
Abstract
Theranostics is a novel concept that refers to the integration of diagnostics with therapeutics in order to generate personalized therapies and is emerging as a promising precise therapeutic paradigm. In oncology, the approach is aimed at more accurate diagnosis of cancer, optimization of patient selection to identify those most likely to benefit from a proposed specific therapy allowing the generation of effective therapeutics that enhance patient survival. Perhaps the most promising target to date for theranostics is the deregulation of cancer cell metabolism, involving the uptake of glucose and glutamate, two key nutrients that are necessary to convert into glucosamine to stimulate protein biosynthesis for the growth and survival of cancer cells. We have recently developed a novel technology whereby the chelator ethylenedicysteine (EC) conjugates with glucosamine to create a vehicle platform (ECG), which mimics N-acetylglucosamine (GlcNAc) that targets highly proliferative cancer cells. Moreover, ECG can be further conjugated to diagnostic/therapeutic metals (rhenium, Re, and platinum, Pt) that function as a new theranostic agent suitable for personalized medicine, targeting key pathways in cancer cells such as highly metabolic diffuse large B-cell lymphoma (DLBCL). This chapter summarizes key signaling pathways linked to dysregulated glucose metabolism in DLBCL and how deregulated glucose metabolism can be utilized for developing innovative new technologies with theranostic applications to eradicate cancer.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Diffuse Large B-Cell Lymphoma Pathophysiology
Malignant B-cell lymphoma represents a major health risk in the USA and worldwide [1, 2]. Non-Hodgkin lymphomas (NHLs) are a common, accounting for about 4% of all cancers, but heterogeneous group of human B lymphocytic neoplasms (NHL-B), that primarily arise (~85%) within the B-cell lineage of the immune system. NHL-B represents the fifth most common cancer in the USA (~72,000 new cases/20,000 deaths) as reported in 2016 [3–5]. Notably, while diffuse large B-cell lymphoma (DLBCL) is the most common (30–40%) of histologically defined NHL-B, it is also the most heterogeneous [6, 7].
As an aggressive, diverse group of lymphoid neoplasms, DLBCL is associated with constitutive activation of key NF-κB signaling pathways [8, 9], although other lymphoma histotypes like Hodgkin and MALT lymphomas may show different activation patterns, signaling proteins, and pathological variants in the canonical NF-κB pathway [10–12]. DLBCL has been extensively studied in recent years by various types of microarray gene analyses (supervised or unsupervised) [13, 14] and is considered to consist of at least three definable genetic “signatures” or occasionally overlapping subtypes, based on various methods of gene expression profiling [15, 16]. Although these putative DLBCL subtypes appear valid, considerable phenotypic genotypic overlaps clearly occur [17]. The Rosenwald/Staudt group, for instance, has equated the expression of MUM1/IRF4 and CD138 immunologic markers with constitutive activation of the NF-κB1 pathway as a specific gene array “signature” that defines an activated B-cell type (ABC-like DLBCL). In contrast, the nonrandom t(14;18) bcl2 cytogenetic translocation, expressing germinal center (GC) markers bcl-6 and CD10, defines a GC B-cell subtype (GCB-like DLBCL) [18–20]. In the era that preceded the standard frontline combination chemotherapy rituximab, cyclophosphamide, hydroxydaunomycin, Oncovin, and prednisone (R-CHOP), these DLBCL subtypes were reported to have disparate clinical outcomes with significantly different 5-year survival rates [21, 22], although better differential biomarkers are still needed.
Notably, studies on DLBCL-associated oncogenes have recently revealed new molecular insights regarding the role of bcl-6 in the pathogenesis of the GCB-DLBCL. For instance, Dalla-Favera’s group has shown that bcl-6 gene expression is regulated through the CD40-NF-κB canonical signaling pathway. They have also shown that in GCB-DLBCL, CD40 appears to activate the transcriptional factor IRF4 (MUM1) gene through the p50/p65 members of canonical NF-κB pathway. Once activated IRF4 binds to the bcl-6 promoter and directly represses its transcription [23]. This suggests that resistance to CD40-NF-κB-IRF4 signaling is an essential mechanism of bcl-6 deregulation in GCB-DLBCL. The data also suggest that while CD40-NF-κB pathway is active in GCB- and ABC-type DLBCL, activation occurs by different mechanisms [24, 25].
Moreover, several studies have examined the potential role(s) of TNFR and related signaling pathways on DLBCL growth and survival (G/S) [26, 27], whose mechanisms are also shared by normal B lymphocytes. However, the difference between normal and malignant cells is that these G/S signaling pathways are aberrantly dysregulated in the latter [28, 29], the hallmark of aggressive NHL-B pathogenesis. These studies initially identified the CD40 signalosome, consisting of the TNFR, CD40, and its cognate ligand (CD40L, CD154) as constitutively expressed in DLBCL cell lines and primary lymphoma cells from patients, along with the signaling components (TRAFs 2,6: IKK complex, c-rel/p65) of the canonical NFkB1 pathway [30]. When the CD40 signalosome was disrupted by antibodies to CD40 or CD154, the signalosome structure and NF-κB1 signaling pathway were interdicted, and cell death was induced through apoptosis [31–33]. Further studies on aberrant survival mechanisms in DLBCL led to the discovery of the role of another TNF superfamily member, the B-cell survival factor BLyS/BAFF, with constitutive expression of their receptor BR3 in aggressive NHL-B (DLBCL and MCL). These studies further revealed that constitutively activated BLyS/BR3 receptor-ligand interactions result in noncanonical (alternative) NF-κB2 pathway signaling, which in analogy to the CD40/CD154 cascade, the BLyS/BR3 signalosome provides a constitutive positive ligand-/receptor-mediated feedback to the NF-κB2 pathway [31].
While earlier studies [33, 34] had also revealed that in addition to constitutive expression of the canonical NF-κB1 pathway, there is evidence that at least some members of the alternative NF-κB2 pathway (e.g., p52 and RelB) are also constitutively activated and expressed in DLBCLs and other aggressive NHL-B (e.g., MCL) [34, 35]. A key study examined a series of validated tissue microarrays (TMA) in DLBCL (ABC and GCB types) cell lines and primary patient samples and found that both NF-κB1 and NF-κB2 signaling pathways were constitutively activated in both ABC and GCB subsets, but that the pattern of activation and NF-κB dimer utilization was characteristically different [36]. These studies suggest that multiple interactive cell signaling pathways, including both canonical and alternative variant “hybrid” NF-κB pathways [37–39], contribute to G/S regulatory mechanisms in DLBCLs. While the canonical/classical NF-κB1 pathway has been studied in many cell types [40, 41], the alternative NF-κB2 pathway has only recently begun to receive attention [31, 42, 43]. Most of these studies, however, have been mainly performed in genetically engineered mice, which basically provided a general outline of some of the regulatory interactions [44].
Examples of genetically altered murine models demonstrate that the alternative NF-κB pathway activation is controlled through a negative feedback mechanism involving increased protein levels of negative regulators of the adaptor proteins TRAF2/3, which inhibit the key upstream NF-κB2 kinase, NIK. Overexpression of wild-type NIK leads to B-cell hyperplasia caused by the amplification of BLyS-induced alternative NF-κB signals. Interruption of the interaction between TRAF3 and NIK induces constitutive BLyS-independent activation of the alternative pathway and leads to a large accumulation of mature B cells in lymphoid organs and disruption of structural integrity. Other studies have proposed a model where interactions between TRAF2 and TRAF3 constitutively block B-cell survival via the inhibition of activation of the alternative NF-κB2 pathway, suggesting a mechanism by which NIK accumulation is prevented, since NIK bears a TRAF3 interaction site that can result in NIK degradation by TRAF3 [45–48]. Interestingly, when BLyS occupies BR3, it sequesters TRAF3 and prevents interactions with TRAF2. This blocks TRAF3/2 interactions that would lead to greater NIK accumulation, subsequent NF-κB2 processing, and hence increased B-cell survival [49]. The results from these findings imply that deregulated NIK expression may contribute to B-cell malignancies, particularly aggressive lymphomagenesis, even if NIK protein remains undetectable at the protein level [50].
Moreover, there is compelling transgenic mouse model that obtained evidence of an important role of CD40 in B-cell lymphomagenesis, resulting from constitutively active CD40 receptor expression, which leads to B-cell-specific enforced activation of the noncanonical NF-κB pathway. Consistent with these findings, LMP1-/CD40-expressing mice developed a high incidence of B-cell lymphomas, indicating that interactions of the signaling pathways induced by constitutive CD40 signaling are sufficient to initiate a neoplastic B-cell process, likely leading to the development of B-cell lymphomas.
