
Abstract
Ab initio molecular dynamics (AIMD) approach to examine excited-state dynamics of ultrafast photoreactions was introduced, and its applications to photoisomerization of azobenzene and stilbene were described. AIMD with a surface hopping scheme was used to examine photoisomerization mechanism of nπ*-excited azobenzene. The rotation pathway was shown to be a preferred one compared with the inversion pathway. A small change in the NN stretching frequency of trans-azobenzene upon nπ* excitation was also reproduced by highly accurate calculations, which support the rotation pathway. To examine the photoreaction of ππ*-excited cis-stilbene, SF-TDDFT was used in AIMD simulations. It was shown that ππ*-excited cis-stilbene propagates primarily toward the twisted structural region due to dynamical effects, with partial branching to the 4,4-dihydrophenanthrene (DHP) structure. AIMD simulations were also carried out for ππ*-excited 1,1’-dimethylstilbene, and it was shown that the perpendicular structure around S1/S0 conical intersections has possibly the long lifetime.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
18.1 Introduction
Photochemistry provides a very broad frontier of quantum chemistry. Last 50 years a laser technology and spectroscopic experiments have clarified many aspects in photoreaction mechanism and dynamics. Especially, transient absorption and emission spectra have provided information of excited-state dynamics of molecules in detail, and sophisticated experiments have extended our understanding of the elementary reaction processes in electronically excited states. Of course, the entire picture of a photoreaction could not be drawn only from the experiments, and nowadays quantum chemical approach plays an essential role to understand the photoreaction mechanism. Owing to advanced ab initio wavefunction theory and density functional theory (DFT), vertical excitation energies can be evaluated with high accuracy, and global potential energy surfaces (PESs) in the excited states can be explored, which clarify the reaction pathways through a couple of excited states. Azobenzene and stilbene are basic textbook molecules in photochemistry. Both molecules show cis-trans photoisomerization, which leads to photoswitch functions in nanotechnology. The first excited state of stilbene is ππ* state, while the first and second excited states of azobenzene are nπ* and ππ* states, respectively. In stilbene, the reaction pathway is restricted to a rotation about the central C = C bond, while in azobenzene, the rotation and inversion pathways are possible. In this chapter, our recent theoretical studies on photoisomerization of azobenzene [1, 2] and stilbene [3, 4] were described.
Ab initio molecular dynamics (AIMD) approach has become a powerful theoretical tool to elucidate the ultrafast photoreaction process. AIMD is a classical trajectory method where atomic positions and velocities are propagated by solving Newton’s equations of motion based on ab initio energy gradients [5,6,7,8]. The target of AIMD approach has been extended to photochemical reactions where non-radiative transitions play a significant role [9,10,11]. In our group, AIMD program code for excited-state dynamics was developed and applied to photochemical reactions in solutions [12,13,14] and to dissociative recombination reactions with the surface hopping scheme [15,16,17] based on a Tully’s surface hopping scheme [18] in which the electronic wavefunction is expanded in terms of adiabatic eigenfunctions, and the expansion coefficients were developed by solving time-dependent Schrödinger equation using non-adiabatic coupling terms. Following Tully’s fewest switches algorithm, the hopping probability between the electronic adiabatic states is evaluated from the electronic amplitudes and non-adiabatic coupling terms, and the hopping is invoked by comparing the hopping probability and a random number. In AIMD approach, all nuclear degrees of freedom are automatically included, and thus, a small time step is required to describe the fast atomic motions. If such fast motions are not important in a given reaction, a relatively large time step could be used by fixing those internal degrees of freedom through dynamics simulation. For this purpose, the RATTLE algorithm [19] was used in our AIMD simulation. A general AIMD code was developed in our group, which is named SPPR [20].
In AIMD simulations, choice of electronic structure theory is a key issue for reliability of dynamics. For electronically excited states, multi-configurational character and dynamic correlation effects are important factors. In ab initio wavefunction theory, we should employ multi-configurational-based theories since the target is not only the Franck–Condon region but also non-adiabatic regions (including conical intersection (CI) regions [21,22,23,24,25,26]) where more than two electronic states could approach each other. The mostly used multi-configurational wavefunction for multiple states is the state-averaged complete active space self-consistent field (SA-CASSCF) method. SA-CASSCF with the appropriate active space can describe the ground and excited electronic states in balance with qualitatively accuracy, but quantitative evaluation of the energetics is difficult because of no dynamic correlation effects included. To include dynamic correlation effects, one can move to multi-reference perturbation theory [27, 28] or multi-reference configuration interaction theory. The multi-reference methodology can reproduce the excited-state PESs with high accuracy, but it requires high computational costs. The alternative approaches, DFT and time-dependent DFT (TDDFT), are very attractive methods. The dynamic correlation effects are included as correlation functional, and thus, they can provide potential energy surfaces including electron correlation effects, with a relatively low cost. However, the usual linear response TDDFT calculations have a disadvantage for study of excited-state reactions [29]; TDDFT provides a discontinuous PES at the crossing point between the reference ground singlet state (S0) and the first excited singlet state (S1), as the reference state becomes the excited state after passing an intersystem crossing point. Furthermore, TDDFT cannot describe doubly excited electronic states. This limitation is serious for describing a process of rotations about double bonds, because the ground state and the doubly excited state cross each other. To overcome this limitation, the spin-flip (SF)-TDDFT method has been developed so that an open-shell triplet configuration is employed as the reference state and the ground and excited states are described as the response state [30,31,32]. In the SF-TDDFT method, five types of electronic states are obtained as solutions [33], i.e., the ground state, the open-shell singlet and triplet states of HOMO-LUMO single excitation, the singlet excited state of HOMO-LUMO double excitation, and spin-mixed states of singlet, triplet and quintet. In the recent AIMD simulation with SF-TDDFT [3], the target state was followed by monitoring orbital coefficients and configuration interaction coefficients along the trajectory, but such approach sometimes did not work in the region where the singlet and triplet states approach each other [3]. Very recently, a new approach, TSF-index method [34], was proposed to trace the target state in S1/S0 minimum energy conical intersection (MECI) search calculations with SF-TDDFT.
In this chapter, we report applications of AIMD methodology to photoisomerization of azobenzene by SA-CASSCF method [1] and to photoisomerization of stilbene by SF-TDDFT methods [3, 4]. The theoretical results are compared with available experimental data, and the reaction pathways and dynamics for the respective photoisomerizations are discussed.
18.2 Photoisomerization of Azobenzene in nπ* Excitation
18.2.1 Past Experimental and Theoretical Studies
The photoisomerization of azobenzene has attracted a great deal of interests in both science and industry since this feature leads to its utilization for light-driven nano-scaled devices such as optical switches, and a number of experimental studies have been reported [35,36,37,38,39,40,41,42,43]. The earlier study on the absorption spectra indicated that the first and second excited states of both cis- and trans-azobenzene were attributed to nπ* and ππ* excitations, respectively [35, 37]. Quantum yields of the isomerizations for cis- and trans-azobenzene were reported to be different between nπ* and ππ* excitations [35,36,37]. Rau proposed that the photoisomerization of trans-azobenzene should follow different pathways in nπ* and ππ* excitations, through a comparison of the simple azobenzene and the sterically hindered azobenzenes [38, 39]; nπ* excitation invokes the inversion of the NNC bond angle within a planar geometry, while ππ* excitation invokes the rotation of two phenyl rings around the NN bond (Fig. 18.1). There have been a number of transient absorption experimental studies for ππ* excitation of trans- and cis-azobenzene [41,42,43]. Tahara and co-workers [40,41,42] carried out the time-resolved Raman, fluorescence and absorption measurements with ππ* excitation of trans-azobenzene in solution. They clarified that isomerization proceeds in nπ* state after electronic relaxation, and proposed that the isomierization pathway is the same as that of direct nπ* excitation. They also found that the NN stretching frequency shows only a slight decrease in the S1(nπ*) state [40].

