
Abstract
Cardiac remodeling and its role in disease progression is a multimechanistic and complex process. Disruption of normal extracellular matrix homeostasis is the most important event responsible for cardiac remodeling, altering heart structure and function. Therefore, targeting extracellular matrix remodeling enzymes such as matrix metalloproteinases has received much interest in terms of developing novel therapeutics strategies. Recent findings of microRNAs (endogenous, non-coding, ~22 nucleotide small RNA) in the cardiac tissue as dynamic modifiers of disease pathogenesis have provided glimpses of undiscovered regulatory mechanisms underlying cardiovascular diseases. The implication of several microRNAs in targeting extracellular matrix components (microRNA-29: collagen, fibrillin, elastin) and matrix metalloproteinases (microRNA-21: MMP-2; microRNA-320: MMP-9) has been well documented. The combined strategy of manipulating extracellular matrix remodeling through targeting matrix metalloproteinases by microRNAs has produced encouraging results in preclinical studies. This chapter reviews the potential of microRNA as therapeutics tool for cardiovascular diseases through direct and indirect interactions with the matrix metalloproteinases.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Cardiovascular diseases (CVD) are the leading cause of human morbidity and mortality worldwide. Adverse myocardial remodeling in response to hemodynamic load and cardiac stress in association with the hormonal response is a significant cause of CVD. In almost all CVD, an increase in cardiac fibroblasts number (due to the transformation of bone marrow (BM)-derived monocytes, BM progenitors, and fibrocytes) and extracellular matrix (ECM) deposition occurs [1]. The ECM forms a milieu surrounding cells that reciprocally influences many aspects of normal cell behavior, including proliferation, adhesion, migration, and survival; thereby assuring the harmonic structure and function of the heart. To do so, ECM undergoes dynamic remodeling regulated by a number of ECM synthesizing as well as degrading enzymes such as matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs). MMPs are zinc-dependent endopeptidase that plays a fundamental role in remodeling of the ECM under both normal and pathological conditions. The proteolytic activity of MMPs is strictly controlled by tissue inhibitors of metalloproteinases (TIMPs) [2, 3]. Considerable evidence implicates that the expression level of MMPs deregulates in cardiovascular diseases such as hypertrophy, atherosclerosis, leading to myocardial infarction and heart failure. Nearby, 60 MMP inhibitors have been pursued as clinical candidates targeting multiple disorders such as cardiovascular diseases, arthritis, and cancer. Since, the actual molecular mechanisms underlying their role in onset and progression of disease are still uncertain, clinical developments of most of the drugs have been discontinued due to nontargeted approach. The aim of this chapter is to review the involvement of a novel class of small noncoding RNAs (microRNA) known to acts as fine-tuner of the protein expression, in the regulation of MMPs and their use as a potential therapeutic tool in CVD.
2 Extracellular Matrix Remodeling in Cardiovascular Diseases
Every cell influences their surroundings and gets influenced from the same. In the human body, fibroblasts are present in every tissue bridging the voids and mediating the connections between other cell types. In myocardium, although cardiac myocytes constitute nearly three-fourth of normal myocardial tissue volume, cardiac fibroblast accounts for more than two-third of cell population [2]. Fibroblast is cells of mesenchymal origin, predominantly responsible for synthesizing and secreting a diverse group of proteins and nonprotein components which constitute a dynamic, noncellular, and three-dimensional support structure called the ECM. Nearly 300 proteins which constitute the ECM are fibrillar, non-fibrillar as well as glycosylated in nature. They include 43 subunits of collagens, 36 glycosylated proteins, and nearly 200 glycoproteins complex. The nonprotein component includes water, minerals, and polysaccharides such as glycosaminoglycans [3]. Depending upon their origin, composition, location, and interactions among different cellular components (epithelial-endothelial elements, fibroblast, myocytes, and adipocyte), ECM forms different specialized structures unique to each tissue. In the cardiac matrix, collagen (types I, III, IV, V, and VI), glycoproteins (fibronectin, laminins, periostin, fibromodulin, and vitronectin), proteoglycans (versican, lumican, and biglycan), and glycosaminoglycans (hyaluronic acid and dermatan sulfate) are the major constituents. The components such as collagen IV, laminin, nidogen, and heparan sulfate synthesized by epithelial and endothelial cells forms the basement membrane. Whereas, the fibrillar collagens, fibronectin, and proteoglycans primarily synthesized by stromal cells (fibroblasts, pericytes) forms the interstitial matrix. Combined together in a systematic manner, the ECM component regulates the cardiac cells behavior biochemically, biophysically, and biomechanically. The biochemical regulations enable the cells to interact with their surroundings through direct and indirect signaling. Whereas, biophysical and biomechanical regulation is the support provided for tissue architecture and integrity which further decides cell proliferation, differentiation, migration, polarity, adhesion, and survival. These dynamic interactions between ECM and intracellular events are mediated by a family of transmembrane receptors known as integrins, discoidin domain receptors, and syndecans [4]. The substitution of even a single amino acid in any ECM component can alter the cell structure as well as its behavior. Therefore, under normal development and physiology, ECM modifying enzymes maintains a precise equilibrium between ECM synthesis and degradation. This equilibrium insures harmonic structure and function of every tissue including myocardium. However, if unconstrained, it leads to accumulation of ECM resulting in fibrosis or disruption of ECM network altering heart structure and function. As discussed above, the arterial wall consists of collagen types I and III, macrophages, and smooth muscle cells. Precursors released during biosynthesis of collagen type I and III such as carboxy-terminal or the amino-terminal propeptides (PICP, PINP, PIIICP, PIIINP) as well as during degradation such as carboxy-terminal or the amino-terminal telopeptide for type I and III collagen (CITP, NITP, CIIITP, PIIINP) have been studied as serological marker of ECM turnover in cardiac patients. A reduced serum level of CITP as well as increased level of CITP, PICP, and TIMP1 has been observed in hypertensive patients’ suggesting both elevated synthesis and degradation occurs during high blood pressure [5,6,7]. Similarly, elevated levels of PIIINP and PICP (fibrosis) have been reported in patients with congestive heart failure and acute myocardial infarction (MI) [8]. These studies vividly illustrate that dysregulation in ECM rebuilding and remodeling contributes to several pathological conditions as severe as heart failure and MI. Majorly, ECM remodeling is mediated by families of metalloproteinases viz., matrix metalloproteinase (MMPs), meprins, a disintegrin and metalloproteinase (ADAMs), and a disintegrin and metalloprotease with thrombospondin motifs (ADAMTS) [9, 10]. In addition, serine, cysteine, heparanases, sulphatases, and cathepsin proteases also degrade ECM resulting in the release of growth factors and cytokines. Alterations in the level of proteases may lead to different types of heart diseases. Therefore, regulating MMPs may prevent the onset or progression of disease.
