
Abstract
The goal of antibiotic drug development is to define optimized dosing regimens that produce maximal therapeutic efficacy with minimal toxic effects. Pharmacokinetics describes the relationship between dose administered and time course of drug concentration and is characterized by systemic input and disposition kinetic processes. An understanding of drug pharmacokinetics is therefore necessary to optimize drug therapy and provide dose recommendation in populations of interest. Here, we review the basic principles defining drug pharmacokinetics, followed by an overview of the methods associated with data analysis and parameter estimation. We then briefly explore the pharmacodynamic indices relevant to antibiotic dosing and discuss how physiological alterations in critically ill patients may influence drug pharmacokinetics. Finally, we provide some examples of pharmacokinetic dosing considerations for specific classes of antibiotic drugs.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


1.1 Introduction
Pharmacokinetics (PK) describes the time course of drug concentration following dosing [1, 2]. It is broadly characterized by the transfer of drug into, within, and out of the body as:
-
1.
Input—drug movement from the site of administration to the systemic circulation
-
2.
Disposition—drug distribution and elimination from the systemic circulation
These kinetic processes are commonly referred to as the Absorption, Distribution, Metabolism, and Elimination (ADME) of a drug.
The ultimate goal of drug development is to identify the optimal dosing regimens that produce maximum treatment effect. Therapeutic benefit is achieved when drug exposures exceed a given threshold for efficacy, yet remain below the toxicity threshold [1]. An understanding of drug PK is therefore important as it provides the link between dose administered and the time course of pharmacodynamic (PD) or toxicokinetic (TK) response [3,4,5].
This chapter provides a brief overview of basic PK principles. The methods used for parameter estimation is then discussed, as applied to research and clinical settings. Finally, the implications of altered PK in critically ill patients are presented, with specific reference to antibiotic dosing.
1.2 Linear Pharmacokinetics
For most drugs, a proportional relationship is observed between concentration at steady-state (C ss) or area under the concentration-time curve (AUC) and administered dose. The PK of these drugs is described as linear or dose independent and is characterized by first-order processes. For drugs exhibiting linear PK, semi-log concentration-versus-time plots will be parallel at different doses.
In contrast, nonlinearity occurs when the relationships between dose administered and C ss, AUC or other PK parameters are not directly proportional. These drugs demonstrate dose-dependent PK that is described by mixed-order, saturable, Michaelis–Menten, or capacity-limited processes. Example antibiotics showing nonlinear PK include dicloxacillin, which is saturated by active renal secretion [6], and amoxicillin, for which absorption decreases with increasing dose [7].
1.3 Clearance
Clearance (CL) is the key PK parameter and is defined as the “volume of blood, plasma or serum from which drug is irreversibly removed per unit time.” It is therefore expressed in volume/time units. Drug clearance may occur via several different organs or pathways of elimination, including hepatic metabolism, renal, and biliary excretion. Total drug removal therefore comprises the sum of all clearance components (Eq. 1.1):
where CLmet, CLren, CLbil, and CLoth represent the metabolic, renal, biliary, and other mechanisms that constitute total (CL tot) clearance.
Physiologically, the rate of drug elimination across an organ is equal to the product of blood flow rate (Q) and the arterial-venous concentration difference (C A – C V). The extraction ratio (E) provides a measure of organ efficiency with respect to drug removal and is based on mass-balance considerations (Eq. 1.2):
Thus, organs that are highly efficient in eliminating drug will have venous concentrations (C V) that approximate zero and an extraction ratio approaching unity. In contrast, organs that are incapable of drug removal will have an extraction ratio approaching zero, as a consequence of equivalent arterial and venous drug concentrations (i.e., C A – C V = 0). The organ clearance of drug is defined as the product of the blood flow rate and extraction ratio (Eq. 1.3):
Practically, however, the estimation of organ drug clearance using the above formula is challenging. Firstly, the experimental determination of arterial and venous drug concentrations is difficult, particularly in humans. Secondly, blood flow rates may not remain constant over a given study interval, thereby constraining its accurate measurement.
The importance of drug clearance from a pharmacological perspective is demonstrated by its relationship to the rate of maintenance dosing. Clearance is “the proportionality constant that relates the rate of drug elimination to its corresponding concentration at a given time in a relevant biological fluid” (Eq. 1.4):
Steady-state average drug concentrations (C ss ave.) are achieved when the rate of drug input equals its rate of elimination and is the basis for maintenance dosing (Eq. 1.5):
The clinical impact of (Eq. 1.5) in achieving defined target steady-state concentrations is demonstrated in Fig. 1.1.