Scheme of rotation and inversion mechanism for cis-trans isomerization of azobenzene
There have been also a number of theoretical studies on the photoisomerization of azobenzene [1, 44,45,46,47,48,49,50,51,52,53,54,55,56,57,58]. First, Monti et al. [44] showed that in the S1(nπ*) state there is a large activation barrier along the rotation pathway while there is no barrier along the inversion pathway at the singly excitation configuration interaction (CIS) level, indicating that the photoisomerization of trans- and cis-azobenzene is likely to proceed via the inversion pathway in nπ* excitation. In 1999, CASSCF method was employed to determine the rotation and inversion pathways between trans- and cis-azobenzene in the respective S1(nπ*) and S2(ππ*) states, followed by the multi-reference perturbation theory energy calculations along the CASSCF reaction pathways; it was concluded that the inversion is probably the preferred pathway for nπ* excitation while the rotation is the preferred one for ππ* excitation [45]. Ishikawa et al. [46] examined the two-dimensional PES of the S1(nπ*) state as a function of the CNNC torsion and NNC bond angles at the multi-reference single and double excitation configuration interaction (MR-SDCI) level, and proposed that the rotation pathway is preferred in the S1(nπ*) state based on the location of the S1/S0 conical intersection along the rotation pathway. After the Ishikawa’s report, all theoretical studies indicate that the rotation is the preferred pathway for nπ* excitation in the gas phase [48, 53].
On-the-fly dynamics simulations were also performed for the photoisomerization of azobenzene on the basis of semi-empirical molecular orbital calculations with the surface hopping method [49, 52, 56, 58] and with the multiple spawning method [51]. Recently AIMD simulations at the CASSCF level [47, 48, 55, 57] and Car–Parrinello molecular dynamics simulations [54] were also performed for the photoisomerization of azobenzene in nπ* excitation. In next section, we introduced a surface hopping AIMD simulation at SA-CASSCF level [1].
18.2.2 AIMD Study on Photoisomerization of Azobenzene
Hence, we summarized a work of AIMD simulations for cis-trans photoisomerization of azobenzene in the lowest excited state S1(nπ*) reported by our group in 2009 [1]. In the AIMD simulations, the SA-CASSCF/STO-3G level was employed where S0, S1(nπ*), S2(nπ*), S3(n2π*2), and S4(ππ*) states were included with equal weight, and the active space was defined as six electrons in four orbitals (n, n, π, π*). The RATTLE algorithm was applied to solve Newton’s equations of motion with geometrical constraint that all CH bond lengths in two phenyl rings were fixed to the equilibrium distances in the ground state. All electronic structure calculations were performed by MOLPRO [59]. In total, 200 trajectories have been run starting from cis-azobenzene over 240 fs, while 100 trajectories have been run starting from trans-azobenzene over 3 ps. The initial atomic coordinates and velocities were determined by the canonical sampling (T = 300 K) based on normal modes of vibration in the ground state. Among 200 trajectories for cis-azobenzene, the isomerization to trans-azobenzene occurs in 90 trajectories while the remaining 110 trajectories go back to the region of cis-azobenzene in the ground state, and thus, the quantum yield for cis to trans photoisomerization is estimated to be 0.45. For reactive trajectories, the reaction pathway can be classified into three patterns: (i) clockwise rotation, (ii) counterclockwise rotation, and (iii) counterclockwise (S1) +clockwise (S0) rotation. In all cases, only the central N–N part shows a drastic motion within 40 fs, while two phenyl rings do not move so much from the initial position because of a relatively heavy mass. The rotational direction of the N–N part is clockwise in the first pathway while it is counterclockwise in the second and third pathways. After the CNNC dihedral angle d CNNC reaches ~ 90° or -90°, surface hopping to S0 occurs and both phenyl rings start to move toward a structure of trans-azobenzene. In the first and second pathways two phenyl rings keep the direction of motion after hopping to S0, while in the third pathway they turn its direction in the ground state. Atomic motions along the AIMD trajectory do not correspond to the conventional rotation pathway where two phenyl rings rotate around the central NN bond. In the simple picture of rotation mechanism, a structural transformation accompanies a movement of two heavy phenyl rings in a long distance (rotation with a large moment of inertia), while in the pathway presented here, the rotation proceeds in two steps, i.e., (1) rotation of the central N–N part and (2) rotation of two phenyl rings with much shorter distance. Because two phenyl rings are deviated from the central C–N–N–C plane in cis-azobenzene, the initial rotation of NN part makes molecular structure close to the S1/S0 MECI point without a movement of phenyl rings. Then two phenyl rings rotate about the C–N bond, resulting in the structure of cis or trans form of azobenzene in the S0 state. The similar reaction mechanism was also reported in dynamics simulations by Doltsinis et al. [54] and Thiel et al. [56].
In a typical trajectory starting from the S1(nπ*) state, surface hopping to the S0 state occurs at around 40 fs. Before hopping to S0, one NNC bond angle increases to 140° while the other decreases to 100°, and then they show fluctuations around the respective positions, indicating that this is not the inversion pathway. On the other hand, the CNNC dihedral angle immediately increases or decreases, and reaches to 90° or -90°, where surface hopping occurs. This feature indicates that the reaction proceeds along the rotation pathway, but as discussed above, this is not a conventional rotation pathway. It is interesting to note that both NNCC dihedral angles show twist motions in the direction opposite to change in the CNNC dihedral angle. The numbers of trajectories assigned to the respective patterns are 13, 64, and 13 for the clockwise rotation, counterclockwise rotation, and counterclockwise (S1) +clockwise (S0) rotation, respectively, and thus counterclockwise rotation is the most preferred pathway. We also performed the steepest descent path calculations from the Franck–Condon cis structure in the S1 state, and found that it is the counterclockwise rotation pathway.
Here we discuss AIMD trajectories starting from trans-azobenzene. The reaction rate for trans-azobenzene is much slower than that for cis-azobenzene because it takes much longer time for trans-azobenzene to deviate from the planar structure. Thus, trajectories from trans-azobenzene were calculated over 3 ps. As the results, isomerization to cis-azobenzene occurs in 24 trajectories, while in 62 trajectories, the molecule goes back to the region of trans-azobenzene in the ground state. The remaining 14 trajectories continue to stay in the S1 state during the simulation time of 3 ps. Thus, the quantum yield for trans to cis photoisomerization is estimated as 0.28. Compared to the reaction pathway in cis to trans isomerization, atomic motions along the trajectory show a conventional rotation pathway where two phenyl rings rotate around the NN bond. For the reactive trajectory, surface hopping to S0 occurs at t = 800–900 fs, and then CNNC dihedral angle starts to change drastically and the molecule transforms from trans to cis structure very quickly within 50 fs. For the unreactive trajectory, both NNCC dihedral angles show violent fluctuations but they do not correspond to the rotational motion of phenyl rings. Both phenyl rings keep their positions, while the central N atoms show a bicycle-pedal motion.