3 Matrix Metalloproteinases-Mediated Cardiac Remodeling
MMPs are the major class of proteases involved in ECM remodeling, therefore; playing a key role in biological (morphogenesis, angiogenesis, and wound healing) and pathological (tumor growth, arthritis, tissue ulceration, and cardiovascular disease) processes [11]. In 1962, a study on collagen remodeling during tadpole tail metamorphosis leads to the discovery of MMPs [12]. Since then, 25 vertebrate MMPs and 23 human homologues have been identified. Although human carries 24 MMP genes, chromosome 1 possesses two identical genes for MMP-23 making a total 23 MMP proteins [13]. On the basis of substrate specificity, domain organization and sequence similarity they are classified into four groups namely: (a) archetypal MMPs that includes collagenases (MMP-1, -8, -13), stromelysins (MMP-3, -10), and other archetypal MMPs (MMP-12, -19, -20, -27), (b) matrilysins (MMP-7, -26), (c) gelatinases (MMP-2, 9), and (d) furin-activatable MMPs such as secreted MMPs (MMP-11, -21, and -28), membrane-type MMPs (MMP-14, -15, -16, -17, -24, and -25), and type II transmembrane MMPs (MMP-23A and -23B). Generally, MMPs possesses four domains structure: a ∼80-residue pro-peptide at the amino terminus, a ∼165-residue catalytic domain with zinc localized at the active site, a ∼200-residue hemopexin-like carboxyl-terminal domain linked to the catalytic domain by 15–65 residue linker region and a transmembrane domain. MMP-7 and MMP-26 lacks hemopexin domain whereas, MMP-23 has unique cysteine-rich, proline-rich, and IL-1 type II receptor-like domains instead of a hemopexin domain. Due to the interaction of a cysteine residue of the pro-peptide domain with the zinc ion of the catalytic site, the MMPs are proteolytically inactive [14]. The proteolytic activity of MMPs is strictly controlled and activated at three different levels: (a) transcriptional and posttranscriptional level (by several cytokines (IL-1, TNF-α), growth factors (TGF-β), glucocorticoids, interferons), (b) zymogen activation (by other already activated MMPs, several serine proteinases or auto-proteolysis), and (c) by tissue inhibitors of metalloproteinases (TIMPs) [15, 16]. These regulatory measures insure the presence of MMPs in precise amount, at the right time and precise location, and in appropriate state (activated or inactive). However, cardiac or hemodynamic load (hypertension), hyperhomocysteinemia, myocarditis, and RAAS-mediated cardiac stress alters the level of MMPs and their regulators leading to morphological, structural, and functional changes of the heart, referred to as cardiac remodeling. Cardiac remodeling includes degradation or deposition of extracellular matrix (ECM), myocardiocytes hypertrophy, abnormal angiogenesis, coronary collateralization, alterations in receptor signaling cascade, apoptosis, abnormal differentiation, and survival of cardiac stem cell, differential expressions of miRNAs and epigenetic modifications.
4 Matric Metalloproteinases and Cardiovascular Diseases
In heart, the function of MMPs extends well beyond ECM degradation, such as, stimulation of multiple biological effects through liberating and mobilizing growth factors, cytokines [17]. Since all cardiac remodeling and myocardial architecture is associated with a change in the ECM composition; MMPs and their tissue inhibitors (TIMPs) are involved in the pathogenesis of a wide spectrum of cardiovascular disorders including atherosclerosis, restenosis, cardiomyopathy, congestive heart failure, myocardial infarction, and aortic aneurysm. Out of 23 MMPs identified in human, MMP-1, -2, -3, -7, -8, -9, -12, -13, -14, and TIMPs-1, -2, -3, -4 have been evaluated to respond to cardiac tissue repair stimuli [18]. The studies on patients tissue extracts with dilated cardiomyopathy revealed a decrease in MMP-1,-12, and increase in MMP-3 level [19, 20]. Similarly, post-MI studies on murine detected an increased level of MMP-8, -9, -13, and -14 [21,22,23]. Out of 23 MMPs, MMP-9 is surfaced as a leading candidate for having direct effects on cardiac remodeling. Its level increases post-MI, its inhibition improves post-MI outcomes and patients with the highest MMP-9 levels at baseline showed the greatest cardiovascular mortality [24]. A study on rat MI model also demonstrated time-dependent increase in relative TIMP-1 and TIMP-2 mRNA levels and decrease in TIMP-4 level [25]. Later, several clinical studies have utilized plasma samples to profile TIMP levels [26, 27].