On the other hand, the noncanonical NF-κB2 pathway has been occasionally implicated in B lymphoid malignancies associated to some cases of chromosomal abnormalities leading to the production of truncated p100 proteins with diminished NF-κB inhibitory ability [45–47]. For instance, two studies in primary multiple myeloma (MM) samples and cell lines exhibited genetic aberrations that affect mediators of NF-κB activation, mostly involving the alternative NF-κB2 pathway. The aberrations led to the absence of negative regulators of NF-κB, such as TRAF3, TRAF2, and c-IAP1/2, or to overexpression of NIK [51, 52]. These studies imply that deregulation of the TRAF3-NIK axis could also play an important role in B-cell lymphomagenesis [50]. Although the data further indicate that elevated canonical and noncanonical NF-κB activity by deregulation of NIK directly contributes to disease progression in primary MM, they also suggest that TRAF3 can function as an important suppressor of lymphoid neoplasia through the negative regulation of both the canonical and noncanonical NF-κB pathways [53, 54].
It must be noted that despite its B-cell lineage, MM cells represent a neoplasm mimicking plasma cells, which is quite different from DLBCL, although some plasmablastoid lymphomas can be quite plasmacytoid. In keeping with this notion, there is little information on the intrinsic nature of NF-κB2 pathway in DLBCL. However, a report by Kim et al. [55] demonstrated the activation of NF-κB2 components by BLyS in DLBCL subsets, while others have described the involvement of several oncogenes in DLBCL and identified NFAT expression as a candidate oncogene in the ABC subtype. Similarly, a variety of genetic abnormalities were identified in DLBCL that were associated with either the ABC or GCB-DLBCL subsets [56, 57].
Although deregulation of NF-κB signaling can be a key mediator of transcription factor (TF) heteromer formation, which targets NF-κB-regulated G/S genes in DLBCLs, other regulators like NFAT (distantly related to NF-κBs [58]) are known to be central for chromatin structural remodeling, which in turn actively modulates gene transcription. The NFAT family of proteins are also Ca2+-inducible transcription factors that prominently stimulate the expression of a wide range of immune response genes in activated T cells [59]. However, we have shown that NFAT plays key regulatory roles in B lymphocytes, particularly in aggressive NHL-B [34, 60]. NFAT-dependent promoters and enhancers rapidly undergo extensive chromatin remodeling to form deoxyribonuclease I (DNAse 1)-hypersensitive sites (HSSs) [61, 62]. NFAT is likely to be at least a driving force involved in chromatin remodeling, which has recently been described as a major NFAT function [63], since NFAT sites are necessary and sufficient to activate DNA-1-driven chromatin HSSs. Chromatin remodeling may well be a primary function of NFAT elements, since even high-affinity NFAT binding to promoter targets results in relatively weak transcriptional activation without bonding between NFAT and transcription factor (TF) associates [64]. The activating protein 1 (AP-1) is the most common TF partner that is directly recruited by NFAT, at the HSS domains, and dimeric NFAT-AP-1 and DNA response elements are very efficient in removing nucleosomes [62]. NFAT-AP-1 complexes recruit both histone acetyltransferases (HATs) and the ATP-dependent SWI-SNF family of chromatin remodelers [65, 66], which together provide the necessary functional proteins required to modify and rearrange nucleosomes.
NFAT may also help to organize chromatin domains and enable enhancer-promoter communication [67]. In activated T cells, inducible intrachromosomal looping occurs between the tumor necrosis factor (TNFα) gene promoter [68] and two NFAT-dependent enhancers located within 9 kb upstream (-) and 3 kb downstream (+) of the promoter region [69]. This topology places the TNF gene and the adjacent lymphotoxin (LT) genes in separate loops, thereby allowing independent regulation of the TNFα gene within multigene loci. This new data supports earlier studies proposing that NFAT functions through the disruption of nucleosomes within enhancers, mobilizing nucleosomes across extensive chromatin domains and linking enhancers and promoters. These studies identify NFAT as a factor that creates a chromatin environment, which is permissive for both the recruitment and aggregation of factors to coordinately control transcriptional processes at promoter and enhancer regions [70]. The specific role that NFAT plays in the multifaceted process of locus activation is still unclear, but its role as an effective orchestrator of essential steps in creating an accessible chromatin environment is compelling [63].
NFAT functions have recently also been linked to tumor immunity [71, 72] that includes the development of T-cell dysfunctions such as CD4+ T-cell anergy [73] and CD8+ T-cell exhaustion, which occur in a variety of cancers [74]. It is conceivable that NFAT regulates PD-1 expression in anergic T cells [75], and thus, the inhibition of NFAT or the kinase-regulating NFAT pathway would enhance T-cell function [76, 77]. These findings support the premise that targeting NFAT pathway in cancer patients could lead to tumor cell killing, either directly by abrogating NFAT-dependent cell survival or indirectly by blocking T-cell activation. Consistent with this hypothesis, a recent study by Ron Levy’s group [78] demonstrated that a combination treatment with anti-PD-L1 antibodies and ibrutinib, a Bruton’s tyrosine kinase (BTK) inhibitor, leads impressive antitumor effects in animal lymphoma models, as well as in breast and colon cancer models. Their findings lead to the speculation that ibrutinib concomitantly targets BTK and interleukin-2-inducible kinase (ITK) to inhibit NFAT activation and thus could play a key role in T-cell-mediated therapies [79].
2 Linking Deregulated Signaling Pathways to Cancer Cell Metabolism in DLBCL
Thioredoxin (TRX) is an integral antioxidant system, which maintains the intracellular redox state and hence a strong candidate to coordinately target a family of proteins to restore sensitivity to chemotherapy [80]. One family member is thioredoxin-1 (TRX-1), a low-molecular-weight (10–12,000) cellular redox protein, which is present in the nucleus and cytoplasm to regulate the activity of various enzymes, including those that counteract oxidative stress within the cell [81]. Intracellular TRX-1 exerts most of its antioxidant properties through scavenging of reactive oxygen species (ROS). Moreover, it plays an important role in the regulation of redox-sensitive transcription factors [82] and acts as a proto-oncogene that stimulates tumor growth and inhibits both programmed and drug-induced cell death [83]. Its increased expression is associated with enhanced HIF-1α (hypoxia-induced factor 11α) levels and transactivation in cancer cells [84], which result in high production of vascular endothelial growth factor (VEGF) and enhanced tumor angiogenesis [85]. Additionally, its overexpression has been correlated with aggressive tumor growth, poorer prognosis, and shortened patient survival [86].
TRX-1 appears to have an important role in maintaining the transformed phenotype of some human cancers as well as their resistance to chemotherapeutic drugs. These functions make it a rational target for cancer drug development, and recent experiments support such potential. To that end, the Leukemia/Lymphoma Molecular Profiling Project (LLMPP) used a microarray technology to define a molecular profile for each of 240 patients with DLBCL and developed a molecular outcome predictor score that accurately determines patient survival. The study found that DLBCL patients with the worst prognosis, according to the outcome predictor score, had decreased expression of TXNIP, a protein that naturally inhibits TRX-1 activity [87]. However, these studies had little or no follow-up on the pathophysiologic impact of TRX-1- and TXNIP-controlled reduction-oxidation (redox) state in B-cell lymphomas.
TXNIP is known to regulate the cellular redox state by binding to and inhibiting thioredoxin in a redox-dependent fashion [88]. Recent studies, however, demonstrated that TXNIP is also a potent negative regulator of glucose uptake [89, 90]. In response to glucose uptake, cells activate a key TF complex that includes the Mondo member A and the Max-like protein (MondoA:Mix), which then enters the nucleus, binds the TXNIP promoter, and upregulates TXNIP gene transcription. How TXNIP blocks glucose uptake is not completely clear, but maintenance of energy homeostasis is clearly regulated through TXNIP. Furthermore, conventional TXNIP-deficient mice revealed that mitochondria were functionally and structurally altered, leading to reduced oxygen consumption and enhanced anaerobic glycolysis [91, 92]. On the other hand, tissue-specific knockout mice showed that TXNIP is essential for maintaining hematopoietic stem cell (HSC) quiescence and homeostatic interactions between HSCs and the bone marrow niche. In addition, targeted deletion of TXNIP causes cardiac dysfunction in response to pressure overload, primarily due to the dysregulation of mitochondria, which switches from oxidative phosphorylation to anaerobic glycolysis [92].