By fitting variations of S1 population averaged over trajectories to exponential function, the lifetimes of S1 for cis- and trans-azobenzenes are estimated as 0.06 and 1.86 ps, respectively. These values are in qualitative agreement with the experimental lifetimes, 0.17 and 2.6 ps, respectively. In this simulation, the quantum yield for cis to trans photoisomerization was calculated to be 0.45, which is almost twice as large as that for trans to cis photoisomerization, 0.28. These results are in good agreement with the experimental results.
18.2.3 Vibrational Spectroscopy of Trans-Azobenzene in nπ* State
The excited-state PES exploration and AIMD simulations indicated that the nπ*-excited azobenzene prefers the rotation pathway. On the other hand, the transient Raman spectra showed that the NN stretching frequency of trans-azobenzene is almost unchanged in the S1(nπ*) state, indicating that the NN bond order does not change during the isomerization process [40]. In order to gain insight to the photoisomerization mechanism of trans-azobenzene, the NN stretching frequency in the S1 state was investigated by theoretical calculations with several methodology including DFT, SA-CASSCF, and CASPT2. In SA-CASSCF calculations, the active space was 14 electrons in 12 orbitals involving two n orbitals and π/π* orbitals of nitrogens, and four sets of π/π* orbitals of phenyl rings. In CASPT2 calculations, the reference SA-CASSCF wavefunction was determined so that the active space includes two n orbitals and π/π* orbitals of nitrogens, and the lowest 17 orbitals are treated as frozen orbitals in the perturbation calculations. The rotation pathway was also calculated as a function of a CNNC torsion angle d CNNC and the inversion pathway as a function of NNC bond angle a NNC on the S1(nπ*) PES, by optimizing geometrical parameters other than the respective reaction coordinates. Then, the NN stretching frequency was calculated along the respective pathways by applying a projection technique [60] at the CASPT2 level. Sapporo-DZP basis sets [61] were employed in most calculations, and only anharmonic vibrational calculations with vibrational self-consistent field (VSCF) and correlation-corrected VSCF (cc-VSCF) methods were carried out with 6-31G*. DFT calculations were performed by GAMESS [62], while CASPT2 calculations were performed by MOLPRO program [59].
For the ground state of trans-azobenzene with C2h symmetry, the calculated geometrical parameters were in good agreements with the experimental values, while the NN stretching harmonic frequency was overestimated by ca. 80 cm−1 relative to the experimental fundamental frequency at the CASPT2 level. The cc-VSCF calculations at the B3LYP/6-31G* level showed that the NN stretching harmonic frequency, 1561 cm−1, was reduced to 1531 cm−1 due to the anharmonicity effect. By using this value as the anharmonicity effect, the NN stretching fundamental frequency in the ground state of trans-azobenzene was estimated to be 1490 cm−1 at the CASPT2 level, which is comparable to the experimental value, 1440 cm−1.
For the S1(nπ*) state, geometry optimization was performed under the C2h symmetry constraint (referred to as (S1-C2h)min). Normal mode analysis indicated that (S1-C2h)min has one imaginary frequency mode which corresponds to a torsional motion of the central -CNNC- (Au) at DFT(CAM-B3LYP) (20i cm−1) and CASPT2 (49i cm−1) levels, although it has no imaginary frequency mode at the DFT(B3LYP) and SA-CASSCF levels. Thus, the planar structure of (S1-C2h)min is unstable with respect to the CNNC torsional motion. The comparison of geometrical parameters between S0 and S1(nπ*) indicates that, by nπ* excitation, the NN bond length is almost unchanged while the NNC bond angle increases by ca. 14° and the CN bond length decreases by ca. 0.06 Å. These geometrical changes can be explained by the nature of molecular orbitals related to nπ* excitation. Figure 18.2 shows natural orbitals relevant to nπ* excitation, i.e., the lone pair n orbital of N atoms and the NN-π* anti-bonding orbital, at the (S0)min structure determined by the SA-CASSCF(6,4) calculations. As shown in Fig. 18.2, the n orbital shows anti-bonding nature for the NN bond, while the π* orbital shows anti-bonding nature for the NN bond and bonding nature for two CN bonds. Thus, due to nπ* excitation, CN bonds should be strengthened while NN bond strength is not so affected. The extension in the NNC bond angle can be explained by the reduction of a repulsive force between bonding electron pairs (NN and CN bonds) and the lone pair of N atoms. The similar finding in geometrical changes of trans-azobenzene due to nπ* excitation was also reported at the semi-empirical OM2/MRCI level [58].

Natural orbitals corresponding to a lone pair orbital of nitrogen and b NN anti-bonding π* orbital of trans-azobenzene related to nπ* excitation determined by the SA-CASSCF(6,4) calculation at (S0)min. Reproduced from Ref. [2], with permission from AIP Publishing
Due to nπ* excitation, the experimental NN stretching frequency decreases by 12 cm−1, while the calculated frequency increases by 116 cm−1 (TDDFT(B3LYP)), 90 cm−1 (TDDFT(CAM-B3LYP)), 76 cm−1 (CASSCF), and 61 cm−1 (CASPT2). By the cc-VSCF calculation at the TDDFT(B3LYP)/6-31G* level, the anharmonicity effect was estimated to be 29 cm−1 (1679 → 1650 cm−1) in the S1 state, which is similar to the anharmonic effect in the ground state, and thus change of NN stretching fundamental frequency upon nπ* excitation should be similar to change in the harmonic frequency.
Before the appearance of ref [2], there was reported only one theoretical work on vibrational frequencies of trans-azobenzene in the S1(nπ*) state [50], which employed state-specific CASSCF calculations to determine minimum energy structures and harmonic frequencies for the ground and S1(nπ*) states under the C2h symmetry restriction, with analytical gradient and analytical Hessian matrix, using the MOLCAS-5.2 program [63]. The calculated frequencies, multiplied by scaling factor 0.91 to take into account the effect of dynamic correlation and anharmonicity, were reported as 1440 cm−1 (S0) and 1438 cm−1 (S1), which were in good agreement with the experimental values. However, our SA-CASSCF frequencies were totally different from their results, and thus, the same calculations as ref [50] were performed by the state-specific CASSCF(14,12) and the atomic natural orbital (ANO) basis sets, using the MOLCAS-6.4 program [63]. Then, the NN stretching frequencies were calculated to be 1701 cm−1 (S0) and 1771 cm−1 (S1), which are reduced to 1548 cm−1 and 1612 cm−1, respectively, by scaling factor of 0.91, and this result is consistent with the CASSCF calculations with Sapporo-DZP basis sets [2].
The increase in the NN stretching frequency due to nπ* excitation can be explained by vibrational mixing concept [64]. The nπ* excitation strengthens CN bonds, leading to an increase of force constants in CN symmetric and anti-symmetric stretching modes. Then, the interaction between the CN symmetric stretching mode and the NN stretching mode of total symmetry representation is invoked, resulting in mixing of these two normal modes. Figure 18.3 shows mechanism of vibrational mixing of the NN stretching mode and the CN symmetric stretching mode due to nπ* excitation. As shown here, the NN stretching mode in the ground state shows almost a pure NN stretching motion while in the S1(nπ*) state the corresponding mode contains a small contribution from a CN symmetric stretching motion; similarly, the original CN symmetric stretching mode contains a small contribution from NN stretching motion with anti-phase. As the result of this mixing, the frequency of the NN stretching mode increases from 1520 to 1581 cm−1, while the frequency of the CN symmetric stretching mode decreases from 1235 to 1207 cm−1.

