Buchmann RC, Fenizia C, Washington Parks R, Edwards D, Fiocchi M, Alcantara LC Jr, Bialuk I, Graham J, Walser JC, McKinnon K, Galvão-Castro B, Gessain A, Venzon D, Jacobson S, Franchini G (2014) Co-dependence of HTLV-1 p12 and p8 functions in virus persistence. PLoS Pathog 10:e1004454
Edwards D, Fukumoto R, de Castro-Amarante MF, Alcantara LC, Galvão-Castro B, Washington Parks R, Pise-Masison C, Franchini G (2014) Palmitoylation and p8-mediated human T-cell leukemia virus type 1 transmission. J Virol 88:2319–2322
Gaudray G, Gachon F, Basbous J, Biard-Piechaczyk M, Devaux C, Mesnard JM (2002) The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol 76:12813–12822
Satou Y, Yasunaga J, Yoshida M, Matsuoka M (2006) HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci U S A 103:720–725
Wolfe ND, Heneine W, Carr JK, Garcia AD, Shanmugam V, Tamoufe U, Torimiro JN, Prosser AT, Lebreton M, Mpoudi-Ngole E, McCutchan FE, Birx DL, Folks TM, Burke DS, Switzer WM (2005) Emergence of unique primate T-lymphotropic viruses among central African bushmeat hunters. Proc Natl Acad Sci U S A 102:7994–7999
Richard L, Mouinga-Ondémé A, Betsem E, Filippone C, Nerrienet E, Kazanji M, Gessain A (2016) Zoonotic transmission of two new strains of human T-lymphotropic virus type 4 in hunters bitten by a gorilla in central Africa. Clin Infect Dis 63:800–803
Gessain A, Rua R, Betsem E, Turpin J, Mahieux R (2013) HTLV-3/4 and simian foamy retroviruses in humans: discovery, epidemiology, cross-species transmission and molecular virology. Virology 435:187–199
Panfil AR, Martinez MP, Ratner L, Green PL (2016) Human T-cell leukemia virus-associated malignancy. Curr Opin Virol 20:40–46
Matsuoka M, Yasunaga J (2013) Human T-cell leukemia virus type 1: replication, proliferation and propagation by Tax and HTLV-1 bZIP factor. Curr Opin Virol 3:684–691
Bangham CR, Ratner L (2015) How does HTLV-1 cause adult T-cell leukaemia/lymphoma (ATL)? Curr Opin Virol 14:93–100
Giam CZ, Semmes OJ (2016) HTLV-1 infection and adult T-cell leukemia/lymphoma-a tale of two proteins: Tax and HBZ. Virus 8:E161
Satake M, Yamaguchi K, Tadokoro K (2012) Current prevalence of HTLV-1 in Japan as determined by screening of blood donors. J Med Virol 84:327–335
Murphy EL (2016) Infection with human T-lymphotropic virus types-1 and -2 (HTLV-1 and -2): implications for blood transfusion safety. Transfus Clin Biol 23:13–19
Xie J, Ge S, Zhang Y, Lin Y, Ni H, Zhang J, Chen C (2015) The prevalence of human T-lymphotropic virus infection among blood donors in southeast China, 2004–2013. PLoS Negl Trop Dis 9:e0003685
Du J, Chen C, Gao J, Xie J, Rong X, Xu X, Wang Y, Wang F, Li J, Lu Z, Guo W, Li G, Wang Z, Xu D, Weng J, Zhao Z, Weng W, Li H, Du Y, Li S, Zhen C, Liu B, Guo T (2014) History and update of HTLV infection in China. Virus Res 191:134–137
Zeng Y, Lan X, Wang B, Fan J, Chen W, Yang T, Liang J, Xu X, Wang Y, Sui Y, Hu R, Hinuma Y (1985) Seroepidemiological studies on human T-cell leukemia antibody in China. Chin J Virol 1:344–348
Au WY, Lo JY (2005) HTLV-1-related lymphoma in Hong Kong Chinese. Am J Hematol 78:80–81
Wang CH, Chen CJ, Hu CY, You SL, Chu CT, Chou MJ, Essex M, Blattner WA, Liu CH, Yang CS (1988) Seroepidemiology of human T-cell lymphotropic virus type I infection in Taiwan. Cancer Res 48:5042–5044
