
Abstract
Epithelial-mesenchymal transition (EMT) is an evolutionary conserved morphogenetic program necessary for the shaping of the body plan during development. It is guided precisely by growth factor signaling and a dedicated network of specialised transcription factors. These are supported by other transcription factor families serving auxiliary functions during EMT, beyond their general roles as effectors of major signaling pathways. EMT transiently induces in epithelial cells mesenchymal properties, such as the loss of cell-cell adhesion and a gain in cell motility. Together, these newly acquired properties enable their migration to distant sites where they eventually give rise to adult epithelia. However, it is now recognized that EMT contributes to the pathogenesis of several human diseases, notably in tissue fibrosis and cancer metastasis. The RUNX family of transcription factors are important players in cell fate determination during development, where their spatio-temporal expression often overlaps with the occurrence of EMT. Furthermore, the dysregulation of RUNX expression and functions are increasingly linked to the aberrant induction of EMT in cancer. The present chapter reviews the current knowledge of this emerging field and the common themes of RUNX involvement during EMT, with the intention of fostering future research.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Epithelial-mesenchymal transition (EMT)
- RUNX
- Auxiliary transcription factors
- Atrioventricular valve
- Mammary gland development
- Lacrimal gland repair
- Cancer metastasis
- Osteomimicry
1 Introduction
Metazoa are composed of three major cell types: epithelial, mesenchymal and neural. Epithelial cells are in close contact with one another and organized in sheets with apico–basal polarity. Within this arrangement, epithelial cells communicate with one another through an ordered series of cell–cell junctional complexes: adherens junctions, desmosomes and tight junctions. Through these contacts, the epithelium serves its function as a selective barrier. In contrast, mesenchymal cells are loosely organized within a three-dimensional extracellular matrix. Their main purpose is to act as connective tissues that provide structural support to the epithelia. Unlike epithelial cells, mesenchymal cells can function independently and are capable of migration, especially during development . The process Epithelial-Mesenchymal Transition (EMT) refers to a cellular program during normal development in which epithelial cells lose their junctional complexes to acquire mesenchymal properties, such as migratory and invasive capabilities. Following EMT, cells will often undergo the reverse process, referred to as Mesenchymal-Epithelial Transition (MET). EMT is a highly conserved morphogenetic process in evolution for the shaping of the body plan in metazoans. EMT and MET operate sequentially throughout embryogenesis and during organogenesis. A round of EMT–MET is reactivated in the adult stage during wound healing and EMT in the kidney epithelium induces fibrosis in the stroma. An EMT–MET round has also been proposed to be activated during the progression of carcinoma.
There is commonality between the process of EMT and the RUNX transcription factor family. Like the EMT process, RUNX proteins are also regulators of development that have been conserved through evolution and are frequently targeted during human carcinogenesis. The EMT process and RUNX participate in both vertebrate and invertebrate development. However, this chapter is devoted to describing the many instances where they intersect in vertebrate development and human disease.
2 Hallmarks and Drivers of EMT
2.1 The EMT Phenotype
Epithelial cells in contact are characterized by a set of specialized junctions including adherens junctions, which are established through the assembly of protein complexes at the site of E-cadherin clusters. These adhesion structures are the main targets in the execution of the EMT program (Huang et al. 2012).
In epithelial cells, the extracellular region of E-cadherin forms an interface between two adjacent epithelial cells, through cis and trans interactions. The E-cadherin adhesome consists of numerous proteins interacting directly or indirectly with the actin cytoskeleton. β-catenin bound to the carboxy-terminal region of the E-cadherin cytoplasmic domain anchors α-catenin, a crucial protein acting as a mechanosensor. P120-catenins also plays a major role in the dynamics of the junctional complexes (Engl et al. 2014; Yonemura et al. 2010). Although EMT occurs in different tissue context during development, these occurrences share a core set of common features. A principal mark of EMT is the reduction or complete loss of E-cadherin at the cell surface leading to the remodelling or disappearance of adherens junctions. From the onset of EMT to the full-blown mesenchymal stage, the disassembly of the adherens complex is timed with the dissolution of other epithelial junctional complexes, namely tight junctions and the desmosomes (Yilmaz and Christofori 2009; Huang et al. 2012; Lamouille et al. 2014). The initial stages of EMT include the transcriptional repression of E-cadherin and protein degradation (Cano et al. 2000; Batlle et al. 2000; van Roy and Berx 2008). As the cell takes on a more mesenchymal phenotype, E-cadherin is typically replaced by other cadherins, such as N-cadherin, cadherin-7 and cadherin 11 (Nakagawa and Takeichi 1995; Vallin et al. 1998). These cadherins sustain weaker intercellular adhesion (Chu et al. 2006). Collectively, the release from rigid epithelial junctional network and the switch to weaker cell-cell adhesion enables more temporal contacts suitable for greater cell motility (Theveneau and Mayor 2012). Furthermore, a cell that has undergone EMT commonly expresses vimentin, a cytoskeletal protein necessary for migration and the condensation of actin stress fibres that power its movement (Mendez et al. 2010). The migratory cell expresses metalloproteinases (e.g. MMP-2, -9 and -13) for the breaking down of the extracellular matrix as it migrates (Nistico et al. 2012).
The detachment of epithelial cells would trigger a form of program cell death, termed anoikis, which is a critical mechanism that safeguards them against anchorage-independent cell growth (Paoli et al. 2013). During EMT, this program is momentarily silenced through the activation of a pro-survival genetic program, rendering cells resistant to anoikis and apoptosis (De Craene and Berx 2013).
2.2 Coordinators of EMT
EMT is controlled by several evolutionarily conserved signaling pathways during development, which collaborate to achieve precise spatio-temporal coordination. The best characterized among these are the transforming growth factor-beta/bone morphogenic protein (TGF-β/BMP), Notch , wingless-Int (Wnt ), Hedgehog (Hh), epidermal growth factor (EGF), Fibroblast growth factor (FGF) pathways. The importance of these cardinal developmental signals in the regulation of EMT has been extensively studied using in vitro and in vivo models and reviewed in depth in dedicated reviews (Thiery et al. 2009; Lamouille et al. 2014). Reflecting their central roles, mutations or disruptions of these pathways would result in the aberrant activation of EMT in human diseases, such as cancer (De Craene and Berx 2013).
Acting downstream of these principal growth signals is an integrated network of specialized transcription factors, which executes the transcriptomic and epigenetic changes as the cells passage through EMT. Like the upstream signaling cascades, molecular mechanisms employed by these EMT drivers have been studied in detail (reviewed in (Lamouille et al. 2014)). A concise summary is provided here for the purpose of the current discussion.
2.2.1 SNAIL
In the vertebrate, the SNAIL family of zinc finger transcription factors are represented by SNAI1 (also called SNAIL), SNAI2 (also called SLUG) and SNAI3. SNAIL transcription factors repress epithelial genes through the binding of an E-box binding sequence, which has been extensively characterised on the E-cadherin (CDH1) promoter (Cano et al. 2000; Batlle et al. 2000). In additional to their transcriptional effects, SNAIL proteins recruit the polycomb repressor complex 2 to target gene loci to repress gene expression epigenetically via histone modification (Herranz et al. 2008; Lamouille et al. 2014). Importantly, this would leave epithelial genes actively repressed but poised for reactivation. It is speculated that the maintenance of a bivalent state is key to the prompt reversion to an epithelial phenotype during MET, and the inherent plasticity associated with cells undergoing EMT (Lamouille et al. 2014).
2.2.2 TWIST
A number of basic helix-loop-helix (bHLH) transcription factors participate in EMT, including TWIST1, TWIST2, E12, E47 and inhibitor of differentiation (ID). These activating and deactivating bHLH proteins form homo- and heterodimers with one another to mediate signals from diverse pathways (Peinado et al. 2007). This includes the HIF-1α pathway, which promotes EMT in a hypoxic environment, such as in a tumor (Yang et al. 2008). Of the bHLH proteins, TWIST1/2 are best studied and mediate the repression of epithelial genes while activating mesenchymal ones (reviewed in (Castanon and Baylies 2002; Thiery et al. 2009; Lamouille et al. 2014)). TWIST proteins are highly conserved in evolution and are essential for the initiation of mesoderm development during gastrulation (Castanon and Baylies 2002). TWIST1 recruits epigenetic modifiers such as SET8 and BMI-1 to silence the E-cadherin while activating N-cadherin promoters (Yang et al. 2010, 2012). A further important function of TWIST proteins is their repression of the INK4A tumor suppressor gene , which is part of the pro-survival effects of EMT (Ansieau et al. 2008; Yang et al. 2010). Importantly, this pro-survival mechanism is often hijacked by cancer cells during tumorigenesis.
2.2.3 ZEB
The zinc finger E-box-binding homeobox (ZEB) transcription factors, ZEB1 and 2, also target the E-boxes in gene promoters to repress epithelial and activate mesenchymal genes (Peinado et al. 2007). In addition, ZEB1/2 remodel the chromatin structure of target loci by interacting with co-repressor CtBP, which recruits the chromatin remodelling complex SNFI/SWI to silence their target genes (Sanchez-Tillo et al. 2010). Conversely, ZEB1/2 interact with co-activators such as p300 and p300/CBP associated factors to activate transcription (Postigo et al. 2003).