Concentration-time profile of a hypothetical drug administered at 100 mg by single intravenous (line), single oral (dashed line), or multiple oral (dotted line) dosing. The latter illustrates use of maintenance dosing to achieve average steady-state plasma drug concentrations (Cpss ave.), i.e., at five times the elimination half-life (T 1/2). Drug disposition is described by a one-compartment model, with clearance 1 L/h, volume of distribution 5.77 L, and absorption rate constant 3 h−1. Adapted from [8]
An alternative approach to estimating clearance is by using the AUC, which is a measure of the total systemic exposure of drug (Eq. 1.6):
Thus, for drugs that are administered intravenously, clearance represents the reciprocal of dose-normalized AUC or systemic exposure.
1.4 Volume of Distribution
The volume of distribution (V d) is a “proportionality constant that relates dose administered to the achieved systemic drug concentration” (Eq. 1.7):
This parameter is therefore the hypothetical or “apparent” volume into which a drug distributes to equal its concentration in blood, plasma, or serum. It is expressed in units of volume. Hydrophilic drugs are water soluble and are primarily distributed in the systemic circulation. As a result, these drugs have relatively small volumes of distribution, and thereby achieve high target concentrations. Example antibiotics that demonstrate low apparent volumes of distribution include the aminoglycosides such as gentamicin, tobramycin, and amikacin (V d ranging from 14 L to 21 L) [9, 10]. In contrast, lipophilic drugs such as rifampicin or metronidazole (V d ~ 70 L) are distributed widely throughout the body and attain lower concentrations in the systemic circulation [11].
Pharmacologically, a “loading” dose is often administered to rapidly achieve defined target steady-state blood (plasma or serum) concentrations. Thus, Eq. 1.7 is useful for calculating this loading dose, provided that the drug volume of distribution is known (Fig. 1.2).

Demonstration of loading dose to quickly achieve average steady-state plasma drug concentrations (Cpss ave.). A loading dose of 1 g vancomycin was administered by intravenous infusion (1 h), followed by maintenance dosing of 500 mg every 6 h. Vancomycin pharmacokinetics is described for a 70 kg adult with creatinine clearance of 100 mL/min, using a model with clearance 2.99 L/h, central distribution volume 0.675 L/kg, peripheral distribution volume 0.732 L/kg and inter-compartmental clearance 2.28 L/h [8]
For doripenem and meropenem, typical loading doses of 1000–2000 mg (V d 15–20 L) provide exposures in the 65–135 mg/L desired total drug concentration range [12].
1.5 Half-Life
For drugs that demonstrate linear (dose-proportional) PK, the half-life (t 1/2) is defined as the “time that it takes for its concentrations to halve.” The dimension of half-life is in units of time. Half-life is directly proportional to drug volume of distribution but inversely proportional to its clearance (Eq. 1.8). While clearance and volume of distribution are used to determine half-life, these two PK parameters are independent of each other.
where ln corresponds to the natural logarithm and is applicable to drugs displaying exponential kinetics. Alternatively, the half-life is calculated using elimination rate constant (k el) that has units of per unit time (Eq. 1.9). This parameter is obtained by determining the terminal slope of a log concentration-versus-time plot. Thus, if dosing is discontinued following intravenous infusion, the concentration will decline exponentially to <10% after four half-lives.
The time course of drug accumulation is calculated using the elimination rate constant and dosing interval, τ (Eq. 1.10):
For drugs administered via constant infusion, the concentration will approximate >90% of steady-state following four half-lives. Thus, a hypothetical drug with a 6 h half-life will achieve twice the steady-state concentration to monotherapy, if dosing occurs every t 1/2 (i.e., 6 h). The dosing interval is determined by three factors that include administered dose, half-life, and drug potency (relating to efficacy, toxicity, or both) or EC50 [3].
1.6 Plasma Protein Binding
Only unbound (and not total) drug concentrations are available for metabolism, tissue distribution, or interaction with receptors to produce a pharmacological response. In general, most acidic drugs bind predominantly to albumin, while basic drugs bind to α1-acid glycoprotein or β-lipoproteins. In vitro, the concentration of unbound drug changes with alterations in free fraction. However, in vivo the unbound concentration remains unchanged despite alterations in free fraction or total drug. This is because the steady-state unbound concentration is dependent only on the maintenance dose rate and free clearance (see Eq. 1.5). Dose modification is therefore not required with changes in protein binding since only unbound concentration produces a given pharmacological effect.
1.7 Absorption
Extravascular routes of drug administration include dosing via any method that is not intravenous, such as oral, subcutaneous, intramuscular, intranasal, intradermal, or topical. Absorption is defined by the “movement of drug from the site of administration to the systemic circulation.” Thus, any delay or loss of drug during absorption may contribute to variability in response or compromised therapeutic effect.
Bioavailability describes both the rate and extent of absorption from site of dosing to the systemic circulation. The extent of drug absorption (F) is defined by the ratio of its AUC in blood, plasma, or serum after extravascular dosing, relative to that following intravenous administration (Eq. 1.11).
The rate of drug absorption is determined by the time at which maximal concentration is achieved (T max). Thus, oral formulations that are designed as slow, sustained, or controlled release, allow for a delayed T max when prolonged drug action is required.