Lu SC, Kao CL, Chin LT, Chen JW, Yang CM, Chang JH, Hsu SC, Chang AC, Chen BH (2001) Seroprevalence and demographic characteristics of HTLV-I among blood donors in Taiwan: 1996–1999. Int J Hematol 74:333–337
Cook LB, Taylor GP (2015) Treatment of adult T-cell leukaemia/lymphoma: is the virus a target? Curr Opin Infect Dis 28:583–588
Iwanaga M, Watanabe T, Utsunomiya A, Okayama A, Uchimaru K, Koh KR, Ogata M, Kikuchi H, Sagara Y, Uozumi K, Mochizuki M, Tsukasaki K, Saburi Y, Yamamura M, Tanaka J, Moriuchi Y, Hino S, Kamihira S, Yamaguchi K (2010) Human T-cell leukemia virus type I (HTLV-1) proviral load and disease progression in asymptomatic HTLV-1 carriers: a nationwide prospective study in Japan. Blood 116:1211–1219
Dutartre H, Clavière M, Journo C, Mahieux R (2016) Cell-free versus cell-to-cell infection by human immunodeficiency virus type 1 and human T-lymphotropic virus type 1: exploring the link among viral source, viral trafficking, and viral replication. J Virol 90:7607–7617
Nagai Y, Kawahara M, Hishizawa M, Shimazu Y, Sugino N, Fujii S, Kadowaki N, Takaori-Kondo A (2015) T memory stem cells are the hierarchical apex of adult T-cell leukemia. Blood 125:3527–3535
Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, Griffiths GM, Tanaka Y, Osame M, Bangham CR (2003) Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299:1713–1716
Nejmeddine M, Bangham CR (2010) The HTLV-1 virological synapse. Virus 2:1427–1447
Pais-Correia AM, Sachse M, Guadagnini S, Robbiati V, Lasserre R, Gessain A, Gout O, Alcover A, Thoulouze MI (2010) Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat Med 16:83–89
Jin DY, Spencer F, Jeang KT (1998) Human T cell leukemia virus type 1 oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell 93:81–91
Bangham CR (2009) CTL quality and the control of human retroviral infections. Eur J Immunol 39:1700–1712
Nagata K, Ohtani K, Nakamura M, Sugamura K (1989) Activation of endogenous c-fos proto-oncogene expression by human T-cell leukemia virus type I-encoded p40lax protein in the human T cell line, Jurkat. J Virol 63:3220–3226
Tagaya Y, Taniguchi Y, Naramura M, Okada M, Suzuki N, Kanamori H, Nikaido T, Honjo T, Yodoi J (1987) Transcription of IL-2 receptor gene is stimulated by ATL-derived factor produced by HTLV-I+ T cell lines. Immunol Lett 15:221–228
Sugamura K, Fujii M, Kannagi M, Sakitani M, Takeuchi M, Hinuma Y (1984) Cell surface phenotypes and expression of viral antigens of various human cell lines carrying human T-cell leukemia virus. Int J Cancer 34:221–228
Derse D, Mikovits J, Ruscetti F (1997) X-I and X-II open reading frames of HTLV-I are not required for virus replication or for immortalization of primary T-cells in vitro. Virology 237:123–128
Lairmore MD (2014) Animal models of bovine leukemia virus and human T-lymphotrophic virus type-1: insights in transmission and pathogenesis. Ann Rev Anim Biosci 2:189–208
Rodríguez SM, Florins A, Gillet N, de Brogniez A, Sánchez-Alcaraz MT, Boxus M, Boulanger F, Gutiérrez G, Trono K, Alvarez I, Vagnoni L, Willems L (2011) Preventive and therapeutic strategies for bovine leukemia virus: lessons for HTLV. Virus 3:1210–1248