Notably, the energy homeostasis is defective in cancer since the TXNIP gene is repressed in many tumors by posttranscriptional and translational mechanisms [91]. These collective data indicate that active glucose metabolism together with activated TRX-1 plays a key role in the pathophysiology of DLBCL. Since TXNIP controls both redox and glucose levels in cancer, its expression could be a prominent target for the therapy of DLBCL. In agreement with this hypothesis, it has been previously shown that epigenetic histone deacetylase (HDAC) inhibitors like vorinostat and histone methyltransferase inhibitors like 3-deazaneplanocin A (DZNep) can reactivate TXNIP gene expression and inhibit TRX-1 in cancer cells [93, 94]. Of note, one of the main functions of DZNep is to disrupt the polycomb-repressive complex 2 (PRC2) by inhibiting the enhancer of the zeste homolog 2 (EZH2) protein [95]. EZH2 is the catalytic subunit of PRC2, which is a highly conserved histone methyltransferase that targets lysine-27 of histone H3 [96]. This methylated H3-K27 chromatin site is commonly associated with the silencing of differentiation genes in organisms ranging from plants to humans. Studies in human tumors have shown that EZH2 is frequently overexpressed in a wide variety of malignant tissues, including lymphomas [97]. Although the mechanistic contribution of EZH2 to cancer progression is not yet determined, functional links between EZH2-mediated histone methylation and histone acetylation suggest a partnership with the gene-silencing machinery implicated in tumor suppressor loss. Of particular pertinent are key studies using whole-genome sequencing in primary DLBCL, which identified frequently recurrent somatic heterozygous mutations in the EZH2 locus [98, 99]. The EZH2 mutations usually result in a gene gain-of-function that acts dominantly to increase histone methylation, particularly histone H3 Lys-27-trimethylation (H3K27me3) [100]. Taken together, these data suggest that epigenetic repression of TXNIP by the PCR2-EZH2 complex is involved in the TRX-1 gene-dependent hyperactivation of glucose metabolism in DLBCL and that targeting this pathway by small-molecule inhibitors has significant potential to reverse the resistance of DLBCL to chemotherapy [101].
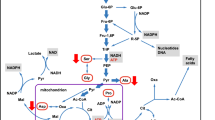
On a different front, the c-myc proto-oncogene has also been shown to be involved in controlling key metabolic pathways in cancer [102, 103]. For instance, MYC overexpression has been recognized in aggressive B-cell lymphomas, primarily due to chromosomal translocations, which inevitably bear an adverse prognosis [104]. The MYC transcriptional network has been also shown to include noncoding microRNA (miRNA) regulators, such as miR-101 and miR-26a, which are linked to the epigenetic control of EZH2 metabolic pathways [105–107]. Previous studies have shown that MYC is transcriptionally regulated by chromatin enhancer functions, which involve the transcription factors NFATc1, NF-κB, and STAT3 [108], underscoring the impact of genetic and/or epigenetic dysregulation of these metabolic pathways in DLBCL (Diagram 2.1).

Epigenetic dysregulation of the metabolic signaling pathways that control cell growth, survival, and chemoresistance mechanisms in DLBCL. Our previous studies have shown that deregulated NF-kB, NFAT, and STAT3 signaling pathways alter the expression of MYC, a key oncogene in DLBCL that is frequently amplified as a result of chromosomal translocations. MYC has recently been shown to negatively regulate miR-101 and miR-26a, which are known to suppress EZH2 expression. Our model hypothesizes that deregulation of EZH2 leads to the epigenetic silencing of the thioredoxin-interacting protein (TXNIP), a key negative regulator of thioredoxin, glucose metabolism, and bcl-6. The result is the hyperactivation of thioredoxin, glucose metabolism, as well as bcl-6, which are highly activated in some DLBCL, which causes uncontrolled tumor cell growth survival, and chemoresistance, a hallmark of lymphomagenesis. Hence, these pathways are rational targets for the design and application of innovative therapies, including theranostic approaches, to specifically reverse the resistance of DLBCL to chemotherapy
The incidence of DLBCL has been rising in recent decades, a situation that underscores the need to improve therapy with greater efficacy and fewer adverse effects. The fundamental problem is that while standard frontline combination chemotherapy of DLBCL with rituximab, cyclophosphamide, hydroxydaunomycin, Oncovin, and prednisone (R-CHOP) achieves lasting therapeutic remissions, it does not usually lead to complete cure. Furthermore, the adverse effects are too toxic for many older patients and pose long-term risks for younger patients. The development of new, affordable, effective, and low-toxicity frontline regimens against DLBCL, which target specific pathways, is feasible but will take many years to achieve and may still be suboptimal if pursued by conventional means.
3 Deregulated Glycolytic Pathway in DLBCL by Monitoring Through Nuclear Imaging
The energy consumed by the cells in the form of adenosine triphosphate (ATP) is generated from two main sources, glycolysis and the tricarboxylic acid (TCA) or Krebs cycle, which are required for normal and malignant cell proliferation and survival. The Warburg effect describes a mechanism by which most cancer cells consume glucose to be converted into ATP via aerobic glycolysis. DLBCL is known to be an aggressive disease, which exhibits high cell proliferation and glucose metabolism rates and influences the response to therapy. As a result of avid glucose consumption, DLBCL cells show higher uptake of fluorine-18F-deoxyglucose (18FDG) by positron-emission tomography (PET) than any other B-cell NHL. Moreover, recent studies link the increased glucose transporter type 1, 2, and 3 (Glut1, 2, and 3) expression and hexokinase II (HKII) activity to the pathogenesis of many hematological malignancies. In support of those studies, we have also found that aggressive B-cell lymphomas express high Glut1, Glut3, and HKII in DLBCL cell lines and primary tumor cells from patients (Fig. 2.1). Several oncogenes and signaling pathways have been implicated in the regulation of cancer cell glycolysis, particularly in DLBCL (Diagram 2.1).

Overexpression of glycolytic pathway proteins in DLBCL. (a) Purified whole cell extracts from normal B lymphocytes obtained from five healthy donors and representative DLBCL cell lines were subjected to Western blot for Glut1, Glut2, HK2, and actin (loading control). (b) HK2 protein expression level in DLBCL cells was compared to normal B lymphocytes. Quantitatively, HK2 protein expression in DLBCL is significantly higher, approximately fourfold higher, in normal B lymphocytes. (c) Tissue microarray (TMA) analysis of HK2 protein expression in 93 cases of primary DLBCL tissue. (d) Table showing the summary of the HK2 expression, low (11%), intermediate (43%), and high (46%) in the TMA
4 Cancer Metabolism and Theranostic Approaches in DLBCL
Theranostics is a novel concept that refers to the integration of imaging diagnostics and therapy, which is emerging as a promising therapeutic paradigm [109]. It is an evolving field related to but different from traditional imaging and therapeutics. It embraces multiple techniques to arrive at in vivo molecular imaging, comprehensive diagnostics, and a personalized treatment regimen. Over the past decade, tremendous effort has been put forth to design and develop methods to produce highly efficient delivery vehicles for theranostic approaches. Liposomes, polymeric nanoparticles (including gold and other metals), dendrimers, carbon nanotubes, and quantum dots are examples of nano-formulations that can be used as multifunctional platforms for cancer theranostics [110]. However, these platforms have their limitations, and they have not been thoroughly developed for effective clinical utilization (Fig. 2.2).

Prognostic significance of interim PET/CT in both the patients who are destined to undergo eight cycles of R-CHOP with interim PET/CT-4 (a) and the patients who are destined to undergo six cycles of R-CHOP with interim PET/CT-3 for OS and PFS, respectively (b). The patients with positive interim PET/CT showed a higher relapse rate (62.8%) than the patients with negative interim PET/CT (12.1%)(P < 0.01) (This figure was adopted from Yang D.H. et al. [12])
In keeping with the unique features of the previously discussed pathways, the most promising target for personalized theranostics is targeting glucose metabolism of cancer cells because unlike normal cells, they metabolize glucose by aerobic glycolysis. Briefly, aerobic glycolysis, also known as the Warburg effect, is characterized by increased glycolysis and lactate production [111], which is often accompanied by increased cellular glucose uptake. Notably, glucose uptake can be imaged in patient tumors by 18FDG-PET [112, 113]. 18FDG-PET is used clinically as a staging tool for diverse types of cancers, including DLBCL [114, 115], and experimental PET tracer probes can distinguish cancer cells from normal cells on the basis of increased glucose metabolism. In addition, positive images of residual 18FDG after therapy are predictive of a poor prognosis and survival of patients with refractory aggressive lymphomas [116, 117]. Initial reports suggested that 18FDG-PET/computed tomography (CT) scans, performed early during treatment (interim PET) after 2–4 courses of CHOP chemotherapy in aggressive B-cell lymphoma (DLBCL in particular), could identify patients who were likely to relapse (Fig. 2.1) [117–119]. However, there are conflicting data on the PET/CT scans performed before treatment (initial PET) in lymphoma patients [120]. On the other hand and irrespective of the interim PET results, the studies indicated that such imaging modalities could swiftly identify lymphoma patients who were likely to respond poorly to induction therapy or frontline treatment, which would prompt an indication for a shift to either intensified regimens or a theranostic approach. Importantly, cancer cells not only consume glucose but also large amounts of glutamine, a key amino acid involved in tumor protein synthesis [121, 122]. Among its various roles, glutamine is a precursor amino acid, which in combination with high glucose levels initiates the hexosamine biosynthetic pathway to synthesize glucosamine [123]. Briefly, fructose-6-phosphate from the glycolytic pathway combines with glutamine in the presence of the initiating enzyme glutamine:fructose-6-phosphate transferase (GFAT) to synthesize glucosamine-6-phosphate. A series of subsequent enzymatic steps lead to the production of uridine diphosphate-N-acetylglucosamine (UDP-GlcNAc), a substrate for O-linked glycosylation that is regulated by the terminating enzyme O-linked GlcNAc transferase (OGT). OGT is the enzyme responsible for the addition of a single N-acetylglucosamine (GlcNAc) residue to the hydroxyl groups of serine and/or threonine residues of target proteins. The hexosamine signaling pathway terminating in O-linked GlcNAc (O-GlcNAc) cycling has been implicated in cellular signaling cascades and regulation of transcription factors involved in cancer biology [124–127]. The biological relevance of the hexosamine biosynthetic signaling pathway has not been completely elucidated, and hence, assessing the impact of altered O-GlcNAc metabolism in tumors such as DLBCL would be useful to determine whether the pathway is a relevant target for the design of personalized theranostics. Analogous to glucose, GlcNAc can be taken up by the cellular glucose transporters [128, 129] and can replace glucose in glucose-depleted cancer cells [130], which supports its relevance as a theranostic probe.