Functionally, an important feature of SNAIL, TWIST and ZEB proteins is the sharing of a core set of key target genes, which allows these transcription factors to individually initiate EMT. In addition to functional redundancy, complex cross regulations exist between these transcription factors, for example ZEB1 is directly regulated by SNAI1 and this could be further enhanced in partnership with TWIST1 (Dave et al. 2011). Lastly, it bears highlighting that the EMT inducers are not functionally identical, as was revealed in the distinct EMT programs driven by Snai2 and Snai1 in normal mammary stem cells and tumor-initiating cells, respectively (Ye et al. 2015).
2.2.4 Secondary Regulators of EMT
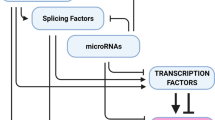
The primary EMT inducing transcription factors are supported by several evolutionarily conserved transcription factor families functioning as auxiliary regulators of EMT. Typically, these supporting transcription factors serve broader functions downstream of major signaling pathways, such as Smad (BMP/TGF-β pathway) and TCF (canonical Wnt pathway) (Derynck et al. 2014; Nawshad et al. 2007). However, the relationship between their general and EMT-promoting roles is often obscure, as is whether these roles are demarcated by spatio-temporal parameters or directed by alternative signaling routes. In this regard, a considerable body of evidence points to the involvement of the RUNX transcription factors during EMT , beyond their established roles in cell fate determination (Fig. 28.1). In the ensuing sections, these evidences are examined in light of normal development and human cancers, where common traits of RUNX involvement in EMT will be discussed.

RUNX proteins as a focal point of key signaling pathways and their partnership with key EMT-inducers to confer the attributes of EMT in normal development and cancer. RTKs receptor tyrosine kinases, Erk1/2 extracellular response kinase 1 and 2, NRs nuclear receptors, R-Smad receptor Smad, NICD Notch intracellular domain
3 RUNX and EMT During Development and Morphogenesis
3.1 Formation of Atrioventricular Valve in the Cardiac Canal
A clear example of RUNX’s participation in EMT during normal development is that of RUNX2 during the formation of cardiac valves in the embryonic heart. The heart is unique amongst organs in that it is derived from three successive rounds of EMT–MET (reviewed in (Thiery et al. 2009; Kovacic et al. 2012)). Cardiac mesodermal cells are specified during gastrulation, and these cells undergo MET to become a transient, two-layered epithelium called the splanchnopleure. During the second round of EMT, the splanchnopleure folds on itself, while an endothelial lining is formed from the dissociated mesenchymal cells. The subsequent MET gives rise to two concentric tubes within which the primordial atrioventricular compartments are formed. During tertiary EMT, cells from the endocardial cushion undergo EMT to invade the underlying cardiac jelly. These mesenchymal cells then proliferate, thicken the endocardiac cushion, and give rise to the atrioventricular (AV) and ventricular outflow tract (OT) valves. This process is dependent on the interplay among Notch (Garg et al. 2005; Nigam and Srivastava 2009), TGFβ2/β3 (Brown et al. 1999), ALK (Mercado-Pimentel et al. 2007), RhoA (Tavares et al. 2006), ErbB (Erickson et al. 1997; Camenisch et al. 2002), and PDGF (Schatteman et al. 1992; Van Den Akker et al. 2005) pathways.
Akin to developmental EMT elsewhere, acting downstream of these pathways is a network of transcription factors that include Snai1/2, RBP-Jκ/CBF1 and Runx2. In the embryonic mouse heart, Runx2 is detectable in the endocardial cells and the mesenchyme of both the AV and OT cushions from 9.5 days post-coitum (d.p.c.; E9.5), and is maintained throughout the mesenchymal transformation (Gitler et al. 2003). The role of Runx2 in the development of the AV canal was investigated in the context of bone morphogenetic protein (BMP) signaling using chicken heart explants (Tavares et al. 2006; Mercado-Pimentel et al. 2007). Runx2 is regulated by Alk2/5 and endoglin early in the development of the AV canal. The knockdown of endoglin by RNAi downregulates Runx2 in the AV cushion but has an opposite effect in the OT cushion, indicative of a complex regulatory mechanism of multiple modulators (Mercado-Pimentel et al. 2007). Other members of the BMP/TGF-β family, namely BMP2 and TGF-β, also participate in the EMT of cardiac endothelial cells (Brown et al. 1999; Boyer et al. 1999; Ma et al. 2005). RUNX2 collaborates with these two signaling pathways in pluripotent mesenchymal precursors and therefore may act as focal points for multiple BMP/TGF-β signals in the development of cardiac valves (Lee et al. 2000). However, despite the dynamic and prominent expression of Runx2 in this tissue, cardiac valvular defects have not been observed in various Runx2-deficient mouse models. It is possible that functional compensation exists with the other Runx family members. Indeed, Runx3 expression is detected in the mouse AV cushion at E10.5. Runx3 is transcriptionally regulated by the Notch pathway and its nuclear effectors, CBF-1/Suppressor of Hairless/Lag-1 (CSL) and mastermind-like protein-1 (MAML-1) (Fu et al. 2011). Once induced, Runx3 sustains the long-term expression of Snai2 to maintain EMT-transformed endothelial cells in a mesenchymal state (Fu et al. 2011). Therefore, Runx3 is enlisted by the Notch pathway to prolong the expression of Snai2 during Notch-induced EMT in the AV cushion.
Interestingly, RUNX2 is also a positive regulator of Snai2 in multiple tissues and may also support Snai2 expression in the AV canal (Lambertini et al. 2010; Niu et al. 2012). However, in the cardiac valve, Notch represses RUNX2 activities via its target genes, Hes1 and Hrt2/Hey2 (Garg et al. 2005; Nigam and Srivastava 2009; McLarren et al. 2000). These Notch effectors physically interact with RUNX2 to prevent the differentiation of valvular cells into osteoblast-like cells, which is causal to calcification in aortic valves (Rajamannan et al. 2003; Garg et al. 2005; Nigam and Srivastava 2009). Collectively, these observations suggest a complex involvement of the Runx proteins in the overall development of the cardiac valves.
3.2 Emergence of Hematopoietic Stem Cells During Definitive Hematopoiesis
Closely related to the EMT of the cardiac endothelial cells is the dissociation of endothelial cells during their transition into hematopoietic stem cells (HSCs) (Kovacic et al. 2012; Kissa and Herbomel 2010). This process, termed Endothelial–Hematopoietic Transition (EHT), occurs in a specialized subpopulation of hemogenic endothelial cells, best characterized at the dorsal aortic floor in the embryonic aortic-gonadal-mesonephric (AGM) region (Medvinsky and Dzierzak 1996; Cumano et al. 1996; Taoudi and Medvinsky 2007). The emergence of HSCs is similarly observed in the vitelline and umbilical arteries (Chen et al. 2009).
Although the regulatory mechanisms underlying EHT are not fully understood, the process nevertheless bears strong resemblance to EMT, wherein is the dissolution of tight junctions (Yue et al. 2012; Zhang et al. 2014), a loss of cell polarity (Wilkinson et al. 2009), and a gain of cell motility and stem cell-like properties (Eilken et al. 2009; Kissa and Herbomel 2010; Boisset et al. 2010; Yue et al. 2012; Kovacic et al. 2012). Furthermore, a similar array of developmental signals is involved in coordinating EHT, including Notch (Hadland et al. 2004; Burns et al. 2005; Richard et al. 2013), TGF-β/BMP (Durand et al. 2007; Wilkinson et al. 2009; Zhang et al. 2014), Wnt (Clements et al. 2011), FGF (Pouget et al. 2014; Lee et al. 2014), ERK (Lan et al. 2014; Zhang et al. 2014), F2r-RhoA/ROCK (Yue et al. 2012) and Hedgehog (Gering and Patient 2005; Wilkinson et al. 2009).
In the mouse, the appearance of adult-repopulating HSCs during definitive hematopoiesis coincides with the spatial-temporal expression of Runx1, which begins at 9.5–10.5 d.p.c. (E9.5–10.5). Runx1 is necessary for the proper maintenance of adult HSCs and is a master regulator of adult hematopoiesis (Voon et al. 2015). Genetic ablation of Runx1 in mouse results in embryonic lethality at E11.5–12.5, characterized by a complete loss of HSC-populated definitive hematopoiesis , and the impairment of vascularization (Okuda et al. 1996; Wang et al. 1996). Numerous studies have shown that Runx1 is indispensable for the emergence of HSCs from the hemogenic endothelium of the dorsal aorta via EHT (North et al. 1999; Yokomizo et al. 2001; Chen et al. 2009; Kissa and Herbomel 2010; Lancrin et al. 2009; North et al. 2009; Adamo et al. 2009; Lam et al. 2010). Here, Runx1 is induced in hemogenic endothelial cells by the Notch signal from sub-aortic mesenchymal cells via Gata2, in order to act in tandem with Notch (Burns et al. 2005; Richard et al. 2013; Kobayashi et al. 2014; Gao et al. 2013). In addition, a number of other signals induce Runx1 in vivo through yet undetermined mechanisms, including BMP (Wilkinson et al. 2009) and mechanical shear force (Adamo et al. 2009; North et al. 2009). The induction of Runx1 in hemogenic endothelial cells coincides strictly with their EHT. In the absence of Runx1, nascent HSCs initially emerge but succumb to a sudden death while exiting the aortic floor (Kissa and Herbomel 2010). Despite its apparent importance in EHT, the precise transcriptional program maintained by Runx1 remains to be elucidated, as is its relationship with the EMT regulators, such as Snail, Twist and Zeb transcription factors. Of note, Snai2 protects hematopoietic stem/progenitors cells against radiation-induced apoptosis by negating the effects of p53/Puma. It is also necessary for modulating the proliferation of HSCs during stress and regeneration (Inoue et al. 2002; Wu et al. 2005). It would be of interest to determine if these EMT-like protective roles trace back to an involvement of Snai2 during EHT in cooperation with Runx1 .