Barez PY, de Brogniez A, Carpentier A, Gazon H, Gillet N, Gutiérrez G, Hamaidia M, Jacques JR, Perike S, Neelature Sriramareddy S, Renotte N, Staumont B, Reichert M, Trono K, Willems L (2015) Recent advances in BLV research. Virus 7:6080–6088
Miura M, Yasunaga J, Tanabe J, Sugata K, Zhao T, Ma G, Miyazato P, Ohshima K, Kaneko A, Watanabe A, Saito A, Akari H, Matsuoka M (2013) Characterization of simian T-cell leukemia virus type 1 in naturally infected Japanese macaques as a model of HTLV-1 infection. Retrovirology 10:118
Hasegawa H, Sawa H, Lewis MJ, Orba Y, Sheehy N, Yamamoto Y, Ichinohe T, Katano H, Tsunetsugu-Yokota Y, Takahashi H, Matsuda J, Sata T, Kurata T, Nagashima K, Hall WW (2006) Thymus-derived leukemia-lymphoma in mice transgenic for the Tax gene of human T-lymphotropic virus type I. Nat Med 12:466–472
Satou Y, Yasunaga J, Zhao T, Yoshida M, Miyazato P, Takai K, Shimizu K, Ohshima K, Green PL, Ohkura N, Yamaguchi T, Ono M, Sakaguchi S, Matsuoka M (2011) HTLV-1 bZIP factor induces T-cell lymphoma and systemic inflammation in vivo. PLoS Pathog 7:e1001274
Zimmerman B, Niewiesk S, Lairmore MD (2010) Mouse models of human T lymphotropic virus type-1-associated adult T-cell leukemia/lymphoma. Vet Pathol 47:677–689
Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL (2012) Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol 12:786–798
Legrand N, Ploss A, Balling R, Becker PD, Borsotti C, Brezillon N, Debarry J, de Jong Y, Deng H, Di Santo JP, Eisenbarth S, Eynon E, Flavell RA, Guzman CA, Huntington ND, Kremsdorf D, Manns MP, Manz MG, Mention JJ, Ott M, Rathinam C, Rice CM, Rongvaux A, Stevens S, Spits H, Strick-Marchand H, Takizawa H, van Lent AU, Wang C, Weijer K, Willinger T, Ziegler P (2009) Humanized mice for modeling human infectious disease: challenges, progress, and outlook. Cell Host Microbe 6:5–9
Marsden MD, Zack JA (2015) Studies of retroviral infection in humanized mice. Virology 479–480:297–309
Pérès E, Bagdassarian E, This S, Villaudy J, Rigal D, Gazzolo L, Duc Dodon M (2015) From immunodeficiency to humanization: the contribution of mouse models to explore HTLV-1 leukemogenesis. Virus 7:6371–6386
Miyazato P, Yasunaga J, Taniguchi Y, Koyanagi Y, Mitsuya H, Matsuoka M (2006) De novo human T-cell leukemia virus type 1 infection of human lymphocytes in NOD-SCID, common γ-chain knockout mice. J Virol 80:10683–10691
Banerjee P, Crawford L, Samuelson E, Feuer G (2010) Hematopoietic stem cells and retroviral infection. Retrovirology 7:8
Tie F, Adya N, Greene WC, Giam CZ (1996) Interaction of the human T-lymphotropic virus type 1 Tax dimer with CREB and the viral 21-base-pair repeat. J Virol 70:8368–8374
Kwok RP, Laurance ME, Lundblad JR, Goldman PS, Shih HM, Connor LM, Marriott SJ, Goodman RH (1996) Control of cAMP-regulated enhancers by the viral transactivator Tax through CREB and the co-activator CBP. Nature 380:642–646
Jin DY, Jeang KT (1997) HTLV-I Tax self-association in optimal trans-activation function. Nucleic Acids Res 25:79–87
Baranger AM, Palmer CR, Hamm MK, Giebler HA, Brauweiler A, Nyborg JK, Schepartz A (1995) Mechanism of DNA-binding enhancement by the human T-cell leukaemia virus transactivator Tax. Nature 376:606–608
Semmes OJ, Jeang KT (1995) Definition of a minimal activation domain in human T-cell leukemia virus type I Tax. J Virol 69:1827–1833