Several drug and physiological properties contribute to the rate and extent of absorption. Prior to reaching the general circulation, drugs must dissolve in solution and pass through various biological membranes. Drug physicochemical properties that may influence absorption include the degree of ionization, partition coefficient, and lipid solubility. Physiological factors comprise blood flow, vascularity, pH, membrane nature, and area of the absorptive surface. For orally administered drugs, additional contributors include gastric motility, food, and hepatic first-pass metabolism.
1.8 Pharmacokinetic Analysis
In general, there are three methods that are routinely used for the analysis of PK data, and comprise non-compartmental, standard two-stage and population modelling approaches. These models aim to quantify the dose–concentration relationship, which in turn, can assist with understanding the association between exposure and response [3, 4].
1.8.1 Non-compartmental Analysis
This approach is model independent and is often utilized to evaluate dose proportionality, drug disposition, and show bioequivalence [13]. Typically, the log trapezoidal rule is used to calculate AUC to infinity or last sampling time and area under the first moment curve (AUMC). Other PK parameters include maximal concentration (C max), volume at steady-state (V ss), Mean residence time (MRT), T max, CL, t 1/2, and k el.
Non-compartmental analysis is usually performed in a small number (10–30) of subjects that have similar disease, renal function, and other pathophysiological demographics. Patients are administered drug at a standard or test dose, followed intensive sampling of blood samples across the initial or steady-state dosing interval. The resulting data is then subject to non-compartmental calculations using statistical packages, or with specific software such as Phoenix WinNonlin®. Once computed, the estimated outputs are often compared to healthy volunteer studies or other patient subgroups using tests for demonstrating statistical significance.
A major disadvantage is that non-compartmental estimation is highly dependent on study design, including subject number, characteristics, and the timing of sample collection. Thus, while this approach may provide information on the statistical differences between studies, extrapolation to other patient groups is not recommended. Furthermore, no assumptions are made regarding drug distribution into other tissues, including the site of disease or infection [14]. Non-compartmental analysis is therefore not suitable for dose recommendation to patients with differing characteristics or pathophysiological status.
1.8.2 Compartmental Modelling
Unlike the model-independent approach, this analysis describes the kinetics of drug transfer into one or more hypothetical compartments [14]. In these models, the systemic circulation is referred to as the central compartment and is used to predict drug concentrations in blood, plasma, or serum. It should be noted that each compartment does not represent a specific organ of the body, unless observed data are directly obtained from that target site. Instead, each compartment characterizes differential rates of drug distribution that appear as biphasic profiles in concentration-versus-time curves. Thus, a rapid distribution of drug following intra- or extravascular dosing is adequately described using a one-compartment model. Here, the term rapid indicates that the rates of drug transfer from blood to all tissues or organs and back is equal and instantaneous. In contrast, slower distribution implies that the equilibrium between vasculature and a set of tissues or organs occurs over a finite period of time. As a consequence, drug disposition is represented by several rates of distribution comprising two or more compartments. Organs with high perfusion, such as the liver, blood, and kidney, may therefore be pooled together to signify a single central compartment. Other less perfused tissues, such as bones, cartilage, and fat, are indicative of a peripheral compartment, where drug distribution and equilibrium occurs at a slower rate.
1.8.3 Standard Two-Stage Approach
This method of data analysis is performed in two stages. The first step estimates PK parameters for each individual using their concentration-versus-time data after dosing. A suitable structural model is used to fit the data, using the method of ordinary least squares [15]. Specialized software packages such as Phoenix® WinNonlin are typically suitable for this purpose. The second stage involves tabulation of PK parameter estimates for all individuals and computation of summary statistics including arithmetic or geometric means, medians, and standard deviations.
In general, the number of subjects routinely used for the two-stage approach is comparable to that for non-compartmental analysis. However, some studies can recruit larger patient numbers with wider demographic ranges to investigate the influence of covariate effects on individual PK estimates. Statistical comparisons can therefore be made between two different pathophysiological groups, such as low and high renal function or healthy versus diseased subjects.
Several limitations exist when analyzing PK data using this method. Firstly, similar to non-compartmental analyses, parameter estimation relies on study design, subject-specific factors, and the frequency of obtaining blood or tissue samples. Secondly, the resulting summary statistics may be influenced by outliers and therefore result in biased estimates. While it is possible to reduce the total number of samples obtained per subject, a poor study design may produce inaccuracies in the estimation of PK parameters. Lastly, interindividual variability includes assay errors, thereby necessitating the development of sensitive and precise analytical methods. These limitations may preclude the applicability of two-stage analyses in designing future dosing recommendations.