Scheme of the vibrational mixing between the NN stretching mode and the CN symmetric stretching mode due to nπ* excitation. Reproduced from Ref. [2], with permission from AIP Publishing
At the CASPT2 level C2h minimum of trans-azobenzene in the S1(nπ*) state has one imaginary frequency mode of Au which is directly related to a reaction coordinate of the rotation pathway, i.e., a torsion angle d CNNC. Then, starting from (S1-C2h)min, the rotation pathway was calculated by choosing d CNNC as a reaction coordinate and optimizing the other geometrical parameters by the CASPT2 method. Along the rotation pathway, vibrational frequencies were calculated with a projection technique [64] at the CASPT2 level. The S1 energy shows a gradual decrease until reaching (S1/S0)CI around d CNNC ~ 95º. It is interesting to note that both r NN and r CN are almost unchanged along the rotation pathway. This result indicates that the NN bond order does not change while proceeding along the rotation pathway. The NN stretching frequency shows only a small decrease (~ 50 cm−1) as d CNNC changes from 180º to 100º; this small decrease is consistent with an almost constant bond length of the central NN bond.
According to chemical intuition, the rotational motion about the central NN bond should weaken the NN bond because π-bonding orbital possibly breaks down. The calculated NN bond lengths and NN stretching frequencies, however, indicate that the NN bond is not weakened along the rotation pathway. Figure 18.4 shows natural orbitals in the active space of SA-CASSCF(6,4) wavefunction. The occupation numbers in these orbitals are also constant, (n)2(π)2(n)1(π*)1. Each orbital almost keeps its shape (i.e., n, π, n, π*) along the rotation pathway against chemical intuition, and this should be the origin of the unchanged bond order of the central NN bond during the rotational motion.

Set of natural orbitals in the active space of SA-CASSCF(6,4) at a d CNNC = 180º ((S1-C2h)min), b 140º, and c 100º, along the rotation pathway [4]
The inversion pathway was also calculated as a function of a bond angle a NNC1 where two carbon atoms of CNNC part are distinguished by C1 and C2. Along the inversion pathway, the S1 energy increases by ca. 0.8 eV at a NNC ~ 180º, the NN bond length decreases by 0.03 Å, and the NN stretching frequency increases by ca. 300 cm−1. The increase in the adiabatic energy clearly indicates that trans-azobenzene prefers the rotation pathway in nπ* excitation. This finding can explain why the isomerization occurs through a rotation of the -N = N- fragment rather than a true torsion along the NN bond [1, 56].
Finally, we summarize significant results from the present calculations based on CASPT2 geometrical structures, vibrational frequencies, and reaction pathways. The energy variations along the reaction pathways indicate that the rotation pathway is preferred to the inversion pathway. Geometrical feature of (S1-C2h)min is shown to be minimum at the CASSCF level, while it changes to a first-order saddle point at the CASPT2 level, indicating that the planar structure of trans-azobenzene is unstable with respect to a torsional motion of the central CNNC dihedral angle. A shift of the NN stretching frequency due to nπ* excitation is relatively small, agreeing to the Raman spectra. A small shift in the NN stretching frequency is explained in terms of natural orbitals related to nπ* excitation and vibrational mixing between the NN stretching mode and the CN symmetric stretching mode. It was also shown that the NN stretching frequency shows a small decrease along the rotation pathway, and thus, the experimentally observed frequency could be assigned to the molecule under a structural transformation along the rotation pathway.
18.3 Photoisomerization of Cis-Stilbene in ππ* Excitation
18.3.1 Past Experimental and Theoretical Studies
Stilbene (SB) is a basic molecule that shows C = C photoisomerization between cis and trans forms. The photoisomerization mechanism of cis-SB has been widely studied both experimentally [65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80] and theoretically [76, 80,81,82,83,84,85,86,87,88,89]. Upon ππ* excitation, cis-SB can transform to trans-SB or 4,4-dihydrophenanthrene (DHP) with a time scale of 1.0 ps [66,67,68,69,70, 74, 77,78,79], and the branching ratio was reported as cis : trans : DHP = 55 : 35 : 10 as shown in Fig. 18.5 [66,67,68]. Steady state [75] and femtosecond time-resolved [79] fluorescence study revealed that there are two decay components with the lifetimes of 0.23 and 1.2 ps. The initial dynamics of ππ*-excited cis-SB was investigated by femtosecond Raman spectroscopy [76]. It showed a temporal change of the vibrational spectrum, which was assigned to the twisting motion of the central C = C bond through quantum chemical calculations [76].

Scheme of photoisomerization of cis-stilbene
Theoretical studies elucidated that ππ*-excited cis-SB evolves on the S1(ππ*) potential energy surface (PES) with the twisting motion of the central C = C bond [73, 81, 83, 87, 89]. Also, an accessible MECI point of S0 and S1 states (denoted as S1/S0-MECI) was located near the minimum on the S1-PES, which corresponds to the C = C bond twisting structure (denoted as twist) [81, 83, 87, 89]. The molecular motion of the cis-trans photoisomerization has been predicted to be a ‘hula-twist’ motion, in which the central C = C bond rotates out of plane; the CH bonds remain out of the plane, while the other atoms reorient to remain coplanar [73]. Minezawa and Gordon investigated the reaction pathways in the relaxation process of ππ*-excited cis-SB by SF-TDDFT [30,31,32]. They located geometries of minima and S1/S0-MECIs for twist ((S1) twist-min and (S1/S0) twist ) and DHP ((S1)DHP-min and (S1/S0)DHP-MECI) regions on the S1-PES, and found that the photocyclization is in competition with the photoisomerization [89], which was later confirmed by the more sophisticated multi-reference method [90].
With respect to dynamics study for cis-SB, Berweger employed CIS/6-31G to generate a three-dimensional constrained PES, and performed dynamics simulations [82]. Dou et al. [85, 86, 88] performed semi-classical electron-radiation-ion dynamics simulations on the relaxation process of cis-SB in the S1 state, and studied three dominant processes, i.e., cis-trans isomerization [86], cis-cis (no isomerization) [85], and cis-DHP photocyclization [88]. However, there has been no dynamics study which focused on the branching mechanism.
In 2012, Berndt et al. measured transient absorption spectra of 1,1′-dimethylstilbene (dmSB) in solution [91]. The lifetime of a transient band observed around 600 nm (CIS* band) was 0.2 ps, which is shorter than the corresponding CIS* band of cis-stilbene (0.92 ps). Another transient band, associated with the perpendicular conformation (denoted as P*), was also observed around 330 nm (P* band). It was found that the lifetime of the P* band of cis-dmSB is 19 ps in hexane and 2.9 ps in acetonitrile, which are much longer than the lifetime of the corresponding P* band of cis-stilbene [77]. They suggested that the longer lifetime of dmSB is attributable to the structural difference between the minimum and the conical intersection around P*. However, there has been no report which studied the relaxation process of dmSB in the S1 state. Both the mechanism of changes in the decay time and the P* structure have never been revealed.