3.3 RUNX and EMT in Other Tissue Contexts
RUNX proteins and EMT feature prominently in a number of other tissues both during development and in the adult. Although direct evidence is still lacking, RUNX proteins are likely part of the EMT genetic program in these tissue contexts. Here we describe two examples.
3.3.1 Mammary Gland
In recent years, compelling evidence points to the involvement of RUNX proteins in embryonic mammary development, postnatal mammary gland morphogenesis, and human breast cancer (Ferrari et al. 2013). Notably, this is a tissue in which EMT is prominent in organogenesis and disease. The mammary epithelium is unique in that much of its development occurs postnatally and its embryonic development is halted with rudimentary glands present at birth. During embryonic development, Runx2 is expressed transiently in the nascent mouse mammary buds at E12–12.5, during lineage segregation (Otto et al. 1997; Ferrari et al. 2015). This is replaced by the expression of Runx1 at E16 before their co-expression in the adult mammary glands (Ferrari et al. 2013).
Postnatal, mammary development resumes through branching morphogenesis and ductal elongation to form the mature mammary gland for milk production. During this period, the temporal and reversible activation of EMT, also termed “partial EMT”, endows stem/progenitor cells at the terminal end buds (TEB) with transient motility to drive ductal elongation (Ewald et al. 2012; Nakaya and Sheng 2013). This is an precisely controlled process that is promoted by EMT inducers, such as Snai1 and Snai2; and restricted by EMT inhibitors, such as Ovol2 (Guo et al. 2012; Ye et al. 2015; Watanabe et al. 2014). In addition to the potential regulation of Snai2, a functional contribution by Runx2 in the “partial EMT” in the TEB is hinted by the co-expression of Runx2 with Snai1, Twist1, Twist2 and other components of the EMT program (Kouros-Mehr and Werb 2006; Lambertini et al. 2010; Niu et al. 2012). Indeed, recent studies have demonstrated that the strict regulation of Runx2 is necessary for proper mammary gland development (Otto et al. 1997; McDonald et al. 2014; Owens et al. 2014; Ferrari et al. 2015). The targeting of Runx2 in mouse mammary glands impaired ductal outgrowth at puberty and disrupted progenitor cell differentiation during pregnancy (Owens et al. 2014). On the other hand, the ectopic expression of Runx2 altered differentiation and led to a gain of EMT phenotype (McDonald et al. 2014; Owens et al. 2014; Ferrari et al. 2015). Although the signaling pathways acting via Runx2 during osteoblast differentiation are well understood, little is known of the cooperating signals in this tissue that could explain these paradoxical phenotypes (Franceschi and Xiao 2003). Nevertheless, these observations are consistent with the need for a strict control of EMT during mammary morphogenesis.
Runx1 is likewise important in the maintenance of mammary gland homeostasis, which is reflected in its recurrent deletion in human luminal breast cancer (Banerji et al. 2012; Network 2012; Ellis et al. 2012). In the mouse, Runx1 is the most highly expressed amongst Runx genes and is present in all subpopulations of mammary epithelial cells except secretary alveolar luminal cells (McDonald et al. 2014; van Bragt et al. 2014). The mammary-specific deletion of Runx1 by MMTV-Cre reduces the proportion of estrogen receptor (ER)-positive luminal cells in virgin adult mice, and this is likely mediated via the interaction between Runx1 and ERα (van Bragt et al. 2014; Stender et al. 2010). Furthermore, Runx1 promotes a mature luminal phenotype while suppressing alveolar luminal cell differentiation through its repression of Elf5 (van Bragt et al. 2014). In addition to being a key driver of alveolar luminal cell differentiation during lactation, Elf5 is an important regulator of Snai2 and EMT of mammary epithelial cells (Chakrabarti et al. 2012). Therefore, it is likely that Runx1 also exerts an influence in partial EMT during mammary gland morphogenesis . Collectively, the associations described herein provide clear reasons to precisely eluciate the roles of Runx proteins during mammary development, especially with respect to “partial EMT”, in future studies.
3.3.2 Lacrimal Gland
The lacrimal gland (LG) is a tubuloacinar exocrine gland that generates most of the aqueous preocular tear film. It is composed of three main cell types: acinar, ductal, and myoepithelial cells. The development of the LG shares common features with that of the mammary gland , with an analogous branching morphogenesis that is governed by factors such as Sox9, fibroblast growth factor (FGF) and BMP signaling (Chen et al. 2014; Dean et al. 2004; Lu et al. 2006). Furthermore, it is likely that LG development progresses by way of a “partial EMT ” similar to that seen in the mammary gland. Indeed, EMT is observed during LG tissue repair following acute inflammation , giving rise to nestin-positive mesenchymal stem-like cells at the sites of the injury, which also expressed vimentin and Snai1 (Zoukhri et al. 2007, 2008; You et al. 2012).
Runx proteins are also similarly involved during LG development and regeneration (Voronov et al. 2013). All Runx proteins are expressed in the developing LG epithelium and their down-regulation in organoid culture has been shown to reduce LG growth, branching and proliferation (Voronov et al. 2013). Of the Runx proteins, Runx1 is highly expressed and its targeting in vivo can impair the timing of LG bud outgrowth during development (Voronov et al. 2013). Of particular note, Runx1 and Runx3 are induced during tissue regeneration and are correlated with epithelial cell proliferation following injury (Voronov et al. 2013); however, it is currently unclear if Runx proteins contribute to the EMT program during LG regeneration. Future studies on this topic will serve as helpful comparisons to understand the involvement of Runx during EMT in exocrine tissues .
4 RUNX and EMT in Disease
4.1 RUNX2 and Bone Metastasis of Cancers
Parallel to its involvement in EMT during development, RUNX proteins have been implicated in the aberrant activation of EMT in cancer in different tissues. The best characterized of these phenomena is the involvement of RUNX2 in the metastases of breast and prostate cancers (Fig. 28.1).
Metastatic breast and prostate cancers are typified by their tropism for the bone, with skeletal colonization accounting for approximately 70% of metastases (Roodman 2004). During dissemination, metastatic carcinoma cells acquire an EMT-like phenotype characterized by increased cell migration and the expression of mesenchymal markers and EMT -promoting transcription factors. Metastatic breast cancer (BCa) and prostate cancer (PCa) cells display osteocyte-like properties that enhance their survival at distant osseous niches , such as the expression of osteopontin (OPN), osteocalcin, bone sialoprotein (BSP) and, importantly, RUNX2 (Koeneman et al. 1999; Barnes et al. 2003; Brubaker et al. 2003; Javed et al. 2005) (Table 28.1).
RUNX2 is a key regulator of osteogenesis (Lian and Stein 2003; Ito et al. 2015). Therefore, its over-expression in breast cancer (BCa) cells offers a mechanistic explanation to the skeletal tropism and osteomimicry of BCa cells (Barnes et al. 2003; Inman and Shore 2003; Khalid et al. 2008; Selvamurugan et al. 2004; Javed et al. 2005; Lau et al. 2006). Indeed, RUNX2 expression is required for the metastasis of BCa in vivo and correlates with the increased grade and poor prognosis in the clinic (Barnes et al. 2004; Javed et al. 2005; Das et al. 2009; Onodera et al. 2010; Ferrari et al. 2013). RUNX2 and its binding partner CBFβ promote BCa cell migration in vitro (Pratap et al. 2005; Chimge et al. 2011; Mendoza-Villanueva et al. 2010), likely via its regulation of invasion-associated genes, such as matrix metalloproteinase (MMP)-13 (Selvamurugan et al. 2004; Mendoza-Villanueva et al. 2010), MMP-9 (Pratap et al. 2005; Mendoza-Villanueva et al. 2010), vascular endothelial growth factor (VEGF) (Zelzer et al. 2001) and BSP (Barnes et al. 2003). In particular, RUNX2 is necessary for the induction of SNAI2, a central coordinator of mammary epithelial stemness and EMT during branch morphogenesis (Chimge et al. 2011; Guo et al. 2012). In metastatic BCa cells, RUNX2 also regulates S100A4, a well-established marker of motility and invasion implicated in cancer EMT and exosome-mediated metastasis (Xu et al. 2015; Hoshino et al. 2015).
Analogous to these observations, RUNX2 enables the osteomimicry of PCa cells , promotes the formation of a pro-surviving osseous niche and is similarly associated with the EMT metastatic phenotype (Yang et al. 2001; Zayzafoon et al. 2004; Baniwal et al. 2009; Lim et al. 2010; Akech et al. 2010; Baniwal et al. 2010; Little et al. 2012). RUNX2 positively regulates EMT drivers such as SMAD3, SNAI2 and SOX9 (Baniwal et al. 2010; Little et al. 2012, 2014). Notably, RUNX2 and androgen receptor cooperatively activate SNAI2 expression to induce EMT-like properties in PCa cells in vitro (Little et al. 2014). The association of RUNX2 with cancer metastasis and aberrant EMT is also observed in thyroid cancers. In this context, the RNAi targeting of RUNX2 suppressed known target genes, such as SNAI2 and VEGF , as well as other EMT-related genes, including TWIST1 and MMP2 (Niu et al. 2012). Interestingly, Twist1 and 2 are shown to functionally antagonize Runx2 during osteoblast differentiation, raising the possibility of intricate cross regulation between Runx2 and the EMT transcription factors during bone metastasis (Bialek et al. 2004).