These observations have led to the recognition of MMPs and TIMPs as potential therapeutic targets through the development of pharmacological reagents. Previous studies revealed that MMP inhibition have reduced the accumulation of myocardial ECM (fibrosis), LV dilation as well as preserved cardiac function and geometry in ischemic cardiomyopathy [25]. A number of pharmacological MMP inhibitors such as trandolapril and ramipril studied in vitro as well as in rat model have found to control MMP level but at the expense of induction or reduction of other MMPs and TIMPS [28,29,30]. Lately, though peptides were found to be selective inhibitors of MMP-9 and MMP-2, their clinical usage is highly questionable because of their susceptibility to proteolysis [31]. In addition, clinical trials of other MMP inhibitors like PG-116800 failed to reduce cardiac remodeling or improve clinical outcomes after MI [32]. Therefore, novel molecular therapies have been investigated to more effectively treat or prevent CVDs. RNA-based epigenetics mechanisms including those controlled by small noncoding micro-RNAs (miRNAs) plays a pivotal role in the regulation of cardiovascular diseases [33]. As a result, miRNAs have emerged as potential diagnostic and therapeutic tool for cardiovascular disease.
5 Small Non-coding RNAs with Big Potential
The existence of miRNAs came into picture nearly two decades ago (1993) when Victor Ambros, Rosalind Lee, and Rhonda Feinbaum discovered that lin-4, a gene involved in development of C. elegans larvae does not code for protein but instead codes a pair of small RNAs [34]. These small RNAs are highly conserved, endogenously expressed, and noncoding RNAs that negatively modulate gene expression by either promoting the degradation of mRNA or downregulating the protein production by translational repression. They are transcribed from either intergenic, intronic, or polycistronic regions of the genome previously thought to be the junk region (~97%). Since their discovery, more than 2000 human miRNAs have been cataloged in mirBase (miRNAs database) (http://www.mirbase.org) predicted to regulate expressions of at least 30% of all human protein-encoding genes. The biogenesis of miRNAs is a complex process where every step is subjected to tight molecular regulation (Fig. 1). It begins within nucleus by transcription of miRNAs genes by RNA polymerase II into ~2000 nucleotides primary transcripts (pri-miRNAs) that can encode one or more miRNAs. A combination of Drosha (an RNase III enzyme) and Pasha also known as DGCR8 (an RNA-binding protein) cleaves primary miRNAs into precursor miRNAs (60–100 nucleotides) which gets transported to cytoplasm in a Ran/GTP/Exportin-5-dependent manner. Once in the cytoplasm, another cleaving enzyme Dicer (RNase III) modifies the precursor miRNAs and generates double-stranded miRNA duplexes of 19–23 nucleotides containing functional guide strand and a passenger strand. The duplex miRNA gets incorporated into RNA-induced silencing complex (RISC) by associating with Argonaute proteins (Ago). The passenger strand gets degraded while the guide strands remains intact and binds the 3′ untranslated region (UTR) of target messenger RNA (mRNA). Pairing across all 22 nucleotides rarely occurs in mammalian cells instead, the seed region of the miRNA (nucleotides 2–7 or 8) appears most important for targeting mRNAs. If the base pair at 3′ UTR matches perfectly, the mRNA gets cleaved and degraded by Ago2-RISC complex. Otherwise, it causes downregulating of protein production through translational repression [35, 36]. miRNAs not only target single gene but often functionally related genes networks thus maintain optimal amount of cellular proteins and hence play a crucial role in the regulation of biological functions. In human cardiac tissue, miRNAs have been identified at all stages of development (for example, miR-1, miR-16, miR-27b, miR-30d, miR-126, miR-133, miR-143, miR-208, and the let-7 family) [37]. Our previous study has characterized several known and novel miRNAs in chicken cardiac tissue at all stages of development. Using next-generation sequencing, we identified a total of 1056 microRNAs, out of which 353 were known (for example, let-7, miR-140, miR-181, miR-30, miR-205, miR-103, and miR-22) while 703 were novel miRNAs [38]. These finding implicate that the miRNAs are highly expressed in non-diseased cardiac tissue from embryogenesis to adult life and play a key role in both normal cardiac maintenance and disease. The role of miRNAs in cardiac remodeling of the adult heart was further established with Dicer deletion through the use of a tamoxifen-inducible Cre recombinase in the postnatal murine myocardium. In juvenile, it lead to premature death within 1 week due to atrial enlargement and mild ventricular remodeling whereas, in adult, it induced myocyte hypertrophy, fibrosis, biventricular enlargement, and induction of fetal transcription [39]. These phenotypes were consistent with the defects during heart development observed in zebrafish embryos devoid of Dicer function [40]. Later, another study came up with the similar outcome but by deletion of the DiGeorge syndrome critical region gene 8 (DGCR8 instead of dicer) using muscle creatine kinase-Cre mice and a conditional floxed allele of the DGCR8 [41]. MicroRNA expression profiling studies revealed that, the 18 most strongly expressed miRNAs and miRNA families account for >90% of all miRNA expressed in the adult mouse heart. Surprisingly, most of them are differentially expressed during cardiac disease, indicating the importance of miRNAs as modifiers of gene expression programs in cardiovascular disease. However, not all differentially expressed miRNAs are implicated in the cardiac disease. For example, miR-214, which is most highly upregulated among the 24 miRNAs studied, failed to induce cardiac phenotypes in transgenic mice in ischemic cardiomyopathy, dilated cardiomyopathy, and aortic stenosis, [42]. Whereas, ablation of miR-133 via double knockout resulted in aberrant proliferation and apoptosis of myocytes, cardiac defects, suggesting their therapeutic application in heart disease [43].