In the following sections, the relaxation mechanism and dynamics of ππ*-excited cis-SB and cis-dmSB were discussed by considering cis-trans photoisomerization and photocyclization to DHP and dmDHP, respectively. The substituent effect for the relaxation mechanism is discussed by comparing the excited-state PES and AIMD results for SB and dmSB. In addition, the origin of CIS* and P* bands in time-resolved absorption experiments is investigated.
18.3.2 Potential Energy Surface of the ππ* State of SB and DmSB
For SB [3] and dmSB [4], equilibrium and TS geometries in the ground and ππ* states, as well as S1/S0-MECI geometries, were optimized at SF-TDDFT(BHHLYP)/6-31G(d) level. The intrinsic reaction coordinate (IRC) pathways and the steepest descent path (meta-IRC) from the Franck–Condon (FC) structure of cis-form in the ππ* state were determined at the same level. All DFT calculations were performed with GAMESS [62].
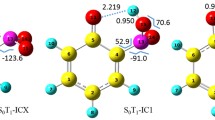
Figure 18.6 shows geometries of cis-form minima in the ground and ππ* states and S1/S0-MECI for SB and dmSB. Definitions of atom numbering are indicated for (S0) cis-min. Three minima in the ππ* state, (S1) cis-min, (S1)DHP-min, and (S1) twist-min, were located for (a) SB and (b) dmSB. In both cases, there is one MECI in a DHP region ((S1/S0)DHP), while there are two in a twist region (S1/S0) twist-1 and (S1/S0) twist-2). Geometries of (S1) twist-min, (S1/S0) twist-1, and (S1/S0) twist-2 are all characterized by a twisted pyramidal structure of the central CC bond. However, in the case of dmSB, the geometry of (S1) twist-min is significantly different from those of (S1/S0) twist-1 and (S1/S0) twist-2. The dihedral angle d C3C1C2C7 that characterizes a pyramidalization is 153.3° for (S1) twist-min, while d C3C1C2C7 is 114.0° and 116.3° for (S1/S0) twist-1 and (S1/S0) twist-2, respectively, indicating that (S1) twist-min shows a structural change between pyramidal and planar. This can be understood as the result of the steric hindrance between phenyl group and methyl group, and such a difference between minimum and MECI geometries was not reported for other molecules, e.g., ethylene, stylene, stilbene, and stiff-stilbene [29, 89, 90, 92]. It is expected that the lifetime of the S1 molecule in the twist region of dmSB should be longer than that of SB.

Geometries of minima in the S0 and S1 states for a SB and b dmSB; S1/S0-MECI geometries for c SB and d dmSB. Definitions of atom numbering are indicated for S0-minimum of SB and dmSB
Figure 18.7 shows geometries and traces of reaction pathways on a two-dimensional configurational space in terms of r C5C6 and d C3C1C2C4 for (a) SB and (b) dmSB [4]. In both SB and dmSB, the meta-IRC pathways from (S1)FC finally lead to a DHP-like geometry, (S1)DHP-min. In the case of SB, the meta-IRC from (S1)FC passes through a cis-stilbene-like structure ((S1) cis-min) in the first stage, and the steepest descent path goes through a very flat region with a large reaction-path curvature. This result is interesting because previous studies predict that the reaction pathway leading from the FC region of cis-stilbene to the twisted structures is preferred [66,67,68]. It is suggested that branching for the twisted structure and the DHP structure occurs in this sharply curved region, and that dynamic effects may be important in determining the branching ratio of the products.

Plots of reaction pathways on the S1-PES, projected on a two-dimensional configurational space in terms of r C5C6 and d C3C1C2C4, for a SB and b dmSB [4]. Minima in the ground state are denoted by a black circle, while minima, TS, and MECI in the S1(ππ*) state are denoted by a red circle, a red triangle, and a red cross mark, respectively. Meta-IRC from the FC point is denoted by a red solid line, while the IRC is denoted by a blue dotted line
In the case of dmSB, the meta-IRC paths from the (S1)FC of cis-dmSB approache (S1)TS1 between (S1)DHP-min and (S1) cis-min, and enters a side of (S1)DHP-min. On the other hand, in SB case, there is no (S1) cis-min and no (S1)TS1 (a kind of shoulder region with a very flat nature); the meta-IRC is initially oriented toward a twist direction, and turns its direction toward (S1)DHP-min. The difference in geometrical feature of the pathways suggests that, in cis-dmSB case, photocyclization is enhanced compared with cis-SB. Since (S1/S0)DHP point is located near (S1)DHP-min, the molecule entering the (S1)DHP-min side would reach the S1/S0-CI region easily, resulting in a non-radiative transition to the ground state. Since the energy barriers for pathways from cis-dmSB to dmDHP or twist-dmSB are very low, the molecule near (S1) cis-min region can easily enter DHP or twist regions.
The difference in the meta-IRC pathways for cis-dmSB and cis-SB was further investigated in the previous paper. In both dmSB and SB, r C5C6 decreases gradually along the meta-IRC paths, and the dihedral angles, d C3C1C2C4, first increase and then decrease almost simultaneously. The difference is the rate of increase in two dihedral angles (slow for dmSB and very fast for SB). It was concluded that a difference in mass of moving fragments, i.e., methyl group (dmSB) and hydrogen atoms (SB) caused the difference in meta-IRC [4].
18.3.3 Ab Initio Molecular Dynamics (AIMD) Simulations for SB and DmSB
For SB [3] and dmSB [4], the excited-state dynamics simulations were started upon excitation to the lowest ππ* singlet state. Each trajectory in the excited state was terminated if the trajectory reached crossing regions between the ground and the first excited states, or if the simulation time reached the maximum (1.5 ps for stilbene and 1.0 ps for dmSB). As discussed in Sect. 18.3.2, there are two types of S1/S0-decay channel in the ππ* state, i.e., DHP- and twist-sides. The branching dynamics toward DHP- and twist-sides in the ππ* state of cis-SB and cis-dmSB were investigated based on the AIMD simulations with SF-TDDFT. 50 and 33 trajectories were calculated for SB and dmSB, respectively. It should be noted that the terminal points of the respective trajectories are not exactly the same as the S1/S0-MECI points; the S1/S0-crossing regions are distributed around the S1/S0-MECI points in configuration space.
In the case of SB, the 50 trajectories are divided into three groups: 13 trajectories (13/50) lead to the formation of DHP, 35 trajectories (35/50) undergo rotation via the torsion of the C1 = C2 bond, and two trajectories (2/50) first began torsional rotation, and then change to DHP. This means that the dynamically important product is different from the terminal geometry of the meta-IRC from (S1)FC, which indicates that this photobranching mechanism in the ππ* state of SB is controlled by dynamics. Experimental quantum yields of the photoreaction cannot be discussed from the present dynamics simulations in a rigorous way, because the trajectories branch into three structures, i.e., DHP, cis-stilbene and trans-stilbene, after relaxing to the ground state. Interestingly, the calculated branching ratio (trans-stilbene : DHP = 35 : 13) is in good agreement with experimental data (trans-stilbene : DHP = 35 : 10), indicating that the dynamics simulations qualitatively reproduce the experimental quantum yield. In the case of dmSB, 33 trajectories are divided into four groups; trajectories entering the DHP-side and reaching the S1/S0-CI region (19/33), trajectories entering the DHP-side and moving to the twist-side (9/33), trajectories entering the twist-side (4/33), trajectories entering the twist-side and moving to the DHP-side (1/33). According to the rate of the destinations of trajectories (DHP : twist ~ 20 : 13), the photocyclization is favorable in the case of dmSB, which is opposite in the case of SB.