1.8.4 Population Modelling
Nonlinear “mixed-effects” modelling is routinely used for PK estimation or simulation, as a means to supporting the clinical development of therapeutics [3, 4, 15,16,17]. The term nonlinear indicates that the relationship between drug concentration (dependent variable) is not proportional to time (independent variable) or PK model parameters. The term “mixed-effects” comprises fixed effects and random effects and are indicative of parameterization. The fixed effects component constitutes a structural model, where parameters do not differ between individuals. In contrast, random effects refer to the estimation of parameters that vary between subjects. Thus, this modelling approach analyses data at both population and individual levels, while simultaneously considering between-subject variability (BSV) and residual unexplained variability (RUV). The residual random error includes variability associated with assays, as well as dosing and sampling or measurement.
Unlike non-compartmental or standard two-stage approaches, population modelling has the ability to include small subject numbers with intensive sampling, or larger patient groups that have very sparse datasets. As a consequence, this method is ideal for populations where frequent sampling is ethically or logistically constrained, such as children [18], neonates [19], or critically ill patient populations [20]. Furthermore, nonlinear mixed-effects modelling is less likely to be influenced by outlier subjects or concentration-time data. A useful feature of population analyses is the capacity to handle censored data that are reported as below the limit of quantitation [21].
A key benefit is the ability of explore the relationships between random interindividual variability and subject-specific covariate effects. The BSV is described by predictable and random components (Eq. 1.12):
where BSVpredictable refers to that portion of the interindividual variability that is potentially explained by inclusion of a covariate effect. Thus, BSVrandom indicates the remaining aspect that cannot be described by covariates or patient demographics. Thus, an informative covariate will lower the random variability associated with a given individual parameter estimate. Clinically, an understanding of the relationships between PK and covariate effects allows for the applicability of individualized dosing strategies.
Once fully developed, covariate PK models can be used to simulate hypothetical patient subgroups, including extrapolation to pediatric [22] or critically ill populations [20]. Examples of optimized antimicrobial dosing include tobramycin in children with cystic fibrosis [18], as well as cefepime [23] and cefpirome [24] in intensive care patients. In addition, population modelling has also provided valuable insights for dose recommendation of gentamicin [25], fluconazole, [26] and aminoglycosides [27] in renal dysfunction. Several software packages have the capability of conducting population analysis including NONMEM®, Monolix®, Phoenix® NLME, S-ADAPT, or WinBUGS®.
1.8.5 Therapeutic Monitoring
From a clinical perspective, the above methods for PK estimation and dose individualization are complex and relatively time consuming. Furthermore, therapeutic drug monitoring rarely provides intensive sampling, with only peak or trough concentrations. Dose adjustment is therefore often undertaken using first principles or educated guesses, rather than applying a formal PK modelling approach. A practical alternative is the use of Bayesian forecasting that incorporate established PK models with covariate-parameter relationships defined a priori. Individualized patient parameters can then be used to obtain a complete PK profile, with the ability to optimize dosing so that target concentrations are achieved [18, 27]. Bayesian methods can therefore allow for the development of improved outcomes and reduced toxicity following therapy in a practical clinical setting. Software packages that are suitable for Bayesian approaches and therapeutic monitoring include TCIWorks or USC-PAK.
1.9 Pharmacodynamic Indices
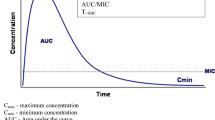
For antimicrobial agents, the ability to inhibit or kill the growth of an infective organism is related to the exposures achieved at a given dose [28]. The PD index is defined by determining the PK exposure relative to an in vitro measure known as the Minimum Inhibitory Concentration (MIC). Kill or inhibition characteristics of antibiotics are described as concentration- or time dependent or a combination of both. Thus, concentration-dependent killing is a measure of the ratio of C max to the defined MIC (i.e., C max/MIC). In contrast, time-dependence is characterized by the duration that an antimicrobial remains above the MIC in a given dosing interval (i.e., T > MIC). The ratio of AUC at 24 h to the MIC (i.e., AUC0–24/MIC) describes drugs with both concentration- and time-dependent killing (Fig. 1.3). Examples of antibiotics classified using these PD indices include the aminoglycosides (concentration-dependent), β-lactams (time-dependent), and fluoroquinolones (concentration- with time-dependence) [20]. While the MIC is routinely used for PD assessment, a possible disadvantage is that it is routinely measured at a single time that ignore potential kinetic differences.

Pharmacokinetic and pharmacodynamic parameters of antibiotics on a concentration-time curve. Key: T > MIC is the time for which a drug’s plasma concentration remains above the minimum inhibitory concentration (MIC) for a dosing period; C max/MIC, the ratio of the maximum plasma antibiotic concentration (C max) to MIC; AUC/MIC, the ratio of the area under the concentration-time curve during a 24 h time period (AUC0–24) to MIC. Adapted from [20]
1.10 Critical Illness
In intensive care patients, pathophysiological changes are common and can influence changes in the time course of drug concentration. The extrapolation of loading or maintenance dose regimens using PK from healthy volunteer studies is therefore inappropriate for maximizing therapeutic benefit (Table 1.1).