Schematic representation of miRNA biogenesis as well as therapeutics. Inside nucleus, either of the intergenic, polycistronic or intronic regions of DNA gets transcribed by RNA Polymerase II (RNA Pol II) into nearly 2 kb long primary transcripts (pri-miRNAs). Drosha/DGCR8 cleaves pri-miRNAs into precursor miRNAs (pre-miRNA) of 60–100 bp which gets transported to cytoplasm by exportin 5. In cytoplasm, Dicer modifies pre-miRNA into double-stranded miRNA duplexes. One of the strand (guide strand) activates the RNA-induced silencing complex (RISC) which in turn causes translational repression as well as mRNA cleavage. mirmimics (left) activates RISC and reduce protein and gene expression while, antagomirs (right) reduce RISC activation resulting in translational repression/mRNA cleavage and in the end increase protein and gene expression
6 Role of microRNAs in Cardiovascular Diseases
miRNAs are highly stable in blood circulation due to their incorporation into microvesicles, exosomes, their association with RNA-binding proteins, and their adherence to circulating lipoproteins. These protective mechanisms enable miRNAs to fulfill biological functions outside the cell and serve as potential prognostic or diagnostic biomarkers in CVD pathologies [44]. Because of their nucleic acid nature they can be detected with high sensitivity and specificity using various molecular techniques such as microarrays and quantitative real-time PCR. Previous studies have shown a number of miRNAs to be unregulated in plasma of cardiovascular disease patients (Fig. 2). miR-208b and miR-499 were highly elevated (1600-fold and 100-fold respectively) in plasma from acute myocardial infarction (AMI) patients as compared with patients presenting with atypical chest pain without AMI [45]. Similarly, elevated miR-208a was detected in plasma of ~90% AMI patients whereas; it was undetectable in patients with non-AMI coronary heart disease or patients with other cardiovascular diseases [46]. However, unlike miR-208a which is specifically expressed in the heart, not all miRNAs are tissue specific. For example, miR-208b and miR-499 are also expressed in skeletal muscle therefore confounding the interpretation of miRNAs as diagnostic biomarkers in cardiac diseases [47]. Although recent advances in prognostics and/or diagnostics have increased the survival rate of patients with AMI, no significant change in mortality rates in patients who survive the initial AMI is observed. In a recent cohort study of 7733 AMI patients, 71% of AMI survival developed heart failure within 5 years [48]. Therefore, an expanded understanding of the function of miRNAs in gene regulatory networks engaged in MMP mediated cardiac remodeling is looked upon as a novel therapeutic strategy for CVD.

7 microRNAs-Mediated Matrix Metalloproteinases Regulation
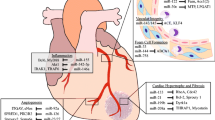
Recent studies have predicted/validated multiple gene targets of several miRNAs implicated in the pathophysiology of cardiovascular diseases [49] (Fig. 3). However, a major deficiency in the available literature on miRNA alterations occurring during cardiovascular diseases is that very few studies have assessed miRNA/MMP interactions. Since all the MMPs have not been explored to the same intensity, especially in the context of miRNA/MMP interactions as it relates to onset and progression of cardiovascular diseases, this part of discussion is skewed in favor of those miRNAs/MMPs and ECM components where the literature is more abundant. Roy et al. published the first report showing that miR-21 regulates MMP-2 expression in cardiac fibroblasts in response to myocardial ischaemia–reperfusion via PTEN (phosphatase and tensin homologue) pathway [50]. Later, Chen et al. found that miR-29b epigenetically regulates MMP-2/MMP-9 genes expression in the context of atherosclerosis. They observed that oxidized low-density lipoprotein (oxLDL) reduces methylation (DNA methyltransferases) of the MMP-2/MMP-9 genes via upregulation of miRNA-29b leading to the increase MMP-2/MMP-9 gene expression which is known to have a profound influence on cardiovascular diseases [51]. The member of miR-29 family suppresses the expression of several ECM proteins and has been predicted to downregulate during myocardial infarction. Abonnenc et al. confirmed the role of miR29 in ECM remodeling through pre-miR-29b and anti-miR-29b transfection in murine cardiac fibroblast (CF) [52]. Based on CF secretome data, they concluded that pre-miR-29b treatment reduced the collagen and MMP-2 secretion while anti-miR-29b did not stimulate their secretion [52, 53]. Lately, Chen et al. examined miRNA changes in different heart-cell compartments using congestive heart-failure experimental model. They observed that with decrease expression of miR-29b, the expression of MMP-2 did not change significantly in fibroblasts of left atrial. However, with increases expression miR-21 in left ventricular fibroblasts, the expression of MMP-2 did change [54]. These studies clearly suggest that aberrant posttranscriptional regulation of matrix metalloproteinases (MMPs) by miRNA is an important factor in cardiovascular diseases. Considering, the possibility of exploiting small noncoding RNAs as therapeutic tool for cardiovascular pathology, few studies used exogenous small interfering RNA (siRNA) which follow the same mechanism as miRNA to targets MMP-2 and -9. As described previously, MMP-2 is known to involve in various vascular complications including plaque destabilization and restenosis. Due to its constitutive expression by vascular smooth muscle cells, MMP-2 can facilitate vascular SMCs migration through hydrolysis of numerous extracellular matrix components leading to atherosclerosis. Transfection of vascular smooth muscle cells with MMP-2 siRNA has significantly decreased MMP-2 gene expression, resulting in the suppression of cell migration [55]. Similarly, atherosclerotic plaque stability has been obtained through silencing of MMP-9 gene using MMP-9 siRNA in apolipoprotein E (ApoE)-/- mice, suggesting that miRNAs and/or siRNAs represents potential pharmacological tool to target MMPs and their associated proteins [56].