The difference in the branching ratio for SB and dmSB can be explained by the difference in reaction pathways [4]. In the case of SB, a dominant change in geometry along the meta-IRC path from (S1)FC is an increase in torsional angle, d C3C1C2C4, and thus, the torsional motion about the central CC bond receives a kinetic energy in the initial stage. Actually most trajectories run toward the twist-side initially, which results in a large branching ratio of twist-side for cis-SB. On the other hand, in the case of dmSB, most trajectories from (S1)FC initially run toward the transition state between (S1)DHP-min and (S1) cis-min. Thus, the trajectories which enter the DHP-side are dominant for cis-dmSB.
The calculated S1-population decay of cis-SB and cis-dmSB in the ππ* state is shown in Fig. 18.8. Here, the trajectories reaching S1/S0-crossing regions are regarded as those hopping to the ground state. The population decay for trajectories in the DHP-side is indicated by a red line, while the decay for those in the twist-side is indicated by a blue line. In the case of SB (Fig. 18.8a), 28/50 trajectories reached the S1/S0-crossing region before 1.0 ps. The population of the ππ* state decreases to 0.44 at 1.0 ps. The time scale of the calculated population decay is in good agreement with the experimental decay of 1.2 ps [79]. The population decay for the trajectories leading to the twist-side (35 trajectories) and for the trajectories leading to DHP-side (13 trajectories) indicated that the lifetime for the DHP-side is relatively longer than that for the twist-side. An evidence from femotosecond time-resolved fluorescence spectra led to the proposal that the photoreaction process of cis-stilbene is a two-step mechanism [79]. The observed fluorescence wavelength, 420 nm [79], nearly coincides with the energy gap between the S0 and S1 states, 3.1 eV, for (S1) cis-min, while the energy difference between the S0 and S1 states in other regions along the IRC is too small to be observed. Thus, the two decay components in the experimental spectra (0.2 ps and 1.2 ps) were attributed to the molecular motion around (S1) cis-min toward DHP-side and twist-side, respectively [3]. For the case of SB, most molecules go into the twist region due to the initial motion in the S1 state, and it takes a relatively long time to enter the DHP side [3]. In the case of dmSB (Fig. 18.8b), the molecule at the DHP-side has a short lifetime while the molecule at the twist-side has a relatively long lifetime in the case of dmSB, which is just opposite to the case of dmSB. In the transient absorption spectra of cis-dmSB [91], the lifetime of the CIS* band at 600 nm and the P* band at 330 nm were reported to be 0.2 ps and 19 ps, respectively, in hexane. Based on the results of AIMD trajectories, the population of the CIS* state should increase initially and gradually decrease through a movement to the DHP side or the twist-side which occurred during the initial 0.2 ps. This time span is in very good agreement with the experimental lifetime of the CIS* band (τ = 0.2 ps) [91]. On the other hand, most trajectories entering the twist-side stay around (S1) twist-min for a long time (at least longer than 1 ps) without reaching the S1/S0-CI region. Thus, the molecule staying at the twist-side should have a long lifetime in the S1 state, corresponding to the P* band observed in the experiment [91].

Decay of S1-population as a function of time derived from AIMD simulations for a SB and b dmSB [4]. S1-population for all the trajectories is plotted in black, while the decays for the trajectories staying at the DHP region and for those at the twist region are plotted in red and in blue, respectively
Next we discussed the experimental transient absorption spectra (Fig. 18.9b) based on AIMD simulations. In the reported time profile of the P* band [91], when normalized to the maximum amplitude of 1.0, the P* signal increases to ~ 0.8 in the first 0.2 ps, exhibits a shoulder feature at 0.2 ~ 0.3 ps, and increases again to 1.0 within 0.6 ps. Then, it gradually decreases, reflecting the lifetime of the P* state. It was also shown that the CIS* signal rapidly decays with a lifetime of 0.2 ps with a distinct shoulder feature observed at 0.2 ~ 0.3 ps. To understand these features seen in the time profiles of the CIS* and P* signals, the time variations of the S1-population for the cis/DHP- and twist-sides in AIMD trajectories are analyzed. To distinguish the cis/DHP- and twist-sides based on geometrical parameters, the structures with d C3C1C2C4 ≤ 68.7° (value at (S1)TS2) are regarded as those located in the cis/DHP-side (red line in Fig. 18.9b), while the structures with d C3C1C2C4 > 68.7° are regarded as those located in the twist-side (blue line in Fig. 18.9a). In AIMD simulations, all trajectories initially enter the cis region, corresponding to appearance of the transient absorption band in the 600-nm region (CIS* band). The S1-population of the cis/DHP-side decreases with a lifetime of ca. 0.3 ps since some trajectories move out to the twist-side or reach the S1/S0-CI regions within the DHP side (consistent with the experimental lifetime of the CIS* band, 0.2 ps). The S1-population of the twist-side increases during 0.6 ps due to moving-in of the trajectories from the cis/DHP-side (in good agreement with the finite rise time in the P* band). In Fig. 18.9a, there is a shoulder feature in the plots of both cis/DHP- and twist-sides during 0.2 ~ 0.3 ps; these shoulders are related to trajectories which once move in the twist-side and then go back to the cis/DHP-side. This feature can explain the shoulder in the experimental time profiles of the CIS* and P* bands in Fig. 18.9b [91].

a Variations of the S1-population from AIMD simulations for dmSB [4]. Rate of S1-population in the cis/DHP-side (d C3C1C2C4 ≤ 68.7°) is plotted in red, while that in twist-side (d C3C1C2C4 > 68.7°) is plotted in blue. The plot for twist-side is normalized so that the maximum value is equal to 1. b Decay of the CIS* band (a red line) and development of the P* band (a cyan line) for cis-dmSB (in acetonitrile). Reprinted from Ref. [91], Copyright (2017), with permission from Elsevier
18.4 Conclusion
Recent progresses in electronic structure theory including ab initio wavefunction theory and DFT have made it possible to explore global excited-state PES involving non-adiabatic region quantitatively. This exploration leads to a new frontier in quantum chemistry. AIMD for photo reaction is possible once energy gradients for excited-state PES are available, and non-adiabatic transitions can be tractable if one gets non-adiabatic coupling terms. Dynamics simulations on the basis of accurate PES can provide a lot of valuable information that cannot be obtained from static calculations of the PES. Collaborations of AIMD approach and spectroscopic experiments can provide a detailed picture of photoreaction processes. In this chapter, we demonstrated the advanced applications of AIMD approach for photoisomerization reactions to elucidate mechanism and dynamics for ultrafast relaxation processes of nπ*-excited azobenzene and ππ*-excited stilbene.