Collectively, the major lesson from these studies is that the aberrant expression of RUNX2 is key to the concurrent activation of EMT and osteomimicry , especially in BCa and PCa. These insights could be instructive in interpreting the involvement of RUNX proteins in other cancer types.
4.2 RUNX3 and EMT – Suppression or Promotion?
In contrast, other RUNX proteins appear to exert an opposite effect to RUNX2. In hepatocellular carcinoma (HCC), RUNX3 is frequently silenced due to promoter methylation, hemizygous deletion and loss of heterozygosity (Mori et al. 2005; Nakanishi et al. 2011; Xiao and Liu 2004). The re-introduction of RUNX3 in EMT-prone HCC cell lines would promote an epithelial-like cell morphology and increased E-cadherin, as well as suppressing mesenchymal markers, N-cadherin and vimentin (Tanaka et al. 2012). In a Runx3- knockout mouse model, increased EMT features and ERK1/2 phosphorylation were associated with defects in the intra-alveolar septa. These phenotypes were partially ameliorated when mice were treated with a pharmacological inhibitor of ERK1/2 (Lee et al. 2011).
Further evidence of a protective role for RUNX3 against aberrant EMT was reported in two independent studies in gastric carcinogenesis. The first reported an inverse correlation between RUNX3 and OPN expression in gastric tumors and cell lines. Concordant with the pro-metastatic role of OPN, RUNX3 expression was associated with better clinical outcome (Cheng et al. 2013). This relationship was confirmed in vitro where RUNX3 suppressed OPN expression and gastric cancer cell migration (Cheng et al. 2013). In the second study, immortalized p53-null/Runx3-deficient gastric cells were found prone to spontaneous EMT due to increased sensitivity to TGF-β, Wnt and EGFR/Ras signaling (Voon et al. 2012; 2013). This led to increased epithelial plasticity, reflected in the expression of the gastric stem cell marker Lgr5, which amplifies canonical Wnt signal (Li et al. 2002; Voon et al. 2012; de Lau et al. 2014). Interestingly, the Runx3-deficient cells were refractory to TGF-β-induced apoptosis but sensitive to TGF-β-mediated EMT, highlighting the importance of RUNX proteins in the context-specific interpretation of TGF-β signaling (Voon et al. 2012).
Overall, the current evidence in the literature supports the notion that RUNX2 promotes cancer metastasis by inducing EMT and osteomimicry , whereas RUNX3 exerts a protective effect. However, there are exceptions to this general observation, underscoring the complexity of cancer biology. For example, a recent study has found that RUNX3 acts as a “metastatic switch” in an oncogenic Kras/p53 mutant mouse model of pancreatic ductal adenocarcinoma (PDA/PDAC) (Whittle et al. 2015). Here, a high expression of Runx3 was correlated with lung and liver metastases and promoted PDA cell migration through its positive regulation of Opn. Intriguingly, Runx3 had the opposite effect on Opn in gastric carcinoma (Cheng et al. 2013; Whittle et al. 2015). This is likely a result of the context-specific nature of Runx protein functions, which stems from their ability to partner a diverse range of transcription factors and co-factors (reviewed in (Blyth et al. 2005; Ito et al. 2015)). These permutations are further compounded by the interplay and cross-regulations between co-expressed Runx family members. Lastly, the proper execution of Runx function is often dependent on gene dosage (Osato and Ito 2005; Ben-Ami et al. 2013; McDonald et al. 2014; Owens et al. 2014; Whittle et al. 2015). Indeed, Whittle et al. observed that Runx3 could serve tumor suppressive or tumor promoting functions in PDAC cells depending on its dosage and the availability of its binding partner, Smad4 (Whittle et al. 2015). In this regard, the dose-dependency of Runx3 function in PDAC is similar to the varied levels of Twist1 required to drive distinct stages of skin carcinogenesis (Beck et al. 2015).
In the clinic, the challenge of combating cancer metastasis is intimately linked to tumor chemoresistance. It is becoming clear that these seemingly distinct phenomena are in fact driven by the same cellular plasticity afforded by aberrant EMT. Furthermore, recent studies have raised the possibility that EMT promotes the spread and survival of cancer cells via a combination of mechanisms, some of which become prominent in the context of medical intervention, such as chemoresistance. As such, it would be profitable to determine the contribution of Runx proteins to these additional EMT -enabled capabilities in future research.
5 Concluding Remarks
The RUNX family of transcription factors and the process of EMT are often regarded as being at the “crossroads of development and disease”. This Chapter undertakes a collation of the published literature where the two topics have overlapped to reveal an intimate connection between RUNX and EMT . In particular, as nuclear effectors of key developmental signals, RUNX proteins function in partnership with EMT initiators, notably SNAI2, to modulate tissue morphogenesis. Consequently, a disruption to normal RUNX functionality often accompanies the aberrant activation of EMT in disease. It remains to be seen if this hypothesis is supported by future findings. In any case, these early impressions provide a framework upon which more definitive studies can be designed to show that RUNX and EMT are in fact working in arms, on the same intersection between development and disease.
References
Adamo, L., Naveiras, O., Wenzel, P. L., McKinney-Freeman, S., Mack, P. J., Gracia-Sancho, J., et al. (2009). Biomechanical forces promote embryonic haematopoiesis. Nature, 459(7250), 1131–1135. doi:10.1038/nature08073.
Akech, J., Wixted, J. J., Bedard, K., van der Deen, M., Hussain, S., Guise, T. A., et al. (2010). Runx2 association with progression of prostate cancer in patients: Mechanisms mediating bone osteolysis and osteoblastic metastatic lesions. Oncogene, 29(6), 811–821. doi:10.1038/onc.2009.389.
Ansieau, S., Bastid, J., Doreau, A., Morel, A. P., Bouchet, B. P., Thomas, C., et al. (2008). Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell, 14(1), 79–89. doi:10.1016/j.ccr.2008.06.005.
Banerji, S., Cibulskis, K., Rangel-Escareno, C., Brown, K. K., Carter, S. L., Frederick, A. M., et al. (2012). Sequence analysis of mutations and translocations across breast cancer subtypes. Nature, 486(7403), 405–409. doi:10.1038/nature11154.
Baniwal, S. K., Khalid, O., Sir, D., Buchanan, G., Coetzee, G. A., & Frenkel, B. (2009). Repression of Runx2 by androgen receptor (AR) in osteoblasts and prostate cancer cells: AR binds Runx2 and abrogates its recruitment to DNA. Molecular Endocrinology (Baltimore, Md), 23(8), 1203–1214. doi:10.1210/me.2008-0470.
Baniwal, S. K., Khalid, O., Gabet, Y., Shah, R. R., Purcell, D. J., Mav, D., et al. (2010). Runx2 transcriptome of prostate cancer cells: Insights into invasiveness and bone metastasis. Molecular Cancer, 9, 258. doi:10.1186/1476-4598-9-258.
Barnes, G. L., Javed, A., Waller, S. M., Kamal, M. H., Hebert, K. E., Hassan, M. Q., et al. (2003). Osteoblast-related transcription factors Runx2 (Cbfa1/AML3) and MSX2 mediate the expression of bone sialoprotein in human metastatic breast cancer cells. Cancer Research, 63(10), 2631–2637.
Barnes, G. L., Hebert, K. E., Kamal, M., Javed, A., Einhorn, T. A., Lian, J. B., et al. (2004). Fidelity of Runx2 activity in breast cancer cells is required for the generation of metastases-associated osteolytic disease. Cancer Research, 64(13), 4506–4513. doi:10.1158/0008-5472.can-03-3851.
Batlle, E., Sancho, E., Franci, C., Dominguez, D., Monfar, M., Baulida, J., & Garcia De Herreros, A. (2000). The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nature Cell Biology, 2(2), 84–89. doi:10.1038/35000034.
Beck, B., Lapouge, G., Rorive, S., Drogat, B., Desaedelaere, K., Delafaille, S., et al. (2015). Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell, 16(1), 67–79. doi:10.1016/j.stem.2014.12.002.
Ben-Ami, O., Friedman, D., Leshkowitz, D., Goldenberg, D., Orlovsky, K., Pencovich, N., et al. (2013). Addiction of t(8;21) and inv(16) acute myeloid leukemia to native RUNX1. Cell Reports, 4(6), 1131–1143. doi:10.1016/j.celrep.2013.08.020.
Bialek, P., Kern, B., Yang, X., Schrock, M., Sosic, D., Hong, N., et al. (2004). A twist code determines the onset of osteoblast differentiation. Developmental Cell, 6(3), 423–435.
Blyth, K., Cameron, E. R., & Neil, J. C. (2005). The RUNX genes: Gain or loss of function in cancer. Nature Reviews Cancer, 5(5), 376–387. doi:10.1038/nrc1607.
Boisset, J. C., van Cappellen, W., Andrieu-Soler, C., Galjart, N., Dzierzak, E., & Robin, C. (2010). In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature, 464(7285), 116–120. doi:10.1038/nature08764.
