
Abstract
Accumulating evidence has shown the presence of cancer stem cells in a wide spectrum of human cancers, which have the ability to self-renew and differentiate, thus leading to tumorigenesis, proliferation, cancer dissemination, drug resistance, and tumor relapse. Cancer cell plasticity allows tumor to invade and grow at primary or distant sites. Epithelial-mesenchymal transition (EMT) is the most important mechanism of cancer cell plasticity and cancer stem cells. Substantial evidence has supported a noncoding RNA network, especially miRNA, in regulating cancer cell plasticity and cancer stem cell biology. Besides, lncRNA is also found to participate in cancer development. Understanding the mechanisms of these processes might be valuable for developing accurate targeted therapies to tackle cancer progression and cancer stem cells.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
6.1 Introduction
Cancer is a group of diseases consist of abnormally growing cells with the potential to invade and metastasize to other parts of the body. Generally, cancer grows when normal cells change as a result of accumulated mutations due to environmental factors or sometimes hereditary mutations. Mutations in normal cells lead to chromosomal instability, proliferation, and finally aggressive metastatic behavior. Owing to mutations, most of human cancers are heterogeneous diseases. There is a high degree of phenotypic and functional diversity between tumors, and even within the same tumor, divergences exist. For instance, breast tumors are diverse in their nature and responsiveness to therapies. According to gene expression molecular pattern, it could be classified into several subtypes including luminal subtype, basal subtype, HER2-overexpression subtype, and normal-like subtype. These subtypes prove to be different in their malignance and responsiveness to treatments [1, 2]. Some cancers also contain a hierarchy in which cancer stem cells (CSCs) differentiate into non-cancer stem cells (or bulk tumor cells) [3].
The cancer cell plasticity describes the ability of cancer cells to transform reversibly between distinct cell states phenotypically and genotypically, contributing to tumor growth in primary and distant sites. For example, some cancer cells, such as breast cancer cells, can transit between epithelial state and mesenchymal state. Reciprocal transition between epithelial state and mesenchymal state, which is a crucial event in embryonic development, has been confirmed to be a hallmark of cancer metastasis [4]. It is reported that, cancer cells in mesenchymal state are more competent than those in epithelial state to invade and form cancer dissemination [4, 5]. Considering the heterogeneity of cancer, Gupta et al. have found that isolated subpopulations of breast cancer cells with given phenotype will finally return to an equilibrium proportions over time, which can be explained by the Markov model, in which they suggest cell transition stochastically between states and any subpopulations of cancer cells can finally return to an equilibrium proportions over time in given conditions [6]. Of note here, cancer cells acquiring drug resistance responsive to therapy is also an important aspect of cancer cell plasticity [7]. Besides, via turning on or off some markers reversibly, cancer cells can transit between distinct states. For example, study in melanoma has revealed that dynamical expression of JRID1B, an H3K4 demethylase, endows cancer cells with tumorigenic ability. The JRID1B-positive and JRID1B-negative cells can transit to each other and the JRID1B-positive cells function in tumor maintenance [8].
The concept of CSCs refers to a subpopulation of cells within tumor possessing the ability to self-renew and differentiate into non-stem progenitor. Increasing evidence has supported the CSC hypothesis that many of human cancers are driven by the CSCs. Self-renewal of CSCs and differentiation into non-stem progeny maintains the cancer cell pool and mediates the cancer metastasis, therapy resistance, and relapse. CSCs’ transition from tumorigenic state to a non-tumorigenic state is one aspect of CSC plasticity. Similar with tumor heterogeneity, there exist different states in the CSC subpopulation. Transition between these states is another important aspect of CSC plasticity [5].
Noncoding RNA (ncRNA) is a functional molecule that is not translated into a protein. Quantities of ncRNAs have been found in recent decades, including ribosomal RNA (rRNA), transfer RNA (tRNA), small nuclear RNA (snRNA), small nucleolar RNA (snoRNA), microRNA (miRNA), long noncoding RNA (lncRNA), and so on. Evidence increasingly indicates that ncRNA has a significant effect on cancer and CSC biology and may act as a potential therapeutic target. In this chapter, we will review the functions and mechanisms of ncRNA, mainly miRNA and lncRNA, in regulating cancer cell plasticity (see Fig. 6.1), tumor progression, and CSC biology.

Overview of epithelial-mesenchymal cancer cell plasticity and the involvement of important miRNA and lncRNA
6.2 Cancer Cell Plasticity, Tumor Progression, and Cancer Stem Cell Biology
Most of the cancers diagnosed are primitively derived from normal tissue cells. After a progression of changes at the cellular, genetic, and epigenetic level, the normal cells are ultimately transformed to acquired uncontrolled cell division ability and therefore tumor forming, which process we usually call carcinogenesis or tumorigenesis. Considering the similarities normal stem cells and cancer stem cells share (ability to self-renew and differentiate), there is accumulating evidence that stem cells and progenitor cells may be the targets of transformation during carcinogenesis [9]. Once transformed, cancer cells grow without control to form a mass in primary tissue and