5 Development of Targeted Molecule ECG that Mimics GlcNAc for Theranostic Approaches
We have devised a metabolic agent that mimics GlcNAc by conjugating the chelator ethylenedicysteine (EC) to two molecules of D-glucosamine [131]. The end result is ECG, a metabolic agent containing two molecules of GlcNAc. At the core of ECG is the chelator, which can bind to diagnostic/therapeutic metals, which can then trace and kill cancer cells (Diagram 2.2).

Synthesis of metalic ECG. D-glucosamine hydrochloride salt was added to ethylenedicysteine, giving rise to ethylenedicysteine with two sugar moiety similar to N-Acetyl-Glucosamine on both sides. The core of ECG is the chelator that binds to diagnostic (99mTc, 68Ga, and Gd) or Therapeutic (187Re or 188Re and Cis-Pt) metals
In terms of diagnostic imaging, the technetium-99m-based 99mTc-ECG radiopharmaceutical has been shown to be an effective imaging agent for various types of cancers in both rodents and humans [131, 132]. The biopharmaceutical company Cell>Point is currently sponsoring Phase III clinical trials for its first 99mTc-ECG product for diagnostic imaging in oncology. The multicenter clinical trial is comparing 99mTc-ECG/single-photon emission CT (SPECT) imaging with FDG-PET imaging to assess and stage patients with non-small cell lung cancer. Remarkably, the Phase I/II results indicate that 99mTc-ECG/SPECT has a higher specificity than 18FDG-PET imaging for detecting tumor metastasis and differentiating between inflammation/infection and tumor recurrence [132]. Following this trial, Cell>Point (Centennial, CO) plans to sponsor Phase IV clinical trials to evaluate 99mTc-ECG in non-Hodgkin lymphoma (Diagram 2.2) and other types of cancer, for the diagnosis and staging of the disease process. Unlike FDG, ECG is not taken up into the brain or inflammatory/infection tissues and therefore has a lower false-positive rate in cancer diagnosis. More importantly, ECG has little or no toxicity to normal tissues in the human body, suggesting that ECG is an excellent vehicle to deliver therapeutic metals to cancer cells. The current interest is to integrate ECG imaging function to its therapeutic potential in a theranostic approach to treat refractory DLBCL and highly metabolic cancers.
To date, platinum-based drugs like cisplatin remain one of the most effective classes of chemotherapeutic agents in clinical use. However, the clinical use of cisplatin is quite limited by dose-dependent adverse effects. More effort should be directed to combat the severe systemic toxicity of traditional platinum anticancer agents by designing therapy systems that exclusively deliver platinum or other metallic complexes to tumor cells. To that end, we have chosen two cold metallic agents, rhenium 187 (Re) and a cis-platinum derivative (Pt), for conjugation with our metabolic agent ECG (see Diagram 2.1). We have tested the feasibility of the metal-ECG conjugation technology and the specific targeting of glucose and glutamine metabolism as a novel theranostic approach in refractory DLBCL (Fig. 2.3) (Diagram 2.3) [133].


Effect of metallic Re-ECG in DLBCL. (a) In vitro viability assays were assessed in 14 representative DLBCL cell lines treated with increasing concentration of Re-ECG. (b) Representative DLBCL cell lines sensitive to Re-ECG or less sensitive to Re-ECG. (c) DLBCL cell lines that are sensitive to Re-ECG are highly proliferative in comparison to DLBCL cell lines that are less sensitive to Re-ECG. (d) Cellular uptake of Re-ECG is more significant in DLBCL cells that are sensitive to Re-ECG (LY-10) in comparison to less sensitive cells (MS). (e) Western blot analysis showing Re-ECG inducing the DNA damage marker pH2AX in two representative DLBCL cell lines. (f) Confocal microscopy analysis showing the induction of the DNA damage marker pH2AX in OCI-LY10 cells treated with Re-ECG. (g) Cellular damage by measuring DNA activity with increasing platinum-ECG concentrations. (h) Western blot analysis showing platinum-ECG inducing the DNA damage marker pH2AX in two representative DLBCL cell lines

99mTc-ECG vs. FDG monitoring during the course of therapy in a lymphoma patient. In FDG-PET images, the lesion appears enlarged and fuzzy because of inflammation caused by chemotherapy, while the lesion appears its actual size and has clearer outlines on 99mTc-ECG-SPECT/CT
6 Theranostic Potential of Metallic ECG in DLBCL
Our model proposes that key growth/survival transcription factors (NFATc1 and p65) in DLBCL, which are activated by upstream signaling pathways, are also regulated by glucose metabolism through the O-linked GlcNAc/hexosamine biosynthetic pathway (Diagram 2.4). Fluxes through the hexosamine biosynthetic pathway involve interaction between the substrate of the glycolytic pathway (fructose-6-phosphate) and glutamine, which in the presence of the initiating enzyme GFAT synthesizes glucosamine-6-phosphate. A series of subsequent enzymatic steps leads to production of UDP-GlcNAc, a substrate for O-linked glycosylation that is regulated by the terminating enzyme OGT. OGT is the enzyme responsible for the addition of a single GlcNAc to the hydroxyl groups of serine and/or threonine residues of target proteins. The metabolic agent ECG mimics GlcNAc and can be taken up easily by cancer cells through glucose transporters and hexosamine pathway; it enters the nucleus via “piggybacking” with OGT-modified nuclear proteins. We propose that increased fluxes through the hexosamine biosynthetic pathway accordingly yield elevations in O-GlcNAcylation status in DLBCL, constituting a new pathophysiologic process in the regulation and activation of key transcription factors that control growth/survival mechanisms. ECG, when conjugated to metallic agents, could be a promising theranostic agent, for treating as well as imaging patients’ tumors.

Glucose Metabolism and the Hexosamine Biosynthetic Pathway Link to Key Growth/Survival Signaling Pathways in DLBCL. This diagram depicts the connection between key growth/survial signaling pathways to the metabolic pathway in DLBCL. The hexosamine biosynthetic pathway give rises to UDP-GlcNAC, which is equivelent to ECG, that modifies key transcription factors, such as NFATc1 and NF-kBp65, allowing these transcription factors to migrate to the nucleus and bind to DNA
7 Concluding Remarks
Molecular imaging in oncology has focused on the identification of tumor-specific markers and the application of these markers to evaluate patient response to radiation therapy, chemotherapy, or chemoradiotherapy. The main application of molecular imaging that dominates until now has been the modality known as 18PET/CT, which is intended to help in the evaluation and management of drug dosage for safety and effectiveness. However, 18PET/CT has fallen short to its premise mostly because of the limitations of the tracer drugs to monitor treatment over the course of treatment. More importantly, the radiotracer should have the ability to assess, noninvasively, disease treatment endpoints, which up to now, almost exclusively, rely on the histopathological diagnosis of biopsies because of the inflammatory process after treatments. In order to develop personalized therapies to achieve optimal diagnosis and early cure, the objective is to design metal-based molecular imaging radiotracers that can image the entire repertoire of metabolically active carbohydrate/sugar substrates (glycome). Chelator-based imaging technology is the cornerstone for theranostic applications, which aim to enable the assessment of target therapies and patient selection. For example, L,L-ethylenedicysteine (EC) is a family of bis-aminoethanethiol (BAT) tetradentate ligands that are known to form stable 99mTc(V)O complexes in which an oxotechnetium core is bound to the thiol-sulfur and the amine-nitrogen atoms. One metal which is being used for detecting cancer which is relatively inexpensive has a long half-life, is easily accessible, and has strong 99mTc which has a complexing property of such N2S2-tetraligand systems that can form label protein linkage or peptide linkage. It has been found that EC is a unique chelator because EC has the potential to be involved in signature pathway events. It’s been observed that EC-homing conjugates are able to mimic pathways and monitor changes in the target expression from pre- to posttreatment. Moreover, target-specific biomarkers that are designed as a universal imaging tracer probe, such as ECG, can assess GP, HBP, and broad glycome status from broad to a specific transitional application in cancer and other metabolic diseases; it is conceivable that the knowledge gained will be helpful to optimize therapies against these disorders.