Boyer, A. S., Ayerinskas, I. I., Vincent, E. B., McKinney, L. A., Weeks, D. L., & Runyan, R. B. (1999). TGFbeta2 and TGFbeta3 have separate and sequential activities during epithelial-mesenchymal cell transformation in the embryonic heart. Developmental Biology, 208(2), 530–545. doi:10.1006/dbio.1999.9211.
Brown, C. B., Boyer, A. S., Runyan, R. B., & Barnett, J. V. (1999). Requirement of type III TGF-beta receptor for endocardial cell transformation in the heart. Science (New York, N.Y.), 283(5410), 2080–2082.
Brubaker, K. D., Vessella, R. L., Brown, L. G., & Corey, E. (2003). Prostate cancer expression of runt-domain transcription factor Runx2, a key regulator of osteoblast differentiation and function. The Prostate, 56(1), 13–22. doi:10.1002/pros.10233.
Burns, C. E., Traver, D., Mayhall, E., Shepard, J. L., & Zon, L. I. (2005). Hematopoietic stem cell fate is established by the Notch-Runx pathway. Genes & Development, 19(19), 2331–2342. doi:10.1101/gad.1337005.
Camenisch, T. D., Schroeder, J. A., Bradley, J., Klewer, S. E., & McDonald, J. A. (2002). Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2-ErbB3 receptors. Nature Medicine, 8(8), 850–855. doi:10.1038/nm742.
Cano, A., Perez-Moreno, M. A., Rodrigo, I., Locascio, A., Blanco, M. J., del Barrio, M. G., et al. (2000). The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nature Cell Biology, 2(2), 76–83. doi:10.1038/35000025.
Castanon, I., & Baylies, M. K. (2002). A Twist in fate: Evolutionary comparison of Twist structure and function. Gene, 287(1–2), 11–22.
Chakrabarti, R., Hwang, J., Andres Blanco, M., Wei, Y., Lukacisin, M., Romano, R. A., et al. (2012). Elf5 inhibits the epithelial-mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2. Nature Cell Biology, 14(11), 1212–1222. doi:10.1038/ncb2607.
Chen, M. J., Yokomizo, T., Zeigler, B. M., Dzierzak, E., & Speck, N. A. (2009). Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature, 457(7231), 887–891. doi:10.1038/nature07619.
Chen, Z., Huang, J., Liu, Y., Dattilo, L. K., Huh, S. H., Ornitz, D., & Beebe, D. C. (2014). FGF signaling activates a Sox9-Sox10 pathway for the formation and branching morphogenesis of mouse ocular glands. Development (Cambridge, England), 141(13), 2691–2701. doi:10.1242/dev.108944.
Cheng, H. C., Liu, Y. P., Shan, Y. S., Huang, C. Y., Lin, F. C., Lin, L. C., et al. (2013). Loss of RUNX3 increases osteopontin expression and promotes cell migration in gastric cancer. Carcinogenesis, 34(11), 2452–2459. doi:10.1093/carcin/bgt218.
Chimge, N. O., Baniwal, S. K., Little, G. H., Chen, Y. B., Kahn, M., Tripathy, D., et al. (2011). Regulation of breast cancer metastasis by Runx2 and estrogen signaling: the role of SNAI2. Breast Cancer Research : BCR, 13(6), R127. doi:10.1186/bcr3073.
Chu, Y. S., Eder, O., Thomas, W. A., Simcha, I., Pincet, F., Ben-Ze’ev, A., et al. (2006). Prototypical type I E-cadherin and type II cadherin-7 mediate very distinct adhesiveness through their extracellular domains. The Journal of Biological Chemistry, 281(5), 2901–2910. doi:10.1074/jbc.M506185200.
Clements, W. K., Kim, A. D., Ong, K. G., Moore, J. C., Lawson, N. D., & Traver, D. (2011). A somitic Wnt16/Notch pathway specifies haematopoietic stem cells. Nature, 474(7350), 220–224. doi:10.1038/nature10107.
Cumano, A., Dieterlen-Lievre, F., & Godin, I. (1996). Lymphoid potential, probed before circulation in mouse, is restricted to caudal intraembryonic splanchnopleura. Cell, 86(6), 907–916.
Das, K., Leong, D. T., Gupta, A., Shen, L., Putti, T., Stein, G. S., et al. (2009). Positive association between nuclear Runx2 and oestrogen-progesterone receptor gene expression characterises a biological subtype of breast cancer. European Journal of Cancer (Oxford, England : 1990), 45(13), 2239–2248. doi:10.1016/j.ejca.2009.06.021.
Dave, N., Guaita-Esteruelas, S., Gutarra, S., Frias, A., Beltran, M., Peiro, S., & de Herreros, A. G. (2011). Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. The Journal of Biological Chemistry, 286(14), 12024–12032. doi:10.1074/jbc.M110.168625.
De Craene, B., & Berx, G. (2013). Regulatory networks defining EMT during cancer initiation and progression. Nature Reviews Cancer, 13(2), 97–110. doi:10.1038/nrc3447.
de Lau, W., Peng, W. C., Gros, P., & Clevers, H. (2014). The R-spondin/Lgr5/Rnf43 module: Regulator of Wnt signal strength. Genes & Development, 28(4), 305–316. doi:10.1101/gad.235473.113.
Dean, C., Ito, M., Makarenkova, H. P., Faber, S. C., & Lang, R. A. (2004). Bmp7 regulates branching morphogenesis of the lacrimal gland by promoting mesenchymal proliferation and condensation. Development (Cambridge, England), 131(17), 4155–4165. doi:10.1242/dev.01285.
Derynck, R., Muthusamy, B. P., & Saeteurn, K. Y. (2014). Signaling pathway cooperation in TGF-beta-induced epithelial-mesenchymal transition. Current Opinion in Cell Biology, 31, 56–66. doi:10.1016/j.ceb.2014.09.001.
Durand, C., Robin, C., Bollerot, K., Baron, M. H., Ottersbach, K., & Dzierzak, E. (2007). Embryonic stromal clones reveal developmental regulators of definitive hematopoietic stem cells. Proceedings of the National Academy of Sciences of the United States of America, 104(52), 20838–20843. doi:10.1073/pnas.0706923105.
Eilken, H. M., Nishikawa, S., & Schroeder, T. (2009). Continuous single-cell imaging of blood generation from haemogenic endothelium. Nature, 457(7231), 896–900. doi:10.1038/nature07760.
Ellis, M. J., Ding, L., Shen, D., Luo, J., Suman, V. J., Wallis, J. W., et al. (2012). Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature, 486(7403), 353–360. doi:10.1038/nature11143.
Engl, W., Arasi, B., Yap, L. L., Thiery, J. P., & Viasnoff, V. (2014). Actin dynamics modulate mechanosensitive immobilization of E-cadherin at adherens junctions. Nature Cell Biology, 16(6), 587–594. doi:10.1038/ncb2973.
Erickson, S. L., O’Shea, K. S., Ghaboosi, N., Loverro, L., Frantz, G., Bauer, M., et al. (1997). ErbB3 is required for normal cerebellar and cardiac development: A comparison with ErbB2-and heregulin-deficient mice. Development (Cambridge, England), 124(24), 4999–5011.
Ewald, A. J., Huebner, R. J., Palsdottir, H., Lee, J. K., Perez, M. J., Jorgens, D. M., et al. (2012). Mammary collective cell migration involves transient loss of epithelial features and individual cell migration within the epithelium. Journal of Cell Science, 125(Pt 11), 2638–2654. doi:10.1242/jcs.096875.
Ferrari, N., McDonald, L., Morris, J. S., Cameron, E. R., & Blyth, K. (2013). RUNX2 in mammary gland development and breast cancer. Journal of Cellular Physiology, 228(6), 1137–1142. doi:10.1002/jcp.24285.
Ferrari, N., Riggio, A. I., Mason, S., McDonald, L., King, A., Higgins, T., et al. (2015). Runx2 contributes to the regenerative potential of the mammary epithelium. Scientific Reports, 5, 15658. doi:10.1038/srep15658.
Franceschi, R. T., & Xiao, G. (2003). Regulation of the osteoblast-specific transcription factor, Runx2: Responsiveness to multiple signal transduction pathways. Journal of Cellular Biochemistry, 88(3), 446–454. doi:10.1002/jcb.10369.
Fu, Y., Chang, A. C., Fournier, M., Chang, L., Niessen, K., & Karsan, A. (2011). RUNX3 maintains the mesenchymal phenotype after termination of the Notch signal. The Journal of Biological Chemistry, 286(13), 11803–11813. doi:10.1074/jbc.M111.222331.
Gao, X., Johnson, K. D., Chang, Y. I., Boyer, M. E., Dewey, C. N., Zhang, J., & Bresnick, E. H. (2013). Gata2 cis-element is required for hematopoietic stem cell generation in the mammalian embryo. The Journal of Experimental Medicine, 210(13), 2833–2842. doi:10.1084/jem.20130733.
Garg, V., Muth, A. N., Ransom, J. F., Schluterman, M. K., Barnes, R., King, I. N., et al. (2005). Mutations in NOTCH1 cause aortic valve disease. Nature, 437(7056), 270–274. doi:10.1038/nature03940.