Schematic representation of miRNAs, involved in development of cardiac hypertrophy and myocardial fibrosis. Cardiac stress leads to the dysregulation of miRNAs (few of them are represented: miR133, miR29, miR21 and miR21) as well as their target genes/proteins (few of them are represented). Rho A Ras homolog gene family, member A, Cdc42 Cell division control protein 42, Nelf-A Negative elongation factor A, WHSC2 Wolf–Hirschhorn syndrome candidate 2, IGFR1 Insulin-like growth factor receptor 1, SGK1 Serum-and glucose- regulated kinase, Col1A1 Collagen type 1 α1, ADAM A disintegrin and metalloproteinase, MMP-2 Matrix metalloprotease-2, ITGB1 Integrin beta-1, ITGA1 Integrin Alpha 1, Spry1 Sprouty RTK Signaling Antagonist 1, PTEN Phosphatase and tensin homolog, ERK Extracellular signal–regulated kinases, MAPK Mitogen-activated protein kinases, Col Collagen, Mef2a Myocyte Enhancer Factor 2A, Fibro Fibronectin, Ras-GAP GTPase-Activating Protein, Cdk9 Cyclin-dependent kinase 9, Rheb Ras homolog enriched in brain
8 microRNA Research and Therapeutics
Despite tremendous efforts in the traditional approach of drug design involving enzymes, cell surface receptors, increasing number of mortality and morbidity due to cardiovascular disease poses a great challenge to the therapeutic strategy adopted until now. Because of their specificity to their targets in a particular cellular pathway, miRNAs are rapidly becoming the most promising pharmacological tool to diagnose and treat cardiovascular disease. Therefore, it is necessary to understand methods, material, and tools required to resolve complex findings of miRNA research. miRNAs represent only about 0.01% of total cell RNA requiring extremely precise and sensitive instruments often with additional purification and enrichment steps. For visualization of individual pre-miRNA or mature miRNA expression in cells or tissues, quantitative real-time PCR (qRT-PCR), northern blotting, and in situ hybridization (ISH) in combination with immunohistochemistry are widely used. Northern blotting and ISH are very simple yet sensitive techniques but consumes relatively more time and sample since they do not involve miRNA amplification. qRT-PCR represents a balance of cost, precision, and sample size using modified way of reverse transcribing individual miRNA from a pool of miRNAs present in isolated total mRNAs. For screening of genome-wide miRNA expression, high-throughput molecular biology techniques such as miRNA microarray, deep sequencing are followed by validation with qRT-PCR. While miRNA microarray and qRT-PCR are two of the most common methods for evaluating known miRs, there are still contrasting reports on the correlation between microarray and qRT-PCR [57, 58]. To study novel miRNAs whose complement has not been ascertained, a more sensitive technique for small RNA sequences, which provides absolute abundance values of miRNAs is next-generation sequencing. It is known that a specific miRNA can regulate the expression of multiple genes (targets) while the expression of individual gene can be regulated by multiple miRNAs [59]. Therefore, for a comprehensive understanding of miRNA function and potential therapeutic use in heart disease, identification and validation of miRNA targets is of fundamental importance. There are several in silico target prediction programs such as TargetScan, PicTar, MiRanda/mirSVR, miRBase/MicroCosm, RNA22, and PITA. These databases use common feature like complementarity between the 3′ UTR of the target mRNA and the 5′-seed of the miRNA, conservation among species, the presence of several miRNA target sites and pattern recognition [60,61,62]. The predicted target needs to be further validated which can be achieved using biochemical methods, proteomic/transcriptome analysis, and RISCome analysis. Biochemical methods include qRT-PCR, western blotting, reporter assays, hybrid PCR, cytoplasm/nucleus ratio of mRNA, affinity purification, biotin-tagged miRNAs, and labeled microRNA pull-down assay (LAMP) [63,64,65,66,67,68]. Proteome analysis includes stable isotype labeling with amino acids in cell culture (SILCA) along with mass spectrometry to identify miRNA–mRNA interactions [69, 70]. Lastly, RISCome analysis includes target identification by sequencing of RNA-induced silencing complex and Argonaute. Other novel high-throughput assays based on immunoprecipitation are RIP-Chip (RNA-Binding Protein Immunoprecipitation-Microarray Profiling), PAR-CLIP (Photoactivatable-Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation), and HITS-CLIP (High-throughput Sequencing of RNAs isolated by cross-linking immunoprecipitation [46, 71,72,73,74,75,76]. Not all these methods have been applied for cardiovascular research yet, but since miRNA research is a growing field, these methods could be adapted because of their accurate and quantitative estimation of miRNA profiles and expression.