Ching YP, Chun ACS, Chin KT, Zhang ZQ, Jeang KT, Jin DY (2004) Specific TATAA and bZIP requirements suggest that HTLV-1 Tax has transcriptional activity subsequent to the assembly of an initiation complex. Retrovirology 1:18
Koga H, Ohshima T, Shimotohno K (2004) Enhanced activation of Tax-dependent transcription of human T-cell leukemia virus type I (HTLV-I) long terminal repeat by TORC3. J Biol Chem 279:52978–52983
Siu YT, Chin KT, Siu KL, Choy EYW, Jeang KT, Jin DY (2006) TORC1 and TORC2 coactivators are required for Tax activation of the human T-cell leukemia virus type 1 long terminal repeats. J Virol 80:7052–7059
Simonis N, Rual JF, Lemmens I, Boxus M, Hirozane-Kishikawa T, Gatot JS, Dricot A, Hao T, Vertommen D, Legros S, Daakour S, Klitgord N, Martin M, Willaert JF, Dequiedt F, Navratil V, Cusick ME, Burny A, Van Lint C, Hill DE, Tavernier J, Kettmann R, Vidal M, Twizere JC (2012) Host-pathogen interactome mapping for HTLV-1 and -2 retroviruses. Retrovirology 9:26
Chan CP, Siu YT, Kok KH, Ching YP, Tang HMV, Jin DY (2013) Group I p21-activated kinases facilitate Tax-mediated transcriptional activation of the human T-cell leukemia virus type 1 long terminal repeats. Retrovirology 10:47
Tang HMV, Gao WW, Chan CP, Siu YT, Wong CM, Kok KH, Ching YP, Takemori H, Jin DY (2013) LKB1 tumor suppressor and salt-inducible kinases negatively regulate human T-cell leukemia virus type 1 transcription. Retrovirology 10:40
Tang HMV, Gao WW, Chan CP, Cheng Y, Deng JJ, Yuen KS, Iha H, Jin DY (2015) SIRT1 suppresses human T-cell leukemia virus type 1 transcription. J Virol 89:8623–8631
Ma G, Yasunaga J, Akari H, Matsuoka M (2015) TCF1 and LEF1 act as T-cell intrinsic HTLV-1 antagonists by targeting Tax. Proc Natl Acad Sci U S A 112:2216–2221
Chan JK, Greene WC (2012) Dynamic roles for NF-κB in HTLV-I and HIV-1 retroviral pathogenesis. Immunol Rev 246:286–310
Jin DY, Giordano V, Kibler KV, Nakano H, Jeang KT (1999) Role of adapter function in oncoprotein-mediated activation of NF-κB. Human T-cell leukemia virus type I Tax interacts directly with IκB kinase γ. J Biol Chem 274 17402–17405
Harhaj EW, Sun SC (1999) IKKγ serves as a docking subunit of the IκB kinase (IKK) and mediates interaction of IKK with the human T-cell leukemia virus Tax protein. J Biol Chem 274:22911–22914
Chu ZL, Shin YA, Yang JM, DiDonato JA, Ballard DW (1999) IKKγ mediates the interaction of cellular IκB kinases with the Tax transforming protein of human T cell leukemia virus type 1. J Biol Chem 274:15297–15300
De Valck D, Jin DY, Heyninck K, Van de Craen M, Contreras R, Fiers W, Jeang KT, Beyaert R (1999) The zinc finger protein A20 interacts with a novel anti-apoptotic protein which is cleaved by specific caspases. Oncogene 18:4182–4190
Pujari R, Hunte R, Thomas R, van der Weyden L, Rauch D, Ratner L, Nyborg JK, Ramos JC, Takai Y, Shembade N (2015) Human T-cell leukemia virus type 1 (HTLV-1) Tax requires CADM1/TSLC1 for inactivation of the NF-κB inhibitor A20 and constitutive NF-κB signaling. PLoS Pathog 11:e1004721
Chin KT, Chun AC, Ching YP, Jeang KT, Jin DY (2007) Human T-cell leukemia virus oncoprotein Tax represses nuclear receptor-dependent transcription by targeting coactivator TAX1BP1. Cancer Res 67:1072–1081