The imaging agent 99mTc-ECG is already in clinical trials for various cancers and has shown great potential to become the next-generation theranostic imaging agents. The premise of therapeutic and diagnostic capabilities of 99mTc-ECG imaging for refractory DLBCL and other types of metabolically active cancers is already in prime time. Such an important approach should have great potential for clinically translatable advances that can have a positive impact on the overall diagnostic and therapeutic process, which will also enhance the quality of life for cancer patients and other diseases of patients.
References
Alexander DD, Mink PJ, Adami HO, Chang ET, Cole P, Mandel JS, et al. The non-Hodgkin lymphomas: a review of the epidemiologic literature. Int J Cancer. 2007;120(Suppl 12):1–39. Epub 2007/04/04. doi:10.1002/ijc.22719. PubMed PMID: 17405121.
Johnson PW. Survival from non-Hodgkin lymphoma in England and Wales up to 2001. Br J Cancer. 2008;99(Suppl 1):S107–9. PubMed PMID: 18813239.
Morton LM, Wang SS, Devesa SS, Hartge P, Weisenburger DD, Linet MS. Lymphoma incidence patterns by WHO subtype in the United States, 1992–2001. Blood. 2006;107(1):265–76. PubMed PMID: 16150940.
Wang M, Burau KD, Fang S, Wang H, Du XL. Ethnic variations in diagnosis, treatment, socioeconomic status, and survival in a large population-based cohort of elderly patients with non-Hodgkin lymphoma. Cancer. 2008;113:3231. PubMed PMID: 18937267.
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96. PubMed PMID: 18287387.
Coiffier B. Current strategies for the treatment of diffuse large B cell lymphoma. Curr Opin Hematol. 2005;12(4):259–65. PubMed PMID: 15928481.
Coiffier B. Treatment of non-Hodgkin’s lymphoma: a look over the past decade. Clin Lymphoma Myeloma. 2006;7(Suppl 1):S7–13. PubMed PMID: 17101073.
Lossos IS. Molecular pathogenesis of diffuse large B-cell lymphoma. J Clin Oncol. 2005;23(26):6351–7. PubMed PMID: 16155019.
Volpe G, Vitolo U, Carbone A, Pastore C, Bertini M, Botto B, et al. Molecular heterogeneity of B-lineage diffuse large cell lymphoma. Genes Chromosomes Cancer. 1996;16(1):21–30. PubMed PMID: 9162193.
Jost PJ, Ruland J. Aberrant NF-kappaB signaling in lymphoma: mechanisms, consequences, and therapeutic implications. Blood. 2007;109(7):2700–7. PubMed PMID: 17119127.
Lam LT, Wright G, Davis RE, Lenz G, Farinha P, Dang L, et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood. 2008;111(7):3701–13. PubMed PMID: 18160665.
Sagaert X, De Wolf-Peeters C, Noels H, Baens M. The pathogenesis of MALT lymphomas: where do we stand? Leukemia. 2007;21(3):389–96. PubMed PMID: 17230229.
Feuerhake F, Kutok JL, Monti S, Chen W, LaCasce AS, Cattoretti G, et al. NFkappaB activity, function, and target-gene signatures in primary mediastinal large B-cell lymphoma and diffuse large B-cell lymphoma subtypes. Blood. 2005;106(4):1392–9. PubMed PMID: 15870177.
Staudt LM, Dave S. The biology of human lymphoid malignancies revealed by gene expression profiling. Adv Immunol. 2005;87:163–208. PubMed PMID: 16102574.
Bea S, Zettl A, Wright G, Salaverria I, Jehn P, Moreno V, et al. Diffuse large B-cell lymphoma subgroups have distinct genetic profiles that influence tumor biology and improve gene-expression-based survival prediction. Blood. 2005;106(9):3183–90. PubMed PMID: 16046532.
Iqbal J, Greiner TC, Patel K, Dave BJ, Smith L, Ji J, et al. Distinctive patterns of BCL6 molecular alterations and their functional consequences in different subgroups of diffuse large B-cell lymphoma. Leukemia. 2007;21:2332. PubMed PMID: 17625604.
Moskowitz CH, Zelenetz AD, Kewalramani T, Hamlin P, Lessac-Chenen S, Houldsworth J, et al. Cell of origin, germinal center versus nongerminal center, determined by immunohistochemistry on tissue microarray, does not correlate with outcome in patients with relapsed and refractory DLBCL. Blood. 2005;106(10):3383–5. PubMed PMID: 16091454.
Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–11. PubMed PMID: 10676951.
Rosenwald A, Wright G, Leroy K, Yu X, Gaulard P, Gascoyne RD, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003;198(6):851–62. PubMed PMID: 12975453.
Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2003;100(17):9991–6. PubMed PMID: 12900505.
Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood. 2005;105(5):1851–61. PubMed PMID: 15550490.
Rosenwald A, Staudt LM. Gene expression profiling of diffuse large B-cell lymphoma. Leuk Lymphoma. 2003;44(Suppl 3):S41–7. PubMed PMID: 15202524.
Saito M, Gao J, Basso K, Kitagawa Y, Smith PM, Bhagat G, et al. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell. 2007;12(3):280–92. PubMed PMID: 17785208.
Parekh S, Polo JM, Shaknovich R, Juszczynski P, Lev P, Ranuncolo SM, et al. BCL6 programs lymphoma cells for survival and differentiation through distinct biochemical mechanisms. Blood. 2007;110(6):2067–74. PubMed PMID: 17545502.
Polo JM, Juszczynski P, Monti S, Cerchietti L, Ye K, Greally JM, et al. Transcriptional signature with differential expression of BCL6 target genes accurately identifies BCL6-dependent diffuse large B cell lymphomas. Proc Natl Acad Sci U S A. 2007;104(9):3207–12. PubMed PMID: 17360630.
Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. PubMed PMID: 15771565.
Zhang G. Tumor necrosis factor family ligand-receptor binding. Curr Opin Struct Biol. 2004;14(2):154–60. PubMed PMID: 15093829.
Eliopoulos AG, Young LS. The role of the CD40 pathway in the pathogenesis and treatment of cancer. Curr Opin Pharmacol. 2004;4(4):360–7. PubMed PMID: 15251129.
Shivakumar L, Ansell S. Targeting B-lymphocyte stimulator/B-cell activating factor and a proliferation-inducing ligand in hematologic malignancies. Clin Lymphoma Myeloma. 2006;7(2):106–8. PubMed PMID: 17026820.
Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death--a new approach to cancer therapy. J Clin Invest. 2005;115(10):2625–32. PubMed PMID: 16200195.
Lin-Lee YC, Pham LV, Tamayo AT, Fu L, Zhou HJ, Yoshimura LC, et al. Nuclear localization in the biology of the CD40 receptor in normal and neoplastic human B lymphocytes. J Biol Chem. 2006;281(27):18878–18887. PubMed PMID: 16644731.
Pham LV, Tamayo AT, Yoshimura LC, Lo P, Terry N, Reid PS, et al. A CD40 Signalosome anchored in lipid rafts leads to constitutive activation of NF-kappaB and autonomous cell growth in B cell lymphomas. Immunity. 2002;16(1):37–50. Epub 2002/02/05. doi: S1074761301002588 [pii]. PubMed PMID: 11825564.
Zhou HJ, Pham LV, Tamayo AT, Lin-Lee YC, Fu L, Yoshimura LC, et al. Nuclear CD40 interacts with c-Rel and enhances proliferation in aggressive B-cell lymphoma. Blood. 2007;110(6):2121–2127. Epub 2007/06/15. doi: blood-2007-02-073080 [pii] 10.1182/blood-2007-02-073080. PubMed PMID: 17567982; PubMed Central PMCID: PMC1976364.
Pham LV, Tamayo AT, Yoshimura LC, Lin-Lee YC, Ford RJ. Constitutive NF-kappaB and NFAT activation in aggressive B-cell lymphomas synergistically activates the CD154 gene and maintains lymphoma cell survival. Blood. 2005;106(12):3940–3947. Epub 2005/08/16. doi: 2005–03-1167 [pii] 10.1182/blood-2005-03-1167. PubMed PMID: 16099873; PubMed Central PMCID: PMC1895110.
Pham LV, Tamayo AT, Yoshimura LC, Lo P, Ford RJ. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol. 2003;171(1):88–95. Epub 2003/06/21. PubMed PMID: 12816986.
Pham LV, Tamayo AT, Zhou H-J, Lin-Lee Y-C, Fu L, Bueso-Ramos C, Medeiros LJ, Ford RJ. Networking NFkB signaling modules from canonical and alternative NFkB pathways regulate growth and survival in large B cell lymphomas. submitted for publication. 2008.