Several demographic factors may influence drug clearance in both healthy adult volunteers and critically ill patients. A theoretical basis exists for the allometric scaling of clearance to total bodyweight, based on evidence for metabolic rates in mammals [29, 30]. However, scaling to total bodyweight does not generally apply in the obese population, for which lean body weight is a more suitable size descriptor [31, 32]. Age or critical illness can also alter the clearance of some drugs, primarily due to renal dysfunction or metabolic insufficiency [20]. Furthermore, patients admitted to intensive care units usually receive several co-administered drugs, as a consequence of multiple changes in normal physiology or organ failure. Drug–drug interactions may therefore contribute to alterations drug clearance, when two or more therapies are used for treatment [20].
In critical illness and sepsis, bacterial or fungal endotoxins may stimulate the production of endogenous mediators, thereby increasing capillary permeability and endothelial damage [33]. This change in capillary structure causes a corresponding transfer of fluid from the vasculature to the interstitial space [34]. As a consequence of leaky capillary development, drug distribution can occur into regions that are usually restricted by the normal vasculature. Thus, critically ill patients could potentially have larger volumes of distribution than expected in a typical population, thereby lowering the concentrations achieved in the systemic circulation [20].
Hypoalbuminemia or elevated to α1-acid glycoprotein often occurs during critical illness, thus modifying overall concentrations of protein in plasma [35]. Higher unbound concentrations are observed for ceftriaxone in intensive care subjects due to hypoalbuminemia, increased volume of distribution and reduced clearance [36].
1.10.1 Antibiotic Dosing Considerations
Aminoglycosides demonstrate concentration-dependent killing, with a post-antibiotic effect that prevents bacterial regrowth even after drug concentrations fall below the MIC [37]. This class of antibiotics often show increased distribution volumes in critical care, with a consequent reduction in attained C max exposures [38,39,40]. Appropriate C max-to-MIC ratios are consistently achieved using maximal weight-based dosing regimens, such as 7 mg/kg for gentamicin or tobramycin [39]. An extended-interval dosing regimen is recommended to optimize aminoglycoside effectiveness, with simultaneous monitoring of trough concentrations to avoid toxicity [20].
β-lactams are hydrophilic drugs that are renally eliminated and have a slow continuous kill characteristic that is time dependent [41]. Thus, treatment with this class of antibiotics must consider high glomerular filtration rates and/or increased distribution volume, which are common in the critically ill [20]. Favorable PK-PD outcomes are obtained with frequent dosing or extended continuous infusions [42, 43]. Altered β-lactam clearance due to renal or hepatic dysfunction, with corresponding increase in biliary elimination is also relevant to the intensive care setting [44, 45].
Carbapenems have comparable PK-PD to β-lactams and show time-dependent bactericidal effect when T > MIC is maintained for 40% of the dosing interval. In critical illness, increased distribution volume and higher clearance is reported for these antibiotics [46]. Optimal activity is suggested using continuous or extended carbapenem infusion, which is suitable for achieving the time-dependent PD index [47].
Colistin is a polymyxin antibiotic that is formed by hydrolysis following administration as the sodium colistin methanesulphate prodrug. These drugs demonstrate concentration-dependent bacterial killing [48, 49].
Fluoroquinolones are highly lipophilic antibiotics that are widely distributed to extra- and intracellular spaces, including neutrophil and lymphocyte penetration [50]. However, the volumes of distribution of most fluoroquinolones are generally less affected in intensive care subjects. The exception is levofloxacin, for which increased loading doses is required in the critically ill setting [51, 52]. These antibiotics display concentration- and some time-dependent killing of the infecting pathogen, with C max- or AUC-to-MIC ratios of 10 and 125 describing optimal microbial eradication, respectively [53, 54].
Glycopeptides are relatively hydrophilic for which the PD indices that produce maximum therapeutic benefit are relatively unknown. The elimination of these antibiotics is predominantly associated with creatinine clearance and significant variability in this PK parameter is observed for vancomycin in acute kidney failure [55,56,57]. As a result, therapeutic monitoring of achieved through concentrations is suggested, with high minimum concentrations (>20 mg/L) of vancomycin potentially increasing the risk of nephrotoxicity [58].
Linezolids are hydrophilic drugs that show extensive tissue distribution and are primarily cleared by hepatic metabolism with a minor component of renal elimination [59, 60]. The PD index is time dependent, with a 600 mg twice daily regimen maintaining target T > MIC at 40–80% throughout the dosing interval [59]. However, critical illness is not expected to influence the PD outcome of linezolid antibiotics, and dose adjustment is not recommended for hepatic or renal dysfunction [59, 60].