Journo C, Filipe J, About F, Chevalier SA, Afonso PV, Brady JN, Flynn D, Tangy F, Israël A, Vidalain PO, Mahieux R, Weil R (2009) NRP/Optineurin cooperates with TAX1BP1 to potentiate the activation of NF-κB by human T-lymphotropic virus type 1 Tax protein. PLoS Pathog 5:e1000521
Shembade N, Harhaj NS, Yamamoto M, Akira S, Harhaj EW (2007) The human T-cell leukemia virus type 1 Tax oncoprotein requires the ubiquitin-conjugating enzyme Ubc13 for NF-κB activation. J Virol 81:13735–13742
Ho YK, Zhi H, Bowlin T, Dorjbal B, Philip S, Zahoor MA, Shih HM, Semmes OJ, Schaefer B, Glover JN, Giam CZ (2015) HTLV-1 Tax stimulates ubiquitin E3 ligase, ring finger protein 8, to assemble lysine 63-linked polyubiquitin chains for TAK1 and IKK activation. PLoS Pathog 11:e1005102
Lavorgna A, Harhaj EW (2014) Regulation of HTLV-1 Tax stability, cellular trafficking and NF-κB activation by the ubiquitin-proteasome pathway. Virus 6:3925–3943
Fujii M, Tsuchiya H, Chuhjo T, Akizawa T, Seiki M (1992) Interaction of HTLV-1 Tax1 with p67SRF causes the aberrant induction of cellular immediate early genes through CArG boxes. Genes Dev 6:2066–2076
Winter HY, Marriott SJ (2007) Human T-cell leukemia virus type 1 Tax enhances serum response factor DNA binding and alters site selection. J Virol 81:6089–6098
Jeang KT, Chiu R, Santos E, Kim SJ (1991) Induction of the HTLV-I LTR by Jun occurs through the Tax-responsive 21-bp elements. Virology 181:218–227
Smith MR, Greene WC (1991) Type I human T cell leukemia virus Tax protein transforms rat fibroblasts through the cyclic adenosine monophosphate response element binding protein/activating transcription factor pathway. J Clin Invest 88:1038–1042
Grassmann R, Dengler C, Müller-Fleckenstein I, Fleckenstein B, McGuire K, Dokhelar MC, Sodroski JG, Haseltine WA (1989) Transformation to continuous growth of primary human T lymphocytes by human T-cell leukemia virus type I X-region genes transduced by a Herpesvirus saimiri vector. Proc Natl Acad Sci U S A 86:3351–3355
Soda Y, Jinno A, Tanaka Y, Akagi T, Shimotohno K, Hoshino H (2000) Rapid tumor formation and development of neutrophilia and splenomegaly in nude mice transplanted with human cells expressing human T cell leukemia virus type I or Tax1. Leukemia 14:1467–1476
Siu YT, Jin DY (2007) CREB-a real culprit in oncogenesis. FEBS J 274:3224–3232
Watanabe T (2017) Adult T-cell leukemia: molecular basis for clonal expansion and transformation of HTLV-1-infected T cells. Blood 129:1071–1081
Reynaud C, Fabre S, Jalinot P (2000) The PDZ protein TIP-1 interacts with the Rho effector rhotekin and is involved in Rho signaling to the serum response element. J Biol Chem 275:33962–33968
Yan P, Fu J, Qu Z, Li S, Tanaka T, Grusby MJ, Xiao G (2009) PDLIM2 suppresses human T-cell leukemia virus type I Tax-mediated tumorigenesis by targeting Tax into the nuclear matrix for proteasomal degradation. Blood 113:4370–4380
Makokha GN, Takahashi M, Higuchi M, Saito S, Tanaka Y, Fujii M (2013) Human T-cell leukemia virus type 1 Tax protein interacts with and mislocalizes the PDZ domain protein MAGI-1. Cancer Sci 104:313–320
Chun ACS, Zhou Y, Wong CM, Kung HF, Jeang KT, Jin DY (2000) Coiled-coil motif as a structural basis for the interaction of HTLV type 1 Tax with cellular cofactors. AIDS Res Hum Retrovir 16:1689–1694