Basak S, Kim H, Kearns JD, Tergaonkar V, O'Dea E, Werner SL, et al. A fourth IkappaB protein within the NF-kappaB signaling module. Cell. 2007;128(2):369–381. PubMed PMID: 17254973.
Dejardin E. The alternative NF-kappaB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem Pharmacol. 2006;72(9):1161–1179. PubMed PMID: 16970925.
Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8(1):49–62. PubMed PMID: 17183360.
Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006(357):re13. PubMed PMID: 17047224.
Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344–362. PubMed PMID: 18267068.
Scheidereit C. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene. 2006;25(51):6685–6705. PubMed PMID: 17072322.
Xiao G, Rabson AB, Young W, Qing G, Qu Z. Alternative pathways of NF-kappaB activation: a double-edged sword in health and disease. Cytokine Growth Factor Rev. 2006;17(4):281–293. PubMed PMID: 16793322.
Ishikawa H, Carrasco D, Claudio E, Ryseck RP, Bravo R. Gastric hyperplasia and increased proliferative responses of lymphocytes in mice lacking the COOH-terminal ankyrin domain of NF-kappaB2. J Exp Med. 1997;186(7):999–1014. PubMed PMID: 9314550.
Beinke S, Ley SC. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem J. 2004;382(Pt 2):393–409. PubMed PMID: 15214841.
Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301–310. PubMed PMID: 12001991.
Neri A, Chang CC, Lombardi L, Salina M, Corradini P, Maiolo AT, et al. B cell lymphoma-associated chromosomal translocation involves candidate oncogene lyt-10, homologous to NF-kappa B p50. Cell. 1991;67(6):1075–1087. PubMed PMID: 1760839.
Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity. 2008;28(3):391–401. PubMed PMID: 18313334.
Cancro MP. Living in context with the survival factor BAFF. Immunity. 2008;28(3):300–301. PubMed PMID: 18342003.
Sasaki Y, Calado DP, Derudder E, Zhang B, Shimizu Y, Mackay F, et al. NIK overexpression amplifies, whereas ablation of its TRAF3-binding domain replaces BAFF:BAFF-R-mediated survival signals in B cells. Proc Natl Acad Sci U S A. 2008;105(31):10883–10888. PubMed PMID: 18663224.
Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12(2):115–130. PubMed PMID: 17692804.
Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12(2):131–144. PubMed PMID: 17692805.
Gilmore TD. Multiple myeloma: lusting for NF-kappaB. Cancer Cell. 2007;12(2):95–97. PubMed PMID: 17692798.
Zarnegar B, Yamazaki S, He JQ, Cheng G. Control of canonical NF-kappaB activation through the NIK-IKK complex pathway. Proc Natl Acad Sci U S A. 2008;105(9):3503–3508. PubMed PMID: 18292232.
Kim SW, Oleksyn DW, Rossi RM, Jordan CT, Sanz I, Chen L, et al. Protein kinase C-associated kinase is required for NF-kappaB signaling and survival in diffuse large B-cell lymphoma cells. Blood. 2008;111(3):1644–1653. PubMed PMID: 18025152.
Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. 2008;105(36):13520–13525. Epub 2008/09/04. doi: 0804295105 [pii] 10.1073/pnas.0804295105. PubMed PMID: 18765795; PubMed Central PMCID: PMC2533222.
Homig-Holzel C, Hojer C, Rastelli J, Casola S, Strobl LJ, Muller W, et al. Constitutive CD40 signaling in B cells selectively activates the noncanonical NF-kappaB pathway and promotes lymphomagenesis. J Exp Med. 2008;205(6):1317–1329. PubMed PMID: 18490492.
Serfling E, Berberich-Siebelt F, Avots A, Chuvpilo S, Klein-Hessling S, Jha MK, et al. NFAT and NF-kappaB factors-the distant relatives. Int J Biochem Cell Biol. 2004;36(7):1166–1170. PubMed PMID: 15109564.
Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17(18):2205–2232. PubMed PMID: 12975316.
Fu L, Lin-Lee YC, Pham LV, Tamayo A, Yoshimura L, Ford RJ. Constitutive NF-kappaB and NFAT activation leads to stimulation of the BLyS survival pathway in aggressive B-cell lymphomas. Blood. 2006;107(11):4540–4548. Epub 2006/02/25. doi: 2005–10-4042 [pii] 10.1182/blood-2005-10-4042. PubMed PMID: 16497967; PubMed Central PMCID: PMC1895801.
Duncliffe KN, Bert AG, Vadas MA, Cockerill PN. A T cell-specific enhancer in the interleukin-3 locus is activated cooperatively by Oct and NFAT elements within a DNase I-hypersensitive site. Immunity. 1997;6(2):175–185. PubMed PMID: 9047239.
Hawwari A, Burrows J, Vadas MA, Cockerill PN. The human IL-3 locus is regulated cooperatively by two NFAT-dependent enhancers that have distinct tissue-specific activities. J Immunol. 2002;169(4):1876–1886. PubMed PMID: 12165512.
Cockerill PN. NFAT is well placed to direct both enhancer looping and domain-wide models of enhancer function. Sci Signal. 2008;1(13):pe15. PubMed PMID: 18385038.
Cockerill PN. Mechanisms of transcriptional regulation of the human IL-3/GM-CSF locus by inducible tissue-specific promoters and enhancers. Crit Rev Immunol. 2004;24(6):385–408. PubMed PMID: 15777160.
Banine F, Bartlett C, Gunawardena R, Muchardt C, Yaniv M, Knudsen ES, et al. SWI/SNF chromatin-remodeling factors induce changes in DNA methylation to promote transcriptional activation. Cancer Res. 2005;65(9):3542–3547. PubMed PMID: 15867346.
Bert AG, Johnson BV, Baxter EW, Cockerill PN. A modular enhancer is differentially regulated by GATA and NFAT elements that direct different tissue-specific patterns of nucleosome positioning and inducible chromatin remodeling. Mol Cell Biol. 2007;27(8):2870–2885. PubMed PMID: 17283044.
Wurster AL, Pazin MJ. BRG1-mediated chromatin remodeling regulates differentiation and gene expression of T helper cells. Mol Cell Biol. 2008. PubMed PMID: 18852284.
Bajpai R, Matulis SM, Wei C, Nooka AK, Von Hollen HE, Lonial S, et al. Targeting glutamine metabolism in multiple myeloma enhances BIM binding to BCL-2 eliciting synthetic lethality to venetoclax. Oncogene. 2015. doi:10.1038/onc.2015.464. PubMed PMID: 26640142.
Tsytsykova AV, Rajsbaum R, Falvo JV, Ligeiro F, Neely SR, Goldfeld AE. Activation-dependent intrachromosomal interactions formed by the TNF gene promoter and two distal enhancers. Proc Natl Acad Sci U S A. 2007;104(43):16850–16855. PubMed PMID: 17940009.
Johnson BV, Bert AG, Ryan GR, Condina A, Cockerill PN. Granulocyte-macrophage colony-stimulating factor enhancer activation requires cooperation between NFAT and AP-1 elements and is associated with extensive nucleosome reorganization. Mol Cell Biol. 2004;24(18):7914–7930. PubMed PMID: 15340054.
Bengsch B, Wherry EJ. The importance of cooperation: partnerless NFAT induces T cell exhaustion. Immunity. 2015;42(2):203–205. doi: 10.1016/j.immuni.2015.01.023. PubMed PMID: 25692694.
Fehr T, Lucas CL, Kurtz J, Onoe T, Zhao G, Hogan T, et al. A CD8 T cell-intrinsic role for the calcineurin-NFAT pathway for tolerance induction in vivo. Blood. 2010;115(6):1280–1287. doi: 10.1182/blood-2009-07-230680. PubMed PMID: 20007805; PubMed Central PMCID: PMC2826238.
Abe BT, Shin DS, Mocholi E, Macian F. NFAT1 supports tumor-induced anergy of CD4(+) T cells. Cancer Res. 2012;72(18):4642–4651. doi: 10.1158/0008-5472.CAN-11-3775. PubMed PMID: 22865456; PubMed Central PMCID: PMC3445721.
Martinez GJ, Pereira RM, Aijo T, Kim EY, Marangoni F, Pipkin ME, et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity. 2015;42(2):265–278. doi: 10.1016/j.immuni.2015.01.006. PubMed PMID: 25680272; PubMed Central PMCID: PMC4346317.
Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 regulates PD-1 expression upon T cell activation. J Immunol. 2008;181(7):4832–4839. PubMed PMID: 18802087; PubMed Central PMCID: PMC2645436.
Lozano T, Villanueva L, Durantez M, Gorraiz M, Ruiz M, Belsue V, et al. Inhibition of FOXP3/NFAT interaction enhances T cell function after TCR stimulation. J Immunol. 2015;195(7):3180–3189. doi: 10.4049/jimmunol.1402997. PubMed PMID: 26324768.