Gering, M., & Patient, R. (2005). Hedgehog signaling is required for adult blood stem cell formation in zebrafish embryos. Developmental Cell, 8(3), 389–400. doi:10.1016/j.devcel.2005.01.010.
Gitler, A. D., Lu, M. M., Jiang, Y. Q., Epstein, J. A., & Gruber, P. J. (2003). Molecular markers of cardiac endocardial cushion development. Developmental Dynamics: An Official Publication of the American Association of the Anatomists, 228(4), 643–650. doi:10.1002/dvdy.10418.
Guo, W., Keckesova, Z., Donaher, J. L., Shibue, T., Tischler, V., Reinhardt, F., et al. (2012). Slug and Sox9 cooperatively determine the mammary stem cell state. Cell, 148(5), 1015–1028. doi:10.1016/j.cell.2012.02.008.
Hadland, B. K., Huppert, S. S., Kanungo, J., Xue, Y., Jiang, R., Gridley, T., et al. (2004). A requirement for Notch1 distinguishes 2 phases of definitive hematopoiesis during development. Blood, 104(10), 3097–3105. doi:10.1182/blood-2004-03-1224.
Herranz, N., Pasini, D., Diaz, V. M., Franci, C., Gutierrez, A., Dave, N., et al. (2008). Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Molecular and Cellular Biology, 28(15), 4772–4781. doi:10.1128/mcb.00323-08.
Hoshino, A., Costa-Silva, B., Shen, T. L., Rodrigues, G., Hashimoto, A., Tesic Mark, M., et al. (2015). Tumour exosome integrins determine organotropic metastasis. Nature. doi:10.1038/nature15756.
Huang, R. Y., Guilford, P., & Thiery, J. P. (2012). Early events in cell adhesion and polarity during epithelial-mesenchymal transition. Journal of Cell Science, 125(Pt 19), 4417–4422. doi:10.1242/jcs.099697.
Inman, C. K., & Shore, P. (2003). The osteoblast transcription factor Runx2 is expressed in mammary epithelial cells and mediates osteopontin expression. The Journal of Biological Chemistry, 278(49), 48684–48689. doi:10.1074/jbc.M308001200.
Inoue, A., Seidel, M. G., Wu, W., Kamizono, S., Ferrando, A. A., Bronson, R. T., et al. (2002). Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell, 2(4), 279–288.
Ito, Y., Bae, S. C., & Chuang, L. S. (2015). The RUNX family: Developmental regulators in cancer. Nature Reviews Cancer, 15(2), 81–95. doi:10.1038/nrc3877.
Javed, A., Barnes, G. L., Pratap, J., Antkowiak, T., Gerstenfeld, L. C., van Wijnen, A. J., et al. (2005). Impaired intranuclear trafficking of Runx2 (AML3/CBFA1) transcription factors in breast cancer cells inhibits osteolysis in vivo. Proceedings of the National Academy of Sciences of the United States of America, 102(5), 1454–1459. doi:10.1073/pnas.0409121102.
Khalid, O., Baniwal, S. K., Purcell, D. J., Leclerc, N., Gabet, Y., Stallcup, M. R., et al. (2008). Modulation of Runx2 activity by estrogen receptor-alpha: Implications for osteoporosis and breast cancer. Endocrinology, 149(12), 5984–5995. doi:10.1210/en.2008-0680.
Kissa, K., & Herbomel, P. (2010). Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature, 464(7285), 112–115. doi:10.1038/nature08761.
Kobayashi, I., Kobayashi-Sun, J., Kim, A. D., Pouget, C., Fujita, N., Suda, T., & Traver, D. (2014). Jam1a-Jam2a interactions regulate haematopoietic stem cell fate through Notch signalling. Nature, 512(7514), 319–323. doi:10.1038/nature13623.
Koeneman, K. S., Yeung, F., & Chung, L. W. (1999). Osteomimetic properties of prostate cancer cells: A hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. The Prostate, 39(4), 246–261.
Kouros-Mehr, H., & Werb, Z. (2006). Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Developmental Dynamics: An Official Publication of the American Association of the Anatomists, 235(12), 3404–3412. doi:10.1002/dvdy.20978.
Kovacic, J. C., Mercader, N., Torres, M., Boehm, M., & Fuster, V. (2012). Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: From cardiovascular development to disease. Circulation, 125(14), 1795–1808. doi:10.1161/circulationaha.111.040352.
Lam, E. Y., Hall, C. J., Crosier, P. S., Crosier, K. E., & Flores, M. V. (2010). Live imaging of Runx1 expression in the dorsal aorta tracks the emergence of blood progenitors from endothelial cells. Blood, 116(6), 909–914. doi:10.1182/blood-2010-01-264382.
Lambertini, E., Franceschetti, T., Torreggiani, E., Penolazzi, L., Pastore, A., Pelucchi, S., et al. (2010). SLUG: A new target of lymphoid enhancer factor-1 in human osteoblasts. BMC Molecular Biology, 11, 13. doi:10.1186/1471-2199-11-13.
Lamouille, S., Xu, J., & Derynck, R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nature Reviews Molecular Cell Biology, 15(3), 178–196. doi:10.1038/nrm3758.
Lan, Y., He, W., Li, Z., Wang, Y., Wang, J., Gao, J., et al. (2014). Endothelial Smad4 restrains the transition to hematopoietic progenitors via suppression of ERK activation. Blood, 123(14), 2161–2171. doi:10.1182/blood-2013-09-526053.
Lancrin, C., Sroczynska, P., Stephenson, C., Allen, T., Kouskoff, V., & Lacaud, G. (2009). The haemangioblast generates haematopoietic cells through a haemogenic endothelium stage. Nature, 457(7231), 892–895. doi:10.1038/nature07679.
Lau, Q. C., Raja, E., Salto-Tellez, M., Liu, Q., Ito, K., Inoue, M., et al. (2006). RUNX3 is frequently inactivated by dual mechanisms of protein mislocalization and promoter hypermethylation in breast cancer. Cancer Research, 66(13), 6512–6520. doi:10.1158/0008-5472.can-06-0369.
Lee, K. S., Kim, H. J., Li, Q. L., Chi, X. Z., Ueta, C., Komori, T., et al. (2000). Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Molecular and Cellular Biology, 20(23), 8783–8792.
Lee, J. M., Shin, J. O., Cho, K. W., Hosoya, A., Cho, S. W., Lee, Y. S., et al. (2011). Runx3 is a crucial regulator of alveolar differentiation and lung tumorigenesis in mice. Differentiation; research in biological diversity, 81(4), 261–268. doi:10.1016/j.diff.2011.02.001.
Lee, Y., Manegold, J. E., Kim, A. D., Pouget, C., Stachura, D. L., Clements, W. K., & Traver, D. (2014). FGF signalling specifies haematopoietic stem cells through its regulation of somitic Notch signalling. Nature Communications, 5, 5583. doi:10.1038/ncomms6583.
Li, Q. L., Ito, K., Sakakura, C., Fukamachi, H., Inoue, K., Chi, X. Z., et al. (2002). Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell, 109(1), 113–124.
Lian, J. B., & Stein, G. S. (2003). Runx2/Cbfa1: A multifunctional regulator of bone formation. Current Pharmaceutical Design, 9(32), 2677–2685.
Lim, M., Zhong, C., Yang, S., Bell, A. M., Cohen, M. B., & Roy-Burman, P. (2010). Runx2 regulates survivin expression in prostate cancer cells. Laboratory Investigation; a journal of technical methods and pathology, 90(2), 222–233. doi:10.1038/labinvest.2009.128.
Little, G. H., Noushmehr, H., Baniwal, S. K., Berman, B. P., Coetzee, G. A., & Frenkel, B. (2012). Genome-wide Runx2 occupancy in prostate cancer cells suggests a role in regulating secretion. Nucleic Acids Research, 40(8), 3538–3547. doi:10.1093/nar/gkr1219.
Little, G. H., Baniwal, S. K., Adisetiyo, H., Groshen, S., Chimge, N. O., Kim, S. Y., et al. (2014). Differential effects of RUNX2 on the androgen receptor in prostate cancer: Synergistic stimulation of a gene set exemplified by SNAI2 and subsequent invasiveness. Cancer Research, 74(10), 2857–2868. doi:10.1158/0008-5472.can-13-2003.
Lu, P., Sternlicht, M. D., & Werb, Z. (2006). Comparative mechanisms of branching morphogenesis in diverse systems. Journal of Mammary Gland Biology and Neoplasia, 11(3–4), 213–228. doi:10.1007/s10911-006-9027-z.
Ma, L., Lu, M. F., Schwartz, R. J., & Martin, J. F. (2005). Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development (Cambridge, England), 132(24), 5601–5611. doi:10.1242/dev.02156.
McDonald, L., Ferrari, N., Terry, A., Bell, M., Mohammed, Z. M., Orange, C., et al. (2014). RUNX2 correlates with subtype-specific breast cancer in a human tissue microarray, and ectopic expression of Runx2 perturbs differentiation in the mouse mammary gland. Disease Models & Mechanisms, 7(5), 525–534. doi:10.1242/dmm.015040.
McLarren, K. W., Lo, R., Grbavec, D., Thirunavukkarasu, K., Karsenty, G., & Stifani, S. (2000). The mammalian basic helix loop helix protein HES-1 binds to and modulates the transactivating function of the runt-related factor Cbfa1. The Journal of Biological Chemistry, 275(1), 530–538.