Jin DY, Teramoto H, Giam CZ, Chun RF, Gutkind JS, Jeang KT (1997) A human suppressor of c-Jun N-terminal kinase 1 activation by tumor necrosis factor α. J Biol Chem 272:25816–25823
Ching YP, Chan SF, Jeang KT, Jin DY (2006) The retroviral oncoprotein Tax targets the coiled-coil centrosomal protein TAX1BP2 to induce centrosome overduplication. Nat Cell Biol 8:717–724
Arnold J, Yamamoto B, Li M, Phipps AJ, Younis I, Lairmore MD, Green PL (2006) Enhancement of infectivity and persistence in vivo by HBZ, a natural antisense coded protein of HTLV-1. Blood 107:3976–3982
Valeri VW, Hryniewicz A, Andresen V, Jones K, Fenizia C, Bialuk I, Chung HK, Fukumoto R, Parks RW, Ferrari MG, Nicot C, Cecchinato V, Ruscetti F, Franchini G (2010) Requirement of the human T-cell leukemia virus p12 and p30 products for infectivity of human dendritic cells and macaques but not rabbits. Blood 116:3809–3817
Mitobe Y, Yasunaga J, Furuta R, Matsuoka M (2015) HTLV-1 bZIP factor RNA and protein impart distinct functions on T-cell proliferation and survival. Cancer Res 75:4143–4152
Zhao T (2016) The role of HBZ in HTLV-1-induced oncogenesis. Virus 8:E34
Ma G, Yasunaga J, Matsuoka M (2016) Multifaceted functions and roles of HBZ in HTLV-1 pathogenesis. Retrovirology 13:16
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Ma G, Yasunaga J, Fan J, Yanagawa S, Matsuoka M (2013) HTLV-1 bZIP factor dysregulates the Wnt pathways to support proliferation and migration of adult T-cell leukemia cells. Oncogene 32:4222–4230
Zane L, Yasunaga J, Mitagami Y, Yedavalli V, Tang SW, Chen CY, Ratner L, Lu X, Jeang KT (2012) Wip1 and p53 contribute to HTLV-1 Tax-induced tumorigenesis. Retrovirology 9:114
Neuveut C, Low KG, Maldarelli F, Schmitt I, Majone F, Grassmann R, Jeang KT (1998) Human T-cell leukemia virus type 1 Tax and cell cycle progression: role of cyclin D-cdk and p110Rb. Mol Cell Biol 18:3620–3632
Kehn K, Fuente Cde L, Strouss K, Berro R, Jiang H, Brady J, Mahieux R, Pumfery A, Bottazzi ME, Kashanchi F (2005) The HTLV-I Tax oncoprotein targets the retinoblastoma protein for proteasomal degradation. Oncogene 24:525–540
Tanaka-Nakanishi A, Yasunaga J, Takai K, Matsuoka M (2014) HTLV-1 bZIP factor suppresses apoptosis by attenuating the function of FoxO3a and altering its localization. Cancer Res 74:188–200
Yuen CK, Chan CP, Fung SY, Wang PH, Wong WM, Tang HMV, Yuen KS, Chan CP, Jin DY, Kok KH (2016) Suppression of type I interferon production by human T-cell leukemia virus type 1 oncoprotein Tax through inhibition of IRF3 phosphorylation. J Virol 90:3902–3912
Yasuma K, Yasunaga J, Takemoto K, Sugata K, Mitobe Y, Takenouchi N, Nakagawa M, Suzuki Y, Matsuoka M (2016) HTLV-1 bZIP factor impairs anti-viral immunity by inducing co-inhibitory molecule, T cell immunoglobulin and ITIM domain (TIGIT). PLoS Pathog 12:e1005372
Belgnaoui SM, Fryrear KA, Nyalwidhe JO, Guo X, Semmes OJ (2010) The viral oncoprotein Tax sequesters DNA damage response factors by tethering MDC1 to chromatin. J Biol Chem 285:32897–32905
Dayaram T, Lemoine FJ, Donehower LA, Marriott SJ (2013) Activation of WIP1 phosphatase by HTLV-1 Tax mitigates the cellular response to DNA damage. PLoS One 8:e55989
Chaib-Mezrag H, Lemaçon D, Fontaine H, Bellon M, Bai XT, Drac M, Coquelle A, Nicot C (2014) Tax impairs DNA replication forks and increases DNA breaks in specific oncogenic genome regions. Mol Cancer 13:205