Taylor A, Harker JA, Chanthong K, Stevenson PG, Zuniga EI, Rudd CE. Glycogen synthase kinase 3 inactivation drives T-bet-mediated downregulation of co-receptor PD-1 to enhance CD8(+) cytolytic T cell responses. Immunity. 2016;44(2):274–286. doi: 10.1016/j.immuni.2016.01.018. PubMed PMID: 26885856; PubMed Central PMCID: PMC4760122.
Sagiv-Barfi I, Kohrt HE, Czerwinski DK, Ng PP, Chang BY, Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci U S A. 2015;112(9):E966–E972. doi: 10.1073/pnas.1500712112. PubMed PMID: 25730880; PubMed Central PMCID: PMC4352777.
Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122(15):2539–2549. doi: 10.1182/blood-2013-06-507947. PubMed PMID: 23886836; PubMed Central PMCID: PMC3795457.
Li C, Thompson MA, Tamayo AT, Zuo Z, Lee J, Vega F, et al. Over-expression of Thioredoxin-1 mediates growth, survival, and chemoresistance and is a druggable target in diffuse large B-cell lymphoma. Oncotarget. 2012. Epub 2012/03/27. doi:463 [pii]. PubMed PMID: 22447839.
Gromer S, Urig S, Becker K. The thioredoxin system--from science to clinic. Med Res Rev. 2004;24(1):40–89. Epub 2003/11/05. doi: 10.1002/med.10051. PubMed PMID: 14595672.
Arner ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267(20):6102–6109. Epub 2000/09/30. doi: ejb1701 [pii]. PubMed PMID: 11012661.
Powis G, Kirkpatrick DL. Thioredoxin signaling as a target for cancer therapy. Curr Opin Pharmacol. 2007;7(4):392–397. Epub 2007/07/06. doi: S1471–4892(07)00091–4 [pii] 10.1016/j.coph.2007.04.003. PubMed PMID: 17611157.
Baker AF, Koh MY, Williams RR, James B, Wang H, Tate WR, et al. Identification of thioredoxin-interacting protein 1 as a hypoxia-inducible factor 1alpha-induced gene in pancreatic cancer. Pancreas. 2008;36(2):178–186. Epub 2008/04/01. doi: 10.1097/MPA.0b013e31815929fe00006676-200803000-00012 [pii]. PubMed PMID: 18376310.
Welsh SJ, Bellamy WT, Briehl MM, Powis G. The redox protein thioredoxin-1 (Trx-1) increases hypoxia-inducible factor 1alpha protein expression: Trx-1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res. 2002;62(17):5089–5095. Epub 2002/09/05. PubMed PMID: 12208766.
Tonissen KF, Di Trapani G. Thioredoxin system inhibitors as mediators of apoptosis for cancer therapy. Mol Nutr Food Res. 2009;53(1):87–103. Epub 2008/11/04. doi: 10.1002/mnfr.200700492. PubMed PMID: 18979503.
Tome ME, Johnson DB, Rimsza LM, Roberts RA, Grogan TM, Miller TP, et al. A redox signature score identifies diffuse large B-cell lymphoma patients with a poor prognosis. Blood. 2005;106(10):3594–3601. Epub 2005/08/06. doi: 2005–02-0487 [pii] 10.1182/blood-2005-02-0487. PubMed PMID: 16081686; PubMed Central PMCID: PMC1895056.
Junn E, Han SH, Im JY, Yang Y, Cho EW, Um HD, et al. Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J Immunol. 2000;164(12):6287–6295. Epub 2000/06/08. doi: ji_v164n12p6287 [pii]. PubMed PMID: 10843682.
Stoltzman CA, Peterson CW, Breen KT, Muoio DM, Billin AN, Ayer DE. Glucose sensing by MondoA:Mlx complexes: a role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc Natl Acad Sci U S A. 2008;105(19):6912–6917. Epub 2008/05/07. doi: 0712199105 [pii] 10.1073/pnas.0712199105. PubMed PMID: 18458340; PubMed Central PMCID: PMC2383952.
Stoltzman CA, Kaadige MR, Peterson CW, Ayer DE. MondoA senses non-glucose sugars: regulation of thioredoxin-interacting protein (TXNIP) and the hexose transport curb. J Biol Chem. 2011;286(44):38027–38034. Epub 2011/09/13. doi: M111.275503 [pii] 10.1074/jbc.M111.275503. PubMed PMID: 21908621; PubMed Central PMCID: PMC3207397.
Zhou J, Yu Q, Chng WJ. TXNIP (VDUP-1, TBP-2): a major redox regulator commonly suppressed in cancer by epigenetic mechanisms. Int J Biochem Cell Biol. 2011;43(12):1668–1673. Epub 2011/10/04. doi: S1357–2725(11)00257–3 [pii] 10.1016/j.biocel.2011.09.005. PubMed PMID: 21964212.
Yoshioka J, Chutkow WA, Lee S, Kim JB, Yan J, Tian R, et al. Deletion of thioredoxin-interacting protein in mice impairs mitochondrial function but protects the myocardium from ischemia-reperfusion injury. J Clin Invest. 2012;122(1):267–279. Epub 2011/12/29. doi: 44927 [pii] 10.1172/JCI44927. PubMed PMID: 22201682; PubMed Central PMCID: PMC3248280.
Butler LM, Zhou X, Xu WS, Scher HI, Rifkind RA, Marks PA, et al. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl Acad Sci U S A. 2002;99(18):11700–11705. Epub 2002/08/22. doi: 10.1073/pnas.182372299182372299 [pii]. PubMed PMID: 12189205; PubMed Central PMCID: PMC129332.
Zhou J, Bi C, Cheong LL, Mahara S, Liu SC, Tay KG, et al. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood. 2011;118(10):2830–2839. Epub 2011/07/08. doi: blood-2010-07-294827 [pii] 10.1182/blood-2010-07-294827. PubMed PMID: 21734239.
Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21(9):1050–1063. Epub 2007/04/18. doi: gad.1524107 [pii] 10.1101/gad.1524107. PubMed PMID: 17437993; PubMed Central PMCID: PMC1855231.
Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343–349. Epub 2011/01/21. doi: nature09784 [pii] 10.1038/nature09784. PubMed PMID: 21248841.
Chase A, Cross NC. Aberrations of EZH2 in cancer. Clin Cancer Res. 2011;17(9):2613–2618. Epub 2011/03/04. doi: 1078–0432.CCR-10-2156 [pii] 10.1158/1078-0432.CCR-10-2156. PubMed PMID: 21367748.
Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109(10):3879–3884. Epub 2012/02/22. doi: 1121343109 [pii] 10.1073/pnas.1121343109. PubMed PMID: 22343534.
Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181–185. Epub 2010/01/19. doi: ng.518 [pii] 10.1038/ng.518. PubMed PMID: 20081860; PubMed Central PMCID: PMC2850970.
Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117(8):2451–2459. Epub 2010/12/31. doi: blood-2010-11-321208 [pii] 10.1182/blood-2010-11-321208. PubMed PMID: 21190999; PubMed Central PMCID: PMC3062411.
Jeong M, Piao ZH, Kim MS, Lee SH, Yun S, Sun HN, et al. Thioredoxin-interacting protein regulates hematopoietic stem cell quiescence and mobilization under stress conditions. J Immunol. 2009;183(4):2495–2505. Epub 2009/07/25. doi: jimmunol.0804221 [pii] 10.4049/jimmunol.0804221. PubMed PMID: 19625652.
Sloan EJ, Ayer DE. Myc, mondo, and metabolism. Genes Cancer. 2010;1(6):587–596. doi: 10.1177/1947601910377489. PubMed PMID: 21113411; PubMed Central PMCID: PMC2992335.
Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, Metabolism, and Cancer. Cancer Discovery. 2015;5(10):1024–1039. doi: 10.1158/2159-8290.CD-15-0507. PubMed PMID: 26382145; PubMed Central PMCID: PMC4592441.
Hu S, Xu-Monette ZY, Tzankov A, Green T, Wu L, Balasubramanyam A, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood. 2013;121(20):4021–4031; quiz 250. doi: 10.1182/blood-2012-10-460063. PubMed PMID: 23449635; PubMed Central PMCID: PMC3709650.
Zhang X, Chen X, Lin J, Lwin T, Wright G, Moscinski LC, et al. Myc represses miR-15a/miR-16-1 expression through recruitment of HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas. Oncogene. 2012;31(24):3002–3008. doi: 10.1038/onc.2011.470. PubMed PMID: 22002311; PubMed Central PMCID: PMC3982396.
Zhang X, Zhao X, Fiskus W, Lin J, Lwin T, Rao R, et al. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell. 2012;22(4):506–523. doi: 10.1016/j.ccr.2012.09.003. PubMed PMID: 23079660; PubMed Central PMCID: PMC3973134.
Zhao X, Lwin T, Zhang X, Huang A, Wang J, Marquez VE, et al. Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell lymphoma survival and clonogenicity. Leukemia. 2013;27(12):2341–2350. doi: 10.1038/leu.2013.94. PubMed PMID: 23538750; PubMed Central PMCID: PMC4015113.