The general strategy of therapy for any disease is to reverse the pathological changes that resulted due to disease. Therefore, those MMPs and miRNAs whose overexpression is responsible for cardiovascular disease should be suppressed and vice versa. The suppression of overexpressed MMPs/miRNAs can be achieved by miRNA mimic technology (miR-mimic) which is an approach for gene silencing by generating artificial double-stranded miRNA-like RNA fragments. These RNA fragments mimic endogenous miRNAs and bind specifically to its target mRNA activating the RNA-induced silencing complex (RISC), downregulating specific mRNAs and thus induce gene suppression [77]. On the contrary, the expression of MMPs can be induced by antimiRs (cholesterol-conjugated, 2-O-methyl–modified antimiRs, called antagomiRs), which are a class of chemically engineered oligonucleotides specifically silencing single endogenous miRNA (Fig. 1). They competitively inhibit specific miRNA by binding to the target mature miRNA and lead to a reduced activation of RISC and consequently to an upregulation of specific mRNA and gene expression [78]. As mentioned above, few studies have demonstrated the value of mimic in downregulating the expression of MMP-9 in mouse model of atherosclerosis as well as in vitro. Besides, to promote miRNAs as a viable therapeutic target there are several difficulties to overcome such as, difference in predicted and actual physiological targets, potential off-target effects of antimiRs, and global effect on ECM of other organs. Further studies are required to elucidate the exact methods by which miRNAs are able to repress translation and initiate mRNA degradation of selected targets. The recent success of the first human clinical trial of a miravirsen (SPC3649) therapeutic for suppression of hepatitis C virus (HCV) replication has raised possibilities for the use miRNAs as a therapeutic target in CVD and clinical trials are eagerly awaited [79].
9 Conclusion
Recent studies have established a cause-and-effect relationship between the induction and activation of MMPs and cardiovascular diseases raising great expectations for MMPs as promising target. Also, miRNAs targeting MMPs and/or their regulators, as directly or indirectly discussed above are sufficient to draw a plausible inference that miRNA regulation of MMPs expression is an important mechanism for causing CVD. Since the activity of MMPs relies upon several complex interconnected molecular interactions, many clinical trials of MMP inhibitors failed to improve clinical outcomes. miRNA-based therapeutics under preclinical trials have shown promising results in numerous animal models of CVD such as cardiac hypertrophy, fibrosis, and MI. Till now, the role of less than 12 MMPs have been investigated post-myocardial infraction and very few MMPs have been predicted to be the targets of miRNAs. Therefore, extensive studies analyzing the complex interactions between specific miRNAs and MMPs in context with CVD will definitely bridge this gap and provide novel opportunities for diagnosis and therapy of CVD.
References
Krenning G, Zeisberg EM, Kalluri R (2010) The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol 225:631–637
Vliegen HW, van der Laarse A, Cornelisse CJ, Eulderink F (1991) Myocardial changes in pressure overload-induced left ventricular hypertrophy. A study on tissue composition, polyploidization and multinucleation. Eur Heart J 12:488–494
Naba A, Clauser KR, Hoersch S et al (2012) The Matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics 11:M111.014647–M111.014647
De Haas HJ, Arbustini E, Fuster V et al (2014) Molecular imaging of the cardiac extracellular matrix. Circ Res 114:903–915
Zannad F, Rossignol P, Iraqi W (2010) Extracellular matrix fibrotic markers in heart failure. Heart Fail Rev 15:319–329
Laviades C, Varo N, Fernandez J et al (1998) Abnormalities of the extracellular degradation of collagen type I in essential hypertension. Circulation 98:535–540
Lindsay MM, Maxwell P, Dunn FG (2002) TIMP-1: a marker of left ventricular diastolic dysfunction and fibrosis in hypertension. Hypertension 40:136–141
López B, Querejeta R, Varo N et al (2001) Usefulness of serum carboxy-terminal propeptide of procollagen type I in assessment of the cardioreparative ability of antihypertensive treatment in hypertensive patients. Circulation 104:286–291
Zannad F, Alla F, Dousset B, Perez A, Pitt B (2000) Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES). Rales Investigators Circ 02:2700–2706
Lin YH, Ho YL, Wang TD et al (2006) The relation of amino-terminal propeptide of type III procollagen and severity of coronary artery disease in patients without myocardial infarction or hibernation. Clin Biochem 39:861–866
Sternlicht MD, Werb Z (2001) How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol 17:463–516
Gross J, Lapiere CM (1962) Collagenolytic activity in amphibian tissues: a tissue culture assay. Proc Natl Acad Sci USA 48:1014–1022
Park HI, Ni J, Gerkema FE et al (2000) Identification and characterization of human endometase (matrix metalloproteinase-26) from endometrial tumor. J Biol Chem 275:20540–20544
Auld DS (2004) Structural zinc sites. In: Messerschmidt A, Bode W, Cygler M (eds) The handbook of metalloproteins, vol 3. Wiley, Chichester, pp 403–415