Medvinsky, A., & Dzierzak, E. (1996). Definitive hematopoiesis is autonomously initiated by the AGM region. Cell, 86(6), 897–906.
Mendez, M. G., Kojima, S., & Goldman, R. D. (2010). Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB Journal : official publication of the Federation of American Societies for Experimental Biology, 24(6), 1838–1851. doi:10.1096/fj.09-151639.
Mendoza-Villanueva, D., Deng, W., Lopez-Camacho, C., & Shore, P. (2010). The Runx transcriptional co-activator, CBFbeta, is essential for invasion of breast cancer cells. Molecular Cancer, 9, 171. doi:10.1186/1476-4598-9-171.
Mercado-Pimentel, M. E., Hubbard, A. D., & Runyan, R. B. (2007). Endoglin and Alk5 regulate epithelial-mesenchymal transformation during cardiac valve formation. Developmental Biology, 304(1), 420–432. doi:10.1016/j.ydbio.2006.12.038.
Mori, T., Nomoto, S., Koshikawa, K., Fujii, T., Sakai, M., Nishikawa, Y., et al. (2005). Decreased expression and frequent allelic inactivation of the RUNX3 gene at 1p36 in human hepatocellular carcinoma. Liver International : official journal of the International Association for the Study of the Liver, 25(2), 380–388. doi:10.1111/j.1478-3231.2005.1059.x.
Nakagawa, S., & Takeichi, M. (1995). Neural crest cell-cell adhesion controlled by sequential and subpopulation-specific expression of novel cadherins. Development (Cambridge, England), 121(5), 1321–1332.
Nakanishi, Y., Shiraha, H., Nishina, S., Tanaka, S., Matsubara, M., Horiguchi, S., et al. (2011). Loss of runt-related transcription factor 3 expression leads hepatocellular carcinoma cells to escape apoptosis. BMC Cancer, 11, 3. doi:10.1186/1471-2407-11-3.
Nakaya, Y., & Sheng, G. (2013). EMT in developmental morphogenesis. Cancer Letters, 341(1), 9–15. doi:10.1016/j.canlet.2013.02.037.
Nawshad, A., Medici, D., Liu, C. C., & Hay, E. D. (2007). TGFbeta3 inhibits E-cadherin gene expression in palate medial-edge epithelial cells through a Smad2-Smad4-LEF1 transcription complex. Journal of Cell Science, 120(Pt 9), 1646–1653. doi:10.1242/jcs.003129.
Network CGA. (2012). Comprehensive molecular portraits of human breast tumours. Nature, 490(7418), 61–70. doi:10.1038/nature11412.
Nigam, V., & Srivastava, D. (2009). Notch1 represses osteogenic pathways in aortic valve cells. Journal of Molecular and Cellular Cardiology, 47(6), 828–834. doi:10.1016/j.yjmcc.2009.08.008.
Nistico, P., Bissell, M. J., & Radisky, D. C. (2012). Epithelial-mesenchymal transition: general principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harbor Perspectives in Biology, 4(2). doi:10.1101/cshperspect.a011908.
Niu, D. F., Kondo, T., Nakazawa, T., Oishi, N., Kawasaki, T., Mochizuki, K., et al. (2012). Transcription factor Runx2 is a regulator of epithelial-mesenchymal transition and invasion in thyroid carcinomas. Laboratory Investigation; a journal of technical methods and pathology, 92(8), 1181–1190. doi:10.1038/labinvest.2012.84.
North, T., Gu, T. L., Stacy, T., Wang, Q., Howard, L., Binder, M., et al. (1999). Cbfa2 is required for the formation of intra-aortic hematopoietic clusters. Development (Cambridge, England), 126(11), 2563–2575.
North, T. E., Goessling, W., Peeters, M., Li, P., Ceol, C., Lord, A. M., et al. (2009). Hematopoietic stem cell development is dependent on blood flow. Cell, 137(4), 736–748. doi:10.1016/j.cell.2009.04.023.
Okuda, T., van Deursen, J., Hiebert, S. W., Grosveld, G., & Downing, J. R. (1996). AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell, 84(2), 321–330.
Onodera, Y., Miki, Y., Suzuki, T., Takagi, K., Akahira, J., Sakyu, T., et al. (2010). Runx2 in human breast carcinoma: its potential roles in cancer progression. Cancer Science, 101(12), 2670–2675. doi:10.1111/j.1349-7006.2010.01742.x.
Osato, M., & Ito, Y. (2005). Increased dosage of the RUNX1/AML1 gene: A third mode of RUNX leukemia? Critical Reviews in Eukaryotic Gene Expression, 15(3), 217–228.
Otto, F., Thornell, A. P., Crompton, T., Denzel, A., Gilmour, K. C., Rosewell, I. R., et al. (1997). Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell, 89(5), 765–771.
Owens, T. W., Rogers, R. L., Best, S. A., Ledger, A., Mooney, A. M., Ferguson, A., et al. (2014). Runx2 is a novel regulator of mammary epithelial cell fate in development and breast cancer. Cancer Research, 74(18), 5277–5286. doi:10.1158/0008-5472.can-14-0053.
Paoli, P., Giannoni, E., & Chiarugi, P. (2013). Anoikis molecular pathways and its role in cancer progression. Biochimica et Biophysica Acta, 1833(12), 3481–3498. doi:10.1016/j.bbamcr.2013.06.026.
Peinado, H., Olmeda, D., & Cano, A. (2007). Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nature Reviews Cancer, 7(6), 415–428. doi:10.1038/nrc2131.
Postigo, A. A., Depp, J. L., Taylor, J. J., & Kroll, K. L. (2003). Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. The EMBO Journal, 22(10), 2453–2462. doi:10.1093/emboj/cdg226.
Pouget, C., Peterkin, T., Simoes, F. C., Lee, Y., Traver, D., & Patient, R. (2014). FGF signalling restricts haematopoietic stem cell specification via modulation of the BMP pathway. Nature Communications, 5, 5588. doi:10.1038/ncomms6588.
Pratap, J., Javed, A., Languino, L. R., van Wijnen, A. J., Stein, J. L., Stein, G. S., & Lian, J. B. (2005). The Runx2 osteogenic transcription factor regulates matrix metalloproteinase 9 in bone metastatic cancer cells and controls cell invasion. Molecular and Cellular Biology, 25(19), 8581–8591. doi:10.1128/mcb.25.19.8581-8591.2005.
Rajamannan, N. M., Gersh, B., & Bonow, R. O. (2003). Calcific aortic stenosis: From bench to the bedside – emerging clinical and cellular concepts. Heart (British Cardiac Society)., 89(7), 801–805.
Richard, C., Drevon, C., Canto, P. Y., Villain, G., Bollerot, K., Lempereur, A., et al. (2013). Endothelio-mesenchymal interaction controls runx1 expression and modulates the notch pathway to initiate aortic hematopoiesis. Developmental Cell, 24(6), 600–611. doi:10.1016/j.devcel.2013.02.011.
Roodman, G. D. (2004). Mechanisms of bone metastasis. The New England Journal of Medicine, 350(16), 1655–1664. doi:10.1056/NEJMra030831.
Sanchez-Tillo, E., Lazaro, A., Torrent, R., Cuatrecasas, M., Vaquero, E. C., Castells, A., et al. (2010). ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene, 29(24), 3490–3500. doi:10.1038/onc.2010.102.
Schatteman, G. C., Morrison-Graham, K., van Koppen, A., Weston, J. A., & Bowen-Pope, D. F. (1992). Regulation and role of PDGF receptor alpha-subunit expression during embryogenesis. Development (Cambridge, England), 115(1), 123–131.
Selvamurugan, N., Kwok, S., & Partridge, N. C. (2004). Smad3 interacts with JunB and Cbfa1/Runx2 for transforming growth factor-beta1-stimulated collagenase-3 expression in human breast cancer cells. The Journal of Biological Chemistry, 279(26), 27764–27773. doi:10.1074/jbc.M312870200.
Stender, J. D., Kim, K., Charn, T. H., Komm, B., Chang, K. C., Kraus, W. L., et al. (2010). Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Molecular and Cellular Biology, 30(16), 3943–3955. doi:10.1128/mcb.00118-10.
Tanaka, S., Shiraha, H., Nakanishi, Y., Nishina, S., Matsubara, M., Horiguchi, S., et al. (2012). Runt-related transcription factor 3 reverses epithelial-mesenchymal transition in hepatocellular carcinoma. International Journal of Cancer/Journal international du cancer, 131(11), 2537–2546. doi:10.1002/ijc.27575.
Taoudi, S., & Medvinsky, A. (2007). Functional identification of the hematopoietic stem cell niche in the ventral domain of the embryonic dorsal aorta. Proceedings of the National Academy of Sciences of the United States of America, 104(22), 9399–9403. doi:10.1073/pnas.0700984104.
Tavares, A. L., Mercado-Pimentel, M. E., Runyan, R. B., & Kitten, G. T. (2006). TGF beta-mediated RhoA expression is necessary for epithelial-mesenchymal transition in the embryonic chick heart. Developmental Dynamics: An Official Publication of the American Association of the Anatomists, 235(6), 1589–1598. doi:10.1002/dvdy.20771.
Theveneau, E., & Mayor, R. (2012). Cadherins in collective cell migration of mesenchymal cells. Current Opinion in Cell Biology, 24(5), 677–684. doi:10.1016/j.ceb.2012.08.002.