Baydoun HH, Cherian MA, Green P, Ratner L (2015) Inducible nitric oxide synthase mediates DNA double strand breaks in human T-cell leukemia virus type 1-induced leukemia/lymphoma. Retrovirology 12:71
Kuhlmann AS, Villaudy J, Gazzolo L, Castellazzi M, Mesnard JM, Duc Dodon M (2007) HTLV-1 HBZ cooperates with JunD to enhance transcription of the human telomerase reverse transcriptase gene (hTERT). Retrovirology 4:92
Zhao T, Satou Y, Sugata K, Miyazato P, Green PL, Imamura T, Matsuoka M (2011) HTLV-1 bZIP factor enhances TGF-β signaling through p300 coactivator. Blood 118:1865–1876
Mitagami Y, Yasunaga J, Kinosada H, Ohshima K, Matsuoka M (2015) Interferon-γ promotes inflammation and development of T-cell lymphoma in HTLV-1 bZIP factor transgenic mice. PLoS Pathog 11:e1005120
Miyazato P, Matsuo M, Katsuya H, Satou Y (2016) Transcriptional and epigenetic regulatory mechanisms affecting HTLV-1 provirus. Viruses 8:E171
Rowan AG, Suemori K, Fujiwara H, Yasukawa M, Tanaka Y, Taylor GP, Bangham CR (2014) Cytotoxic T lymphocyte lysis of HTLV-1 infected cells is limited by weak HBZ protein expression, but non-specifically enhanced on induction of Tax expression. Retrovirology 11:116
Satou Y, Miyazato P, Ishihara K, Yaguchi H, Melamed A, Miura M, Fukuda A, Nosaka K, Watanabe T, Rowan AG, Nakao M, Bangham CR (2016) The retrovirus HTLV-1 inserts an ectopic CTCF-binding site into the human genome. Proc Natl Acad Sci U S A 113:3054305–3054309
Cook LB, Melamed A, Niederer H, Valganon M, Laydon D, Foroni L, Taylor GP, Matsuoka M, Bangham CR (2014) The role of HTLV-1 clonality, proviral structure, and genomic integration site in adult T-cell leukemia/lymphoma. Blood 123:3925–3931
Sugata K, Yasunaga J, Mitobe Y, Miura M, Miyazato P, Kohara M, Matsuoka M (2015) Protective effect of cytotoxic T lymphocytes targeting HTLV-1 bZIP factor. Blood 126:1095–1105
Kuo YL, Giam CZ (2006) Activation of the anaphase promoting complex by HTLV-1 Tax leads to senescence. EMBO J 25:1741–1752
Melamed A, Laydon DJ, Gillet NA, Tanaka Y, Taylor GP, Bangham CR (2013) Genome-wide determinants of proviral targeting, clonal abundance and expression in natural HTLV-1 infection. PLoS Pathog 9:e1003271
Melamed A, Witkover AD, Laydon DJ, Brown R, Ladell K, Miners K, Rowan AG, Gormley N, Price DA, Taylor GP, Murphy EL, Bangham CR (2014) Clonality of HTLV-2 in natural infection. PLoS Pathog 10:e1004006
Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, Totoki Y, Chiba K, Sato-Otsubo A, Nagae G, Ishii R, Muto S, Kotani S, Watatani Y, Takeda J, Sanada M, Tanaka H, Suzuki H, Sato Y, Shiozawa Y, Yoshizato T, Yoshida K, Makishima H, Iwanaga M, Ma G, Nosaka K, Hishizawa M, Itonaga H, Imaizumi Y, Munakata W, Ogasawara H, Sato T, Sasai K, Muramoto K, Penova M, Kawaguchi T, Nakamura H, Hama N, Shide K, Kubuki Y, Hidaka T, Kameda T, Nakamaki T, Ishiyama K, Miyawaki S, Yoon SS, Tobinai K, Miyazaki Y, Takaori-Kondo A, Matsuda F, Takeuchi K, Nureki O, Aburatani H, Watanabe T, Shibata T, Matsuoka M, Miyano S, Shimoda K, Ogawa S (2015) Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet 47:1304–1315
Yeh CH, Bellon M, Pancewicz-Wojtkiewicz J, Nicot C (2016) Oncogenic mutations in the FBXW7 gene of adult T-cell leukemia patients. Proc Natl Acad Sci U S A 113:6731–6736