Photoisomerization mechanism of nπ*-excited azobenzene was investigated by AIMD simulation with the RATTLE algorithm, based on the SA-CASSCF method. The photoisomerization from cis to trans proceeds much faster than that from trans to cis due to non-planarity of cis-form. It was clarified that cis to trans isomerization occurs via two-step rotation mechanism, accompanying rotations of the central NN part and two phenyl rings, and this process can be classified into two types, namely, clockwise and counterclockwise rotation pathways; on the other hand, trans to cis isomerization occurs via conventional rotation pathway where two phenyl rings rotate around the NN bond. The quantum yields for cis-trans photoisomerization were well reproduced. Experimentally, the transient Raman spectra showed that the NN stretching frequency of trans-azobenzene is almost unchanged in the S1(nπ*) state, indicating that the NN bond order does not change during the isomerization process. To examine this mechanism, a multi-reference second-order perturbation theory was applied to calculate equilibrium structures and vibrational frequencies of trans-azobenzene in the ground and nπ∗-excited states. It was found that the NN stretching frequency exhibits a slight increase at the minimum energy structure in the nπ∗ state, which is explained by the vibrational mixing of the NN stretching mode with the CN symmetric stretching mode. The NN stretching frequency was also calculated at several selected structures along the rotation and inversion pathways in the nπ∗ state; it was shown that the frequency decreases gradually along the rotation pathway while it increases by ca. 300 cm−1 along the inversion pathway. The frequencies and energy variations along the respective pathways indicate that the rotation pathway is more consistent with the experimental observation of the NN stretching frequency in nπ∗ excitation.
The first excited state of stilbene is ππ* state, which is the most important electronic state to realize the photofunctionality of stilbene-like species. From theoretical viewpoint, the description of ππ* state requires dynamic correlation effects in the electronic structure theory, and so SA-CASSCF is not a good choice for AIMD simulations. Recently, SF-TDDFT method was developed so that non-adiabatic region of S0 and S1 states can be described. Then, AIMD simulations with SF-TDDFT were carried out to examine the photoisomerization mechanism of ππ*-excited cis-stilbene. To follow the target state among nearly degenerate electronic states during the dynamics simulations, a state tracking method was developed. It was shown that the meta-IRC from the FC structure of cis-stilbene in the ππ* state reached the S1-minimum of DHP via a cis-stilbene-like structure on a very flat region of the S1-PES. However, AIMD simulations showed that more trajectories reached CI in the twist region that is located between cis and trans forms, compared with CI in DHP region, and the branching ratio was very close to the experiment. The discrepancy between the meta-IRC and AIMD simulations can be understood from geometrical features of the excited-state PES. It was concluded that ππ*-excited cis-stilbene propagates primarily toward the twisted structural region due to dynamic effects, with partial branching to the DHP structural region via the flat-surface region around (S1) cis-min. AIMD simulations were also carried out for ππ*-excited cis-dmSB at the SF-TDDFT level to get insights into the substitution effects on the photoisomerization dynamics of stilbene. For cis-dmSB, the meta-IRC from the FC structure of cis-dmSB is oriented toward the DHP side, which is in contrast to the case of SB. The optimized geometries of minima and MECI suggested that molecules in the DHP region could easily decay to the ground state. On the other hand, S1/S0-MECI and S1-minimum in the twist region have a relatively different geometry from each other, which is consistent with the experimental observation of the long lifetime of the perpendicular structure. AIMD simulations showed that more trajectories enter the well of the DHP side than the well of the twist-side, and that all of the trajectories going to the DHP-side reached the S1/S0-CI region with ~ 0.2 ps on average, while very few trajectories reached S1/S0-CI even after 1 ps in the twist region.
References
Y. Ootani, K. Satoh, A. Nakayama, T. Noro, T. Taketsugu, J. Chem. Phys. 131, 194306 (2009)
Y. Harabuchi, M. Ishii, A. Nakayama, T. Noro, T. Taketsugu, J. Chem. Phys. 138, 064305 (2013)
Y. Harabuchi, K. Keipert, F. Zahariev, T. Taketsugu, M.S. Gordon, J. Phys. Chem. A 118, 11987 (2014)
Y. Harabuchi et al., J. Phys. Chem. A 120, 8804 (2016)
T. Taketsugu, M.S. Gordon, J. Phys. Chem. 99, 8462 (1995)
T. Taketsugu, M.S. Gordon, J. Phys. Chem. 99, 14597 (1995)
T. Taketsugu, M.S. Gordon, J. Chem. Phys. 103, 10042 (1995)
M.S. Gordon, G. Chaban, T. Taketsugu, J. Phys. Chem. 100, 11512 (1996)
T. Vreven et al., J. Am. Chem. Soc. 119, 12687 (1997)
A.L. Kaledin, K. Morokuma, J. Chem. Phys. 113, 5750 (2000)
M. Ben-Nun, J. Quenneville, T.J. Martinez, J. Phys. Chem. A 104, 5161 (2000)
D. Kina, A. Nakayama, T. Noro, T. Taketsugu, M.S. Gordon, J. Phys. Chem. A 112, 9675 (2008)
D. Kina et al., Int. J. Quantum Chem. 109, 2308 (2009)
A. Nakayama, G. Arai, S. Yamazaki, T. Taketsugu, J Chem Phys 139, 214304 (2013)
T. Taketsugu, A. Tajima, K. Ishii, T. Hirano, Astrophys. J. 608, 323 (2004)
M. Kayanuma, T. Taketsugu, K. Ishii, Chem. Phys. Lett. 418, 511 (2006)
M. Kayanuma, T. Taketsugu, K. Ishii, Theor. Chem. Acc. 120, 191 (2007)
J.C. Tully, J. Chem. Phys. 93, 1061 (1990)
H.C. Andersen, J. Comput. Phys. 52, 24 (1983)
Y. Harabuchi et al., SPPR, a developmental version at Hokkaido University (Sapporo, Japan, 2016)
F. Bernardi, M. Olivucci, M.A. Robb, Chem. Soc. Rev. 25, 321 (1996)
D.R. Yarkony, Acc. Chem. Res. 31, 511 (1998)