1.11 Conclusions
In conclusion, this chapter reviews the basic principles that define the pharmacokinetics of drugs following dose administration. An understanding of these theoretical concepts is essential to better consider appropriate dose adjustment with pathophysiological changes in intensive care patients. More specifically, antibiotic dosing considerations, with reference to PK-PD indices are presented. These examples demonstrate how an understanding of the time course of drug concentration can result in recommendations that individualize antibiotic dosing in the critical care setting.
References
Begg EJ (2008) Instant clinical pharmacology, 2nd edn. Blackwell, Christchurch
Rowland M, Tozer TN (1995) Clinical pharmacokinetics: concepts and applications, 3rd edn. Williams & Wilkins, Philadelphia
Al-Sallami HS, Pavan Kumar VV, Landersdorfer CB, Bulitta JB, Duffull SB (2009) The time course of drug effects. Pharm Stat 8(3):176–185. doi:10.1002/pst.393
Holford N (1992) Clinical pharmacokinetics and pharmacodynamics: the quantitative basis for therapeutics. In: Melmon K, Morrelli H, Nierenberg D, Hoffman B (eds) Melmon and morrelli’s clinical pharmacology: basic principles in therapeutics, 3rd edn. McGraw-Hill, New York, p 1141
Welling PG (1995) Differences between pharmacokinetics and toxicokinetics. Toxicol Pathol 23(2):143–147
Nauta EH, Mattie H (1976) Dicloxacillin and cloxacillin: pharmacokinetics in healthy and hemodialysis subjects. Clin Pharmacol Ther 20(1):98–108
Paintaud G, Alvan G, Dahl ML, Grahnen A, Sjovall J, Svensson JO (1992) Nonlinearity of amoxicillin absorption kinetics in human. Eur J Clin Pharmacol 43(3):283–288
Patel K, Kirkpatrick CM (2011) Pharmacokinetic concepts revisited—basic and applied. Curr Pharm Biotechnol 12(12):1983–1990
Bauer LA, Blouin RA (1983) Influence of age on amikacin pharmacokinetics in patients without renal disease. Comparison with gentamicin and tobramycin. Eur J Clin Pharmacol 24(5):639–642
Bauer LA, Blouin RA (1982) Gentamicin pharmacokinetics: effect of aging in patients with normal renal function. J Am Geriatr Soc 30(5):309–311
Ulldemolins M, Rello J (2011) The relevance of drug volume of distribution in antibiotic dosing. Curr Pharm Biotechnol 12(12):1996–2001
Petrosillo N, Giannella M, Lewis R, Viale P (2013) Treatment of carbapenem-resistant Klebsiella pneumoniae: the state of the art. Expert Rev Anti-Infect Ther 11(2):159–177
Bulitta JB, Holford NHG (2014) Non-compartmental analysis. In: Statistics reference online. Wiley. doi:10.1002/9781118445112.stat06889
DiStefano JJ III (1982) Noncompartmental vs. compartmental analysis: some bases for choice. Am J Phys 243(1):R1–R6
Sheiner LB (1984) The population approach to pharmacokinetic data analysis: rationale and standard data analysis methods. Drug Metab Rev 15(1–2):153–171. doi:10.3109/03602538409015063
Mould DR, Upton RN (2013) Basic concepts in population modeling, simulation, and model-based drug development-part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacometrics Syst Pharmacol 2:e38. doi:10.1038/psp.2013.14
Mould DR, Upton RN (2012) Basic concepts in population modeling, simulation, and model-based drug development. CPT Pharmacometrics Syst Pharmacol 1:e6. doi:10.1038/psp.2012.4
Hennig S, Norris R, Kirkpatrick CM (2008) Target concentration intervention is needed for tobramycin dosing in paediatric patients with cystic fibrosis—a population pharmacokinetic study. Br J Clin Pharmacol 65(4):502–510. doi:10.1111/j.1365-2125.2007.03045.x
Stickland MD, Kirkpatrick CM, Begg EJ, Duffull SB, Oddie SJ, Darlow BA (2001) An extended interval dosing method for gentamicin in neonates. J Antimicrob Chemother 48(6):887–893
Roberts JA, Lipman J (2009) Pharmacokinetic issues for antibiotics in the critically ill patient. Crit Care Med 37(3):840–851.; Quiz 859. doi:10.1097/CCM.0b013e3181961bff
Beal SL (2001) Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn 28(5):481–504
Holford N, Heo YA, Anderson B (2013) A pharmacokinetic standard for babies and adults. J Pharm Sci 102(9):2941–2952. doi:10.1002/jps.23574