Pham LV, Tamayo AT, Li C, Bueso-Ramos C, Ford RJ. An epigenetic chromatin remodeling role for NFATc1 in transcriptional regulation of growth and survival genes in diffuse large B-cell lymphomas. Blood. 2010;116(19):3899–3906. Epub 2010/07/29. doi: blood-2009-12-257378 [pii] 10.1182/blood-2009-12-257378. PubMed PMID: 20664054; PubMed Central PMCID: PMC2981542.
Kelkar SS, Reineke TM. Theranostics: combining imaging and therapy. Bioconjug Chem. 2011;22(10):1879–1903. Epub 2011/08/13. doi: 10.1021/bc200151q. PubMed PMID: 21830812.
Fernandez-Fernandez A, Manchanda R, McGoron AJ. Theranostic applications of nanomaterials in cancer: drug delivery, image-guided therapy, and multifunctional platforms. Appl Biochem Biotechnol. 2011;165(7–8):1628–1651. Epub 2011/09/29. doi: 10.1007/s12010-011-9383-z. PubMed PMID: 21947761; PubMed Central PMCID: PMC3239222.
Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11(5):325–337. Epub 2011/04/22. doi: nrc3038 [pii] 10.1038/nrc3038. PubMed PMID: 21508971.
Kubota K. From tumor biology to clinical PET: a review of positron emission tomography (PET) in oncology. Ann Nucl Med. 2001;15(6):471–486. Epub 2002/02/08. PubMed PMID: 11831394.
Weber WA, Schwaiger M, Avril N. Quantitative assessment of tumor metabolism using FDG-PET imaging. Nucl Med Biol. 2000;27(7):683–687. Epub 2000/11/25. doi: S0969–8051(00)00141–4 [pii]. PubMed PMID: 11091112.
Cheson BD. Role of functional imaging in the management of lymphoma. J Clin Oncol. 2011;29(14):1844–1854. Epub 2011/04/13. doi: JCO.2010.32.5225 [pii] 10.1200/JCO.2010.32.5225. PubMed PMID: 21482982.
Juweid ME, Stroobants S, Hoekstra OS, Mottaghy FM, Dietlein M, Guermazi A, et al. Use of positron emission tomography for response assessment of lymphoma: consensus of the Imaging Subcommittee of International Harmonization Project in Lymphoma. J Clin Oncol. 2007;25(5):571–578. Epub 2007/01/24. doi: JCO.2006.08.2305 [pii] 10.1200/JCO.2006.08.2305. PubMed PMID: 17242397.
Hutchings M, Barrington SF. PET/CT for therapy response assessment in lymphoma. J Nucl Med. 2009;50 Suppl 1:21S–30S. Epub 2009/04/22. doi: jnumed.108.057190 [pii] 10.2967/jnumed.108.057190. PubMed PMID: 19380407.
Spaepen K, Stroobants S, Dupont P, Van Steenweghen S, Thomas J, Vandenberghe P, et al. Prognostic value of positron emission tomography (PET) with fluorine-18 fluorodeoxyglucose ([18F]FDG) after first-line chemotherapy in non-Hodgkin’s lymphoma: is [18F]FDG-PET a valid alternative to conventional diagnostic methods? J Clin Oncol. 2001;19(2):414–419. Epub 2001/02/24. PubMed PMID: 11208833.
Jerusalem G, Beguin Y, Fassotte MF, Najjar F, Paulus P, Rigo P, et al. Persistent tumor 18F-FDG uptake after a few cycles of polychemotherapy is predictive of treatment failure in non-Hodgkin's lymphoma. Haematologica. 2000;85(6):613–618. Epub 2000/06/28. PubMed PMID: 10870118.
Yang DH, Min JJ, Song HC, Jeong YY, Chung WK, Bae SY, et al. Prognostic significance of interim (1)F-FDG PET/CT after three or four cycles of R-CHOP chemotherapy in the treatment of diffuse large B-cell lymphoma. Eur J Cancer. 2011;47(9):1312–8. Epub 2011/02/22. doi: S0959–8049(11)00038–4 [pii]10.1016/j.ejca.2010.12.027. PubMed PMID: 21334197.
Cox MC, Ambrogi V, Lanni V, Cavalieri E, Pelliccia S, Scopinaro F, et al. Use of interim [(18)F]fluorodeoxyglucose-positron emission tomography is not justified in diffuse large B-cell lymphoma during first-line immunochemotherapy. Leuk Lymphoma. 2012;53(2):263–269. Epub 2011/08/19. doi: 10.3109/10428194.2011.614704. PubMed PMID: 21846184.
Rajagopalan KN, DeBerardinis RJ. Role of glutamine in cancer: therapeutic and imaging implications. J Nucl Med. 2011;52(7):1005–1008. Epub 2011/06/18. doi: jnumed.110.084244 [pii] 10.2967/jnumed.110.084244. PubMed PMID: 21680688.
Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35(8):427–433. Epub 2010/06/24. doi: S0968–0004(10)00091–5 [pii] 10.1016/j.tibs.2010.05.003. PubMed PMID: 20570523; PubMed Central PMCID: PMC2917518.
Hanover JA, Krause MW, Love DC. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim Biophys Acta. 2010;1800(2):80–95. Epub 2009/08/04. doi: S0304–4165(09)00207–4 [pii] 10.1016/j.bbagen.2009.07.017. PubMed PMID: 19647043; PubMed Central PMCID: PMC2815088.
Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–858. Epub 2011/03/12. doi: 10.1146/annurev-biochem-060608-102511. PubMed PMID: 21391816.
Lynch TP, Ferrer CM, Jackson SR, Shahriari KS, Vosseller K, Reginato MJ. Critical role of O-GlcNAc transferase in prostate cancer invasion, angiogenesis and metastasis. J Biol Chem. 2012. Epub 2012/01/26. doi: M111.302547 [pii] 10.1074/jbc.M111.302547. PubMed PMID: 22275356.
Krzeslak A, Forma E, Bernaciak M, Romanowicz H, Brys M. Gene expression of O-GlcNAc cycling enzymes in human breast cancers. Clin Exp Med. 2011. Epub 2011/05/14. doi: 10.1007/s10238-011-0138-5. PubMed PMID: 21567137.
Ozcan S, Andrali SS, Cantrell JE. Modulation of transcription factor function by O-GlcNAc modification. Biochim Biophys Acta. 2010;1799(5–6):353–364. Epub 2010/03/06. doi: S1874–9399(10)00047–7 [pii] 10.1016/j.bbagrm.2010.02.005. PubMed PMID: 20202486; PubMed Central PMCID: PMC2881704.
Rogacka D, Piwkowska A, Jankowski M, Kocbuch K, Dominiczak MH, Stepinski JK, et al. Expression of GFAT1 and OGT in podocytes: transport of glucosamine and the implications for glucose uptake into these cells. J Cell Physiol. 2010;225(2):577–584. Epub 2010/05/28. doi: 10.1002/jcp.22242. PubMed PMID: 20506529.
Uldry M, Ibberson M, Hosokawa M, Thorens B. GLUT2 is a high affinity glucosamine transporter. FEBS Lett. 2002;524(1–3):199–203. Epub 2002/07/24. doi: S0014579302030582 [pii]. PubMed PMID: 12135767.
Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW, et al. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 2010;24(24):2784–2799. Epub 2010/11/26. doi: gad.1985910 [pii] 10.1101/gad.1985910. PubMed PMID: 21106670; PubMed Central PMCID: PMC3003197.
Yang DJ, Kim CG, Schechter NR, Azhdarinia A, Yu DF, Oh CS, et al. Imaging with 99mTc ECDG targeted at the multifunctional glucose transport system: feasibility study with rodents. Radiology. 2003;226(2):465–473. doi: 10.1148/radiol.2262011811. PubMed PMID: 12563141.
Yang D, Yukihiro M, Yu DF, Ito M, Oh CS, Kohanim S, et al. Assessment of therapeutic tumor response using 99mtc-ethylenedicysteine-glucosamine. Cancer Biother Radiopharm. 2004;19(4):443–456. Epub 2004/09/30. doi: 10.1089/cbr.2004.19.443. PubMed PMID: 15453959.
Yang DJ, Kong F-L, Oka T, Bryant JL. Molecular imaging kits for hexosamine biosynthetic pathway in oncology. Curr Med Chem. 2012;19(20):3310–4.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Pham, L.V., Bryant, J.L., Yang, D., Ford, R.J. (2017). Theranostic Approaches for Pathway-Activated Systems in Oncology. In: Inoue, T., Yang, D., Huang, G. (eds) Personalized Pathway-Activated Systems Imaging in Oncology. Springer, Singapore. https://doi.org/10.1007/978-981-10-3349-0_2
Download citation
DOI: https://doi.org/10.1007/978-981-10-3349-0_2
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-3348-3
Online ISBN: 978-981-10-3349-0
eBook Packages: MedicineMedicine (R0)