Nagase H, Woessner JF (1999) Matrix metalloproteinases. J Biol Chem 274:21491–21494
Fini ME, Cook JR, Mohan R (1998) Brinckerhoff, CE. Regulation of matrix metalloproteinase gene expression. In: Parks WC, Mecham RP (eds) Matrix metalloproteinases. Academic Press, New York, pp 299–356
Mittal B, Mishra A, Srivastava A et al (2014) Matrix metalloproteinases in coronary artery disease. Adv Clin Chem 64:1–72
Rodríguez D, Morrison CJ, Overall CM (2010) Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta Mol Cell Res 1803:39–54
Zamilpa R, Lindsey ML (2010) Extracellular matrix turnover and signaling during cardiac remodeling following MI: causes and consequences. J Mol Cell Cardiol 48:558–563
Thomas CV, Coker ML, Zellner JL et al (1998) Increased matrix metalloproteinase activity and selective upregulation in LV myocardium from patients with end-stage dilated cardiomyopathy. Circulation 97:1708–1715
Lindsey ML, Goshorn DK, Squires CE et al (2005) Age-dependent changes in myocardial matrix metalloproteinase/tissue inhibitor of metalloproteinase profiles and fibroblast function. Cardiovasc Res 66:410–419
Deten A, Volz HC, Holzl A et al (2003) Effect of propranolol on cardiac cytokine expression after myocardial infarction in rats. Mol Cell Biochem 251:127–137
Greene J, Wang M, Liu YE et al (1996) Molecular cloning and characterization of human tissue inhibitor of metalloproteinase 4. J Biol Chem 271:30375–30380
Blankenberg S, Rupprecht HJ, Poirier O et al (2003) Plasma concentrations and genetic variation of matrix metalloproteinase 9 and prognosis of patients with cardiovascular disease. Circulation 107:1579–1585
Peterson JT, Li H, Dillon L, Bryant JW (2000) Evolution of matrix metalloprotease and tissue inhibitor expression during heart failure progression in the infarcted rat. Cardiovasc Res 46:307–315
Kai H, Ikeda H, Yasukawa H et al (1998) Peripheral blood levels of matrix metalloproteases-2 and -9 are elevated in patients with acute coronary syndromes. J Am Coll Cardiol 32:368–372
Wagner DR, Delagardelle C, Ernens I et al (2006) Matrix metalloproteinase-9 is a marker of heart failure after acute myocardial infarction. J Card Fail 12:66–72
Jayasankar V, Woo YJ, Bish LT et al (2004) Inhibition of matrix metalloproteinase activity by TIMP-1 gene transfer effectively treats ischemic cardiomyopathy. Circulation 110(11)
Lindsey ML, Zamilpa R (2012) Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc Ther 30:31–41
Yamamoto D, Takai S, Jin D et al (2007) Molecular mechanism of imidapril for cardiovascular protection via inhibition of MMP-9. J Mol Cell Cardiol 43:670–676
Seeland U, Kouchi I, Zolk O et al (2002) Effect of ramipril and furosemide treatment on interstitial remodeling in post-infarction heart failure rat hearts. J Mol Cell Cardiol 34:151–163
Hudson MP, Armstrong PW, Ruzyllo W et al (2006) Effects of selective matrix metalloproteinase inhibitor (PG-116800) to prevent ventricular remodeling after myocardial infarction: results of the PREMIER (prevention of myocardial infarction early remodeling) trial. J Am Coll Cardiol 48:15–20
Tyagi AC, Sen U, Mishra KP (2011) Synergy of microrna and stem cell: a novel therapeutic approach for diabetes mellitus and cardiovascular diseases. Curr Diabetes Rev 7:367–376
Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854
Davison TS, Johnson CD, Andruss BF (2006) Analyzing micro-RNA expression using microarrays. Methods Enzymol 411:14–34
Zeng Y, Cullen BR (2003) Sequence requirements for micro RNA processing and function in human cells. RNA 9:112–123
Thum T, Galuppo P, Wolf C et al (2007) microRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation 116:258–267
Rustagi Y, Jaiswal HK, Rawal K et al (2015) Comparative characterization of cardiac development specific microRNAs: fetal regulators for future. PLoS ONE 10:10
Da Costa Martins PA, Bourajjaj M, Gladka M et al (2008) Conditional dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation 118:1567–1576
Giraldez AJ, Cinalli RM, Glasner ME et al (2005) microRNAs regulate brain morphogenesis in zebrafish. TL-308. Science 308:833–838
Rao PK, Toyama Y, Chiang HR et al (2009) Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ Res 105:585–594
van Rooij E, Sutherland LB, Liu N et al (2006) A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA 103:18255–18260
Carè A, Catalucci D, Felicetti F et al (2007) MicroRNA-133 controls cardiac hypertrophy. Nat Med 13:613–618
Gilad S, Meiri E, Yogev Y et al (2008) Serum microRNAs are promising novel biomarkers. PLoS ONE 3:9
Corsten MF, Dennert R, Jochems S et al (2010) Circulating microRNA-208b and microRNA-499 reflect myocardial damage in cardiovascular disease. Circ Cardiovasc Genet 3:499–506
Wang G-K, Zhu J-Q, Zhang J-T et al (2010) Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur Heart J 31:659–666
van Rooij E, Quiat D, Johnson BA et al (2009) A family of micrornas encoded by myosin genes governs myosin expression and muscle performance. Dev Cell 17:662–673
Ezekowitz JA, Kaul P, Bakal JA et al (2009) Declining in-hospital mortality and increasing heart failure incidence in elderly patients with first myocardial infarction. J Am Coll Cardiol 53:13–20
Neppl RL, Wang DZ (2014) The myriad essential roles of microRNAs in cardiovascular homeostasis and disease. Genes Dis 1:18–39