Roos JF, Bulitta J, Lipman J, Kirkpatrick CM (2006) Pharmacokinetic-pharmacodynamic rationale for cefepime dosing regimens in intensive care units. J Antimicrob Chemother 58(5):987–993. doi:10.1093/jac/dkl349
Roos JF, Lipman J, Kirkpatrick CM (2007) Population pharmacokinetics and pharmacodynamics of cefpirome in critically ill patients against Gram-negative bacteria. Intensive Care Med 33(5):781–788. doi:10.1007/s00134-007-0573-7
Roberts JA, Field J, Visser A, Whitbread R, Tallot M, Lipman J, Kirkpatrick CM (2010) Using population pharmacokinetics to determine gentamicin dosing during extended daily diafiltration in critically ill patients with acute kidney injury. Antimicrob Agents Chemother 54(9):3635–3640. doi:10.1128/aac.00222-10
Patel K, Roberts JA, Lipman J, Tett SE, Deldot ME, Kirkpatrick CM (2011) Population pharmacokinetics of fluconazole in critically ill patients receiving continuous venovenous hemodiafiltration: using Monte Carlo simulations to predict doses for specified pharmacodynamic targets. Antimicrob Agents Chemother 55(12):5868–5873. doi:10.1128/aac.00424-11
Matthews I, Kirkpatrick C, Holford N (2004) Quantitative justification for target concentration intervention—parameter variability and predictive performance using population pharmacokinetic models for aminoglycosides. Br J Clin Pharmacol 58(1):8–19. doi:10.1111/j.1365-2125.2004.02114.x
Craig WA (2003) Basic pharmacodynamics of antibacterials with clinical applications to the use of beta-lactams, glycopeptides, and linezolid. Infect Dis Clin N Am 17(3):479–501
West GB, Brown JH, Enquist BJ (1997) A general model for the origin of allometric scaling laws in biology. Science 276(5309):122–126
West GB, Brown JH, Enquist BJ (1999) The fourth dimension of life: fractal geometry and allometric scaling of organisms. Science 284(5420):1677–1679
Duffull SB, Dooley MJ, Green B, Poole SG, Kirkpatrick CM (2004) A standard weight descriptor for dose adjustment in the obese patient. Clin Pharmacokinet 43(15):1167–1178. doi:10.2165/00003088-200443150-00007
Han PY, Duffull SB, Kirkpatrick CM, Green B (2007) Dosing in obesity: a simple solution to a big problem. Clin Pharmacol Ther 82(5):505–508. doi:10.1038/sj.clpt.6100381
van der Poll T (2001) Immunotherapy of sepsis. Lancet Infect Dis 1(3):165–174. doi:10.1016/s1473-3099(01)00093-7
Gosling P, Sanghera K, Dickson G (1994) Generalized vascular permeability and pulmonary function in patients following serious trauma. J Trauma 36(4):477–481
Arredondo G, Martinez-Jorda R, Calvo R, Aguirre C, Suarez E (1994) Protein binding of itraconazole and fluconazole in patients with chronic renal failure. Int J Clin Pharmacol Ther 32(7):361–364
Joynt GM, Lipman J, Gomersall CD, Young RJ, Wong EL, Gin T (2001) The pharmacokinetics of once-daily dosing of ceftriaxone in critically ill patients. J Antimicrob Chemother 47(4):421–429
Vogelman B, Craig WA (1986) Kinetics of antimicrobial activity. J Pediatr 108(5 Pt 2):835–840
Beckhouse MJ, Whyte IM, Byth PL, Napier JC, Smith AJ (1988) Altered aminoglycoside pharmacokinetics in the critically ill. Anaesth Intensive Care 16(4):418–422
Buijk SE, Mouton JW, Gyssens IC, Verbrugh HA, Bruining HA (2002) Experience with a once-daily dosing program of aminoglycosides in critically ill patients. Intensive Care Med 28(7):936–942. doi:10.1007/s00134-002-1313-7
Triginer C, Izquierdo I, Fernandez R, Rello J, Torrent J, Benito S, Net A (1990) Gentamicin volume of distribution in critically ill septic patients. Intensive Care Med 16(5):303–306
McKinnon PS, Paladino JA, Schentag JJ (2008) Evaluation of area under the inhibitory curve (AUIC) and time above the minimum inhibitory concentration (T>MIC) as predictors of outcome for cefepime and ceftazidime in serious bacterial infections. Int J Antimicrob Agents 31(4):345–351. doi:10.1016/j.ijantimicag.2007.12.009
Boselli E, Breilh D, Cannesson M, Xuereb F, Rimmele T, Chassard D, Saux MC, Allaouchiche B (2004) Steady-state plasma and intrapulmonary concentrations of piperacillin/tazobactam 4 g/0.5 g administered to critically ill patients with severe nosocomial pneumonia. Intensive Care Med 30(5):976–979. doi:10.1007/s00134-004-2222-8