Thiery, J. P., Acloque, H., Huang, R. Y., & Nieto, M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell, 139(5), 871–890. doi:10.1016/j.cell.2009.11.007.
Vallin, J., Girault, J. M., Thiery, J. P., & Broders, F. (1998). Xenopus cadherin-11 is expressed in different populations of migrating neural crest cells. Mechanisms of Development, 75(1–2), 171–174.
van Bragt, M. P., Hu, X., Xie, Y., & Li, Z. (2014). RUNX1, a transcription factor mutated in breast cancer, controls the fate of ER-positive mammary luminal cells. eLife, 3, e03881. doi:10.7554/eLife.03881.
Van Den Akker, N. M., Lie-Venema, H., Maas, S., Eralp, I., DeRuiter, M. C., Poelmann, R. E., & Gittenberger-De Groot, A. C. (2005). Platelet-derived growth factors in the developing avian heart and maturating coronary vasculature. Developmental Dynamics: An Official Publication of the American Association of the Anatomists, 233(4), 1579–1588. doi:10.1002/dvdy.20476.
van Roy, F., & Berx, G. (2008). The cell-cell adhesion molecule E-cadherin. Cellular and Molecular Life Sciences: CMLS, 65(23), 3756–3788. doi:10.1007/s00018-008-8281-1.
Voon, D. C., Wang, H., Koo, J. K., Nguyen, T. A., Hor, Y. T., Chu, Y. S., et al. (2012). Runx3 protects gastric epithelial cells against epithelial-mesenchymal transition-induced cellular plasticity and tumorigenicity. Stem Cells (Dayton, Ohio), 30(10), 2088–2099. doi:10.1002/stem.1183.
Voon, D. C., Wang, H., Koo, J. K., Chai, J. H., Hor, Y. T., Tan, T. Z., et al. (2013). EMT-induced stemness and tumorigenicity are fueled by the EGFR/Ras pathway. PloS One, 8(8), e70427. doi:10.1371/journal.pone.0070427.
Voon, D. C., Hor, Y. T., & Ito, Y. (2015). The RUNX complex: Reaching beyond haematopoiesis into immunity. Immunology. doi:10.1111/imm.12535.
Voronov, D., Gromova, A., Liu, D., Zoukhri, D., Medvinsky, A., Meech, R., & Makarenkova, H. P. (2013). Transcription factors Runx1 to 3 are expressed in the lacrimal gland epithelium and are involved in regulation of gland morphogenesis and regeneration. Investigative Ophthalmology & Visual Science, 54(5), 3115–3125. doi:10.1167/iovs.13-11791.
Wang, Q., Stacy, T., Binder, M., Marin-Padilla, M., Sharpe, A. H., & Speck, N. A. (1996). Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proceedings of the National Academy of Sciences of the United States of America, 93(8), 3444–3449.
Watanabe, K., Villarreal-Ponce, A., Sun, P., Salmans, M. L., Fallahi, M., Andersen, B., & Dai, X. (2014). Mammary morphogenesis and regeneration require the inhibition of EMT at terminal end buds by Ovol2 transcriptional repressor. Developmental Cell, 29(1), 59–74. doi:10.1016/j.devcel.2014.03.006.
Whittle, M. C., Izeradjene, K., Rani, P. G., Feng, L., Carlson, M. A., DelGiorno, K. E., et al. (2015). RUNX3 controls a metastatic switch in pancreatic ductal adenocarcinoma. Cell, 161(6), 1345–1360. doi:10.1016/j.cell.2015.04.048.
Wilkinson, R. N., Pouget, C., Gering, M., Russell, A. J., Davies, S. G., Kimelman, D., & Patient, R. (2009). Hedgehog and Bmp polarize hematopoietic stem cell emergence in the zebrafish dorsal aorta. Developmental Cell, 16(6), 909–916. doi:10.1016/j.devcel.2009.04.014.
Wu, W. S., Heinrichs, S., Xu, D., Garrison, S. P., Zambetti, G. P., Adams, J. M., & Look, A. T. (2005). Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell, 123(4), 641–653. doi:10.1016/j.cell.2005.09.029.
Xiao, W. H., & Liu, W. W. (2004). Hemizygous deletion and hypermethylation of RUNX3 gene in hepatocellular carcinoma. World Journal of Gastroenterology, 10(3), 376–380.
Xu, H., Li, M., Zhou, Y., Wang, F., Li, X., Wang, L., & Fan, Q. (2015). S100A4 participates in epithelial-mesenchymal transition in breast cancer via targeting MMP2. Tumour Biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. doi:10.1007/s13277-015-3709-3.
Yang, J., Fizazi, K., Peleg, S., Sikes, C. R., Raymond, A. K., Jamal, N., et al. (2001). Prostate cancer cells induce osteoblast differentiation through a Cbfa1-dependent pathway. Cancer Research, 61(14), 5652–5659.
Yang, M. H., Wu, M. Z., Chiou, S. H., Chen, P. M., Chang, S. Y., Liu, C. J., et al. (2008). Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nature Cell Biology, 10(3), 295–305. doi:10.1038/ncb1691.
Yang, M. H., Hsu, D. S., Wang, H. W., Wang, H. J., Lan, H. Y., Yang, W. H., et al. (2010). Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nature Cell Biology, 12(10), 982–992. doi:10.1038/ncb2099.
Yang, F., Sun, L., Li, Q., Han, X., Lei, L., Zhang, H., & Shang, Y. (2012). SET8 promotes epithelial-mesenchymal transition and confers TWIST dual transcriptional activities. The EMBO Journal, 31(1), 110–123. doi:10.1038/emboj.2011.364.
Ye, X., Tam, W. L., Shibue, T., Kaygusuz, Y., Reinhardt, F., Ng Eaton, E., & Weinberg, R. A. (2015). Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature, 525(7568), 256–260. doi:10.1038/nature14897.
Yilmaz, M., & Christofori, G. (2009). EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Reviews, 28(1–2), 15–33. doi:10.1007/s10555-008-9169-0.
Yokomizo, T., Ogawa, M., Osato, M., Kanno, T., Yoshida, H., Fujimoto, T., et al. (2001). Requirement of Runx1/AML1/PEBP2alphaB for the generation of haematopoietic cells from endothelial cells. Genes to Cells: Devoted to Molecular & Cellular Mechanisms, 6(1), 13–23.
Yonemura, S., Wada, Y., Watanabe, T., Nagafuchi, A., & Shibata, M. (2010). alpha-Catenin as a tension transducer that induces adherens junction development. Nature Cell Biology, 12(6), 533–542. doi:10.1038/ncb2055.
You, S., Avidan, O., Tariq, A., Ahluwalia, I., Stark, P. C., Kublin, C. L., & Zoukhri, D. (2012). Role of epithelial-mesenchymal transition in repair of the lacrimal gland after experimentally induced injury. Investigative Ophthalmology & Visual Science, 53(1), 126–135. doi:10.1167/iovs.11-7893.
Yue, R., Li, H., Liu, H., Li, Y., Wei, B., Gao, G., et al. (2012). Thrombin receptor regulates hematopoiesis and endothelial-to-hematopoietic transition. Developmental Cell, 22(5), 1092–1100. doi:10.1016/j.devcel.2012.01.025.
Zayzafoon, M., Abdulkadir, S. A., & McDonald, J. M. (2004). Notch signaling and ERK activation are important for the osteomimetic properties of prostate cancer bone metastatic cell lines. The Journal of Biological Chemistry, 279(5), 3662–3670. doi:10.1074/jbc.M308158200.
Zelzer, E., Glotzer, D. J., Hartmann, C., Thomas, D., Fukai, N., Soker, S., & Olsen, B. R. (2001). Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mechanisms of Development, 106(1–2), 97–106.
Zhang, C., Lv, J., He, Q., Wang, S., Gao, Y., Meng, A., et al. (2014). Inhibition of endothelial ERK signalling by Smad1/5 is essential for haematopoietic stem cell emergence. Nature Communications, 5, 3431. doi:10.1038/ncomms4431.
Zoukhri, D., Macari, E., & Kublin, C. L. (2007). A single injection of interleukin-1 induces reversible aqueous-tear deficiency, lacrimal gland inflammation, and acinar and ductal cell proliferation. Experimental Eye Research, 84(5), 894–904. doi:10.1016/j.exer.2007.01.015.
Zoukhri, D., Fix, A., Alroy, J., & Kublin, C. L. (2008). Mechanisms of murine lacrimal gland repair after experimentally induced inflammation. Investigative Ophthalmology & Visual Science, 49(10), 4399–4406. doi:10.1167/iovs.08-1730.
Acknowledgement
The authors thank Rebecca Jackson for her generous help in the preparation of the manuscript. We are also grateful to Jormay Lim and Isao Kobayashi for their valuable input.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Voon, D.CC., Thiery, J.P. (2017). The Emerging Roles of RUNX Transcription Factors in Epithelial-Mesenchymal Transition. In: Groner, Y., Ito, Y., Liu, P., Neil, J., Speck, N., van Wijnen, A. (eds) RUNX Proteins in Development and Cancer. Advances in Experimental Medicine and Biology, vol 962. Springer, Singapore. https://doi.org/10.1007/978-981-10-3233-2_28
Download citation
DOI: https://doi.org/10.1007/978-981-10-3233-2_28
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-3231-8
Online ISBN: 978-981-10-3233-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)