Moles R, Nicot C (2015) The emerging role of miRNAs in HTLV-1 infection and ATLL pathogenesis. Viruses 7:4047–4074
Kato K, Akashi K (2015) Recent advances in therapeutic approaches for adult T-cell leukemia/lymphoma. Viruses 7:6604–6612
Nasr R, Marçais A, Hermine O, Bazarbachi A (2017) Overview of targeted therapies for adult T-cell leukemia/lymphoma. Methods Mol Biol 1582:197–216
Hermine O, Bouscary D, Gessain A, Turlure P, Leblond V, Franck N, Buzyn-Veil A, Rio B, Macintyre E, Dreyfus F, Bazarbachi A (1995) Treatment of adult T-cell leukemia-lymphoma with zidovudine and interferon α. N Engl J Med 332:1749–1751
Bazarbachi A, Plumelle Y, Carlos Ramos J, Tortevoye P, Otrock Z, Taylor G, Gessain A, Harrington W, Panelatti G, Hermine O (2010) Meta-analysis on the use of zidovudine and interferon-alfa in adult T-cell leukemia/lymphoma showing improved survival in the leukemic subtypes. J Clin Oncol 28:4177–4183
Ishida T, Joh T, Uike N, Yamamoto K, Utsunomiya A, Yoshida S, Saburi Y, Miyamoto T, Takemoto S, Suzushima H, Tsukasaki K, Nosaka K, Fujiwara H, Ishitsuka K, Inagaki H, Ogura M, Akinaga S, Tomonaga M, Tobinai K, Ueda R (2012) Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. J Clin Oncol 30:837–842
Sugata K, Yasunaga J, Kinosada H, Mitobe Y, Furuta R, Mahgoub M, Onishi C, Nakashima K, Ohshima K, Matsuoka M (2016) HTLV-1 viral factor HBZ induces CCR4 to promote T-cell migration and proliferation. Cancer Res 76:5068–5079
Ishida T, Utsunomiya A, Iida S, Inagaki H, Takatsuka Y, Kusumoto S, Takeuchi G, Shimizu S, Ito M, Komatsu H, Wakita A, Eimoto T, Matsushima K, Ueda R (2003) Clinical significance of CCR4 expression in adult T-cell leukemia/lymphoma: its close association with skin involvement and unfavorable outcome. Clin Cancer Res 9:3625–3634
Kreitman RJ, Stetler-Stevenson M, Jaffe ES, Conlon KC, Steinberg SM, Wilson W, Waldmann TA, Pastan I (2016) Complete remissions of adult T-cell leukemia with anti-CD25 recombinant immunotoxin LMB-2 and chemotherapy to block immunogenicity. Clin Cancer Res 22:310–318
Willems L, Hasegawa H, Accolla R, Bangham C, Bazarbachi A, Bertazzoni U, Carneiro-Proietti AB, Cheng H, Chieco-Bianchi L, Ciminale V, Coelho-Dos-Reis J, Esparza J, Gallo RC, Gessain A, Gotuzzo E, Hall W, Harford J, Hermine O, Jacobson S, Macchi B, Macpherson C, Mahieux R, Matsuoka M, Murphy E, Peloponese JM, Simon V, Tagaya Y, Taylor GP, Watanabe T, Yamano Y (2017) Reducing the global burden of HTLV-1 infection: an agenda for research and action. Antivir Res 137:41–48
Acknowledgments
Our research was supported by SK Yee Medical Research Fund (2001) and Hong Kong Research Grants Council (C7011-15R).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Chan, CP., Kok, KH., Jin, DY. (2017). Human T-Cell Leukemia Virus Type 1 Infection and Adult T-Cell Leukemia. In: Cai, Q., Yuan, Z., Lan, K. (eds) Infectious Agents Associated Cancers: Epidemiology and Molecular Biology. Advances in Experimental Medicine and Biology, vol 1018. Springer, Singapore. https://doi.org/10.1007/978-981-10-5765-6_9
Download citation
DOI: https://doi.org/10.1007/978-981-10-5765-6_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-5764-9
Online ISBN: 978-981-10-5765-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)