Lipman J, Gomersall CD, Gin T, Joynt GM, Young RJ (1999) Continuous infusion ceftazidime in intensive care: a randomized controlled trial. J Antimicrob Chemother 43(2):309–311
Cooper BE, Nester TJ, Armstrong DK, Dasta JF (1986) High serum concentrations of mezlocillin in a critically ill patient with renal and hepatic dysfunction. Clin Pharm 5(9):764–766
Green L, Dick JD, Goldberger SP, Angelopulos CM (1985) Prolonged elimination of piperacillin in a patient with renal and liver failure. Drug Intell Clin Pharm 19(6):427–429
Novelli A, Adembri C, Livi P, Fallani S, Mazzei T, De Gaudio AR (2005) Pharmacokinetic evaluation of meropenem and imipenem in critically ill patients with sepsis. Clin Pharmacokinet 44(5):539–549
Li C, Kuti JL, Nightingale CH, Nicolau DP (2006) Population pharmacokinetic analysis and dosing regimen optimization of meropenem in adult patients. J Clin Pharmacol 46(10):1171–1178. doi:10.1177/0091270006291035
Tam VH, Schilling AN, Vo G, Kabbara S, Kwa AL, Wiederhold NP, Lewis RE (2005) Pharmacodynamics of polymyxin B against Pseudomonas aeruginosa. Antimicrob Agents Chemother 49(9):3624–3630. doi:10.1128/aac.49.9.3624-3630.2005
Nation RL, Li J (2007) Optimizing use of colistin and polymyxin B in the critically ill. Semin Respir Crit Care Med 28(6):604–614. doi:10.1055/s-2007-996407
Smith RP, Baltch AL, Franke MA, Michelsen PB, Bopp LH (2000) Levofloxacin penetrates human monocytes and enhances intracellular killing of Staphylococcus aureus and Pseudomonas aeruginosa. J Antimicrob Chemother 45(4):483–488
Kiser TH, Hoody DW, Obritsch MD, Wegzyn CO, Bauling PC, Fish DN (2006) Levofloxacin pharmacokinetics and pharmacodynamics in patients with severe burn injury. Antimicrob Agents Chemother 50(6):1937–1945. doi:10.1128/aac.01466-05
Pea F, Di Qual E, Cusenza A, Brollo L, Baldassarre M, Furlanut M (2003) Pharmacokinetics and pharmacodynamics of intravenous levofloxacin in patients with early-onset ventilator-associated pneumonia. Clin Pharmacokinet 42(6):589–598
Forrest A, Nix DE, Ballow CH, Goss TF, Birmingham MC, Schentag JJ (1993) Pharmacodynamics of intravenous ciprofloxacin in seriously ill patients. Antimicrob Agents Chemother 37(5):1073–1081
Preston SL, Drusano GL, Berman AL, Fowler CL, Chow AT, Dornseif B, Reichl V, Natarajan J, Corrado M (1998) Pharmacodynamics of levofloxacin: a new paradigm for early clinical trials. JAMA 279(2):125–129
Llopis-Salvia P, Jimenez-Torres NV (2006) Population pharmacokinetic parameters of vancomycin in critically ill patients. J Clin Pharm Ther 31(5):447–454. doi:10.1111/j.1365-2710.2006.00762.x
Macias WL, Mueller BA, Scarim SK (1991) Vancomycin pharmacokinetics in acute renal failure: preservation of nonrenal clearance. Clin Pharmacol Ther 50(6):688–694
Udy AA, Covajes C, Taccone FS, Jacobs F, Vincent JL, Lipman J, Roberts JA (2013) Can population pharmacokinetic modelling guide vancomycin dosing during continuous renal replacement therapy in critically ill patients? Int J Antimicrob Agents 41(6):564–568. doi:10.1016/j.ijantimicag.2013.01.018
Hidayat LK, Hsu DI, Quist R, Shriner KA, Wong-Beringer A (2006) High-dose vancomycin therapy for methicillin-resistant Staphylococcus aureus infections: efficacy and toxicity. Arch Intern Med 166(19):2138–2144. doi:10.1001/archinte.166.19.2138
MacGowan AP (2003) Pharmacokinetic and pharmacodynamic profile of linezolid in healthy volunteers and patients with Gram-positive infections. J Antimicrob Chemother 51(Suppl 2):ii17–ii25. doi:10.1093/jac/dkg248
Brier ME, Stalker DJ, Aronoff GR, Batts DH, Ryan KK, O’Grady M, Hopkins NK, Jungbluth GL (2003) Pharmacokinetics of linezolid in subjects with renal dysfunction. Antimicrob Agents Chemother 47(9):2775–2780
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Patel, K., Kirkpatrick, C.M. (2018). Basic Pharmacokinetic Principles. In: Udy, A., Roberts, J., Lipman, J. (eds) Antibiotic Pharmacokinetic/Pharmacodynamic Considerations in the Critically Ill. Adis, Singapore. https://doi.org/10.1007/978-981-10-5336-8_1
Download citation
DOI: https://doi.org/10.1007/978-981-10-5336-8_1
Published:
Publisher Name: Adis, Singapore
Print ISBN: 978-981-10-5335-1
Online ISBN: 978-981-10-5336-8
eBook Packages: MedicineMedicine (R0)