Roy S, Khanna S, Hussain SR et al (2009) MicroRNA expression in response to murine myocardial infarction: MiR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res 82:21–29
Chen K-C, Wang Y-S, Hu C-Y et al (2011) OxLDL up-regulates microRNA-29b, leading to epigenetic modifications of MMP-2/MMP-9 genes: a novel mechanism for cardiovascular diseases. FASEB J 25:1718–1728
Abonnenc M, Nabeebaccus AA, Mayr U et al (2013) Extracellular matrix secretion by cardiac fibroblasts: role of MicroRNA-29b and MicroRNA-30c. Circ Res 113:1138–1147
van Rooij E, Olson EN (2009) Searching for miR-acles in cardiac fibrosis. Circ Res 104:138–140
Chen Y, Wakili R, Xiao J et al (2014) Detailed characterization of microRNA changes in a canine heart failure model: relationship to arrhythmogenic structural remodeling. J Mol Cell Cardiol 1–12
Kim D, Lee D, Jang YL et al (2012) Facial amphipathic deoxycholic acid-modified polyethyleneimine for efficient MMP-2 siRNA delivery in vascular smooth muscle cells. Eur J Pharm Biopharm 81:14–23
Jin Z, Xiong Q, Jia F et al (2015) Investigation of RNA interference suppression of matrix metalloproteinase-9 in mouse model of atherosclerosis. Int J Exp Med 8:5272–5278
Ach RA, Wang H, Curry B (2008) Measuring microRNAs: comparisons of microarray and quantitative PCR measurements, and of different total RNA prep methods. BMC Biotechnol 8:69
Chen Y, Gelfond JAL, McManus LM, Shireman PK (2009) Reproducibility of quantitative RT-PCR array in miRNA expression profiling and comparison with microarray analysis. BMC Genom 10:407
Romaine SPR, Tomaszewski M, Condorelli G, Samani NJ (2015) microRNAs in cardiovascular disease: an introduction for clinicians. Heart 101:921–928
Witkos TM, Koscianska E, Krzyzosiak WJ (2011) Practical aspects of microRNA target prediction. Curr Mol Med 11:93–109
Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20
Krek A, Grün D, Poy MN et al (2005) Combinatorial microRNA target predictions. Nat Genet 37:495–500
Thum T, Catalucci D, Bauersachs J (2008) microRNAs: novel regulators in cardiac development and disease. Cardiovasc Res 79:562–570
Huang Y, Qi Y, Ruan Q et al (2011) A rapid method to screen putative mRNA targets of any known microRNA. Virol J 8:8
Li J, Xia W, Huang B et al (2010) A strategy to rapidly identify the functional targets of microRNAs by combining bioinformatics and mRNA cytoplasmic/nucleic ratios in culture cells. FEBS Lett 584:3198–3202
Orom UA, Lund AH (2007) Isolation of microRNA targets using biotinylated synthetic microRNAs. Methods 43:162–165
Vo NK, Dalton RP, Liu N et al (2010) Affinity purification of microRNA-133a with the cardiac transcription factor, Hand2. Proc Natl Acad Sci USA 107:19231–19236
Hsu RJ, Tsai HJ (2011) Performing the labeled microRNA pull-down (LAMP) assay system: an experimental approach for high-throughput identification of microRNA-target mRNAs. Methods Mol Biol 764:241–247
Baek D, Villén J, Shin C et al (2008) The impact of microRNAs on protein output. Nature 455:64–71
Yang Y, Chaerkady R, Kandasamy K et al (2010) Identifying targets of miR-143 using a SILAC-based proteomic approach. Mol BioSyst 6:1873–1882
Fleissner F, Jazbutyte V, Fiedler J et al (2010) Short communication: asymmetric dimethylarginine impairs angiogenic progenitor cell function in patients with coronary artery disease through a MicroRNA-21-dependent mechanism. Circ Res 107:138–143
Matkovich SJ, Van Booven DJ, Eschenbacher WH, Dorn GW (2011) RISC RNA sequencing for context-specific identification of in vivo microRNA targets. Circ Res 108:18–26
Tan LP, Seinen E, Duns G et al (2009) A high throughput experimental approach to identify miRNA targets in human cells. Nucleic Acids Res 37:20
Wang W-X, Wilfred BR, Hu Y et al (2010) Anti-Argonaute RIP-Chip shows that miRNA transfections alter global patterns of mRNA recruitment to microribonucleoprotein complexes. RNA 16:394–404
Hafner M, Landthaler M, Burger L et al (2010) Transcriptome-wide identification of RNA-binding protein and MicroRNA target sites by PAR-CLIP. Cell 141:129–141
Chi SW, Zang JB, Mele A, Darnell RB (2009) Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460:479–486
van Rooij E, Sutherland LB, Thatcher JE et al (2008) Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA 105:13027–13032
Stenvang J, Petri A, Lindow M et al (2012) Inhibition of microRNA function by antimiR oligonucleotides. Silence 3:1
Lanford RE, Hildebrandt-Eriksen ES, Petri A et al (2010) Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327:198–201
Thum T, Gross C, Fiedler J et al (2008) MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 456:980–984
van Rooij E, Sutherland LB, Qi X et al (2007) Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 316:575–579
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Saxena, S., Rustagi, Y., Jain, A., Dubey, S., Rani, V. (2017). microRNAs-Mediated MMPs Regulation: Novel Mechanism for Cardiovascular Diseases. In: Chakraborti, S., Chakraborti, T., Dhalla, N. (eds) Proteases in Human Diseases. Springer, Singapore. https://doi.org/10.1007/978-981-10-3162-5_24
Download citation
DOI: https://doi.org/10.1007/978-981-10-3162-5_24
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-3161-8
Online ISBN: 978-981-10-3162-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)