A.L. Sobolewski, W. Domcke, C. Dedonder-Lardeux, C. Jouvet, Phys. Chem. Chem. Phys. 4, 1093 (2002)
B.G. Levine, T.J. Martinez, Annu. Rev. Phys. Chem. 58, 613 (2007)
T. Mori, S. Kato, Chem. Phys. Lett. 476, 97 (2009)
S. Nanbu, T. Ishida, H. Nakamura, Chem. Sci. 1, 663 (2010)
H. Nakano, J. Chem. Phys. 99, 7983 (1993)
H. Nakano, Chem. Phys. Lett. 207, 372 (1993)
B.G. Levine, C. Ko, J. Quenneville, T.J. Martinez, Mol. Phys. 104, 1039 (2006)
Y.H. Shao, M. Head-Gordon, A.I. Krylov, J. Chem. Phys. 118, 4807 (2003)
F. Wang, T. Ziegler, J. Chem. Phys. 121, 12191 (2004)
N. Minezawa, M.S. Gordon, J. Phys. Chem. A 113, 12749 (2009)
Y.A. Bernard, Y. Shao, A.I. Krylov, J. Chem. Phys. 136 (2012) 204103/1
S. Maeda, Y. Harabuchi, T. Taketsugu, K. Morokuma, J. Phys. Chem. A 118, 12050 (2014)
G. Zimmerman, L.Y. Chow, U.J. Paik, J. Am. Chem. Soc. 80, 3528 (1958)
D. Gegiou, K.A. Muszkat, E. Fischer, J. Am. Chem. Soc. 90, 3907 (1968)
P. Bortolus, S. Monti, J. Phys. Chem. 83, 648 (1979)
H. Rau, E. Luddecke, J. Am. Chem. Soc. 104, 1616 (1982)
H. Rau In Photochromism: Molecules and Systems; Durr, H., Bounas-Laurent, H., Eds.; Elsevier: Amsterdam, 1990; Vol. 1, p 165
T. Fujino, T. Tahara, J. Phys. Chem. A 104, 4203 (2000)
T. Fujino, S.Y. Arzhantsev, T. Tahara, J. Phys. Chem. A 105, 8123 (2001)
T. Fujino, S.Y. Arzhantsev, T. Tahara, Bull. Chem. Soc. Jpn 75, 1031 (2002)
C.W. Chang, Y.C. Lu, T.T. Wang, E.W.G. Diau, J. Am. Chem. Soc. 126, 10109 (2004)
S. Monti, G. Orlandi, P. Palmieri, Chem. Phys. 71, 87 (1982)
P. Cattaneo, M. Persico, Phys. Chem. Chem. Phys. 1, 4739 (1999)
T. Ishikawa, T. Noro, T. Shoda, J. Chem. Phys. 115, 7503 (2001)
T. Schultz et al., J. Am. Chem. Soc. 125, 8098 (2003)
A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi, G. Orlandi, J. Am. Chem. Soc. 126, 3234 (2004)
C. Ciminelli, G. Granucci, M. Persico, Chem. Eur. J. 10, 2327 (2004)
L. Gagliardi, G. Orlandi, F. Bernardi, A. Cembran, M. Garavelli, Theo. Chem. Acc. 111, 363 (2004)
A. Toniolo, C. Ciminelli, M. Persico, T.J. Martínez, J. Chem. Phys. 123, 234308 (2005)
G. Granucci, M. Persico, Theor. Chem. Acc. 117, 1131 (2007)
I. Conti, M. Garavelli, G. Orlandi, J. Am. Chem. Soc. 130, 5216 (2008)
M. Boeckmann, N.L. Doltsinis, D. Marx, Angew. Chem. Int. Edit. 49, 3382 (2010)
T. Cusati, G. Granucci, M. Persico, J. Am. Chem. Soc. 133, 5109 (2011)
O. Weingart, Z. Lan, A. Koslowski, W. Thiel, J. Phys. Chem. Lett. 2, 1506 (2011)
M. Pederzoli, J. Pittner, M. Barbatti, H. Lischka, J. Phys. Chem. A 115, 11136 (2011)
J.A. Gamez, O. Weingart, A. Koslowski, W. Thiel, J. Chem. Theory Comput. 8, 2352 (2012)
H.J. Werner et al. MOLPRO, version 2012, a package of ab initio programs, 2012
W.H. Miller, N.C. Handy, J.E. Adams, J. Chem. Phys. 72, 99 (1980)
T. Noro, M. Sekiya, T. Koga, Theor. Chem. Acc. 131, 1124 (2012)
M.W. Schmidt et al., J. Comput. Chem. 14, 1347 (1993)
G. Karlström et al., Comput. Mater. Sci. 28, 222 (2003)
T. Hirano, T. Taketsugu, Y. Kurita, The Journal of Physical Chemistry 98, 6936 (1994)
W.M. Moore, D.D. Morgan, F.R. Stermitz, J. Am. Chem. Soc. 85, 829 (1963)
K.A. Muszkat, E. Fischer, J. Chem. Soc. B (1967) 662
T. Wismonskiknittel, G. Fischer, E. Fischer, J. Chem. Soc., Perkin Trans. (1974) 1930
H. Petek et al., J. Phys. Chem. 94, 7539 (1990)
D.C. Todd et al., J. Chem. Phys. 93, 8658 (1990)
S.T. Repinec, R.J. Sension, A.Z. Szarka, R.M. Hochstrasser, J. Phys. Chem. 95, 10380 (1991)
J.M. Rodier, X.P. Ci, A.B. Myers, Chem. Phys. Lett. 183, 55 (1991)
S. Pedersen, L. Banares, A.H. Zewail, J. Chem. Phys. 97, 8801 (1992)
W. Fuss, C. Kosmidis, W.E. Schmid, S.A. Trushin, Angew. Chem. Int. Edit. 43, 4178 (2004)
K. Ishii, S. Takeuchi, T. Tahara, Chem. Phys. Lett. 398, 400 (2004)
T. Nakamura, S. Takeuchi, N. Suzuki, T. Tahara, Chem. Phys. Lett. 465, 212 (2008)
S. Takeuchi et al., Science 322, 1073 (2008)
S.A. Kovalenko, A.L. Dobryakov, I. Ioffe, N.P. Ernsting, Chem. Phys. Lett. 493, 255 (2010)
M. Sajadi, A.L. Dobryakov, E. Garbin, N.P. Ernsting, S.A. Kovalenko, Chem. Phys. Lett. 489, 44 (2010)
T. Nakamura, S. Takeuchi, T. Taketsugu, T. Tahara, Phys. Chem. Chem. Phys. 14, 6225 (2012)
A.L. Dobryakov, I. Ioffe, A.A. Granovsky, N.P. Ernsting, S.A. Kovalenko, J. Chem. Phys. 137, 244505 (2012)
M.J. Bearpark et al., J. Phys. Chem. A 101, 3841 (1997)
C.D. Berweger, W.F. van Gunsteren, F. Muller-Plathe, J. Chem. Phys. 108, 8773 (1998)
Y. Amatatsu, Chem. Phys. Lett. 314, 364 (1999)
V. Molina, M. Merchan, B.O. Roos, Spectrochimica Acta. Part A 55, 433 (1999)
Y.S. Dou, R.E. Allen, Chem. Phys. Lett. 378, 323 (2003)
Y.S. Dou, R.E. Allen, J. Chem. Phys. 119, 10658 (2003)
J. Quenneville, T.J. Martinez, J. Phys. Chem. A 107, 829 (2003)
Y.S. Dou, R.E. Allen, J. Mod. Opt. 51, 2485 (2004)
N. Minezawa, M.S. Gordon, J. Phys. Chem. A 115, 7901 (2011)
I.N. Ioffe, A.A. Granovsky, J. Chem. Theory Comput. 9, 4973 (2013)
F. Berndt et al., Chem. Phys. Lett. 544, 39 (2012)
Y. Amatatsu, Chem. Phys. Lett. 631–632, 47 (2015)
Acknowledgements
We are sincerely grateful to Dr. Yusuke Otani, Dr. Akira Nakayama, Dr. Takeshi Noro, Dr. Satoshi Maeda, Prof. Mark Gordon, Dr. Satoshi Takeuchi, and Dr. Tahei Tahara for collaborations and discussions on the target photoreaction dynamics. This work is supported by a grant from JSPS KAKENHI with Grant Number 26288001. A part of calculations was performed using the Research Center for Computational Science, Okazaki, Japan.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Taketsugu, T., Harabuchi, Y. (2018). Ab Initio Molecular Dynamics Study on Photoisomerization Reactions: Applications to Azobenzene and Stilbene. In: Wójcik, M., Nakatsuji, H., Kirtman, B., Ozaki, Y. (eds) Frontiers of Quantum Chemistry. Springer, Singapore. https://doi.org/10.1007/978-981-10-5651-2_18
Download citation
DOI: https://doi.org/10.1007/978-981-10-5651-2_18
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-5650-5
Online ISBN: 978-981-10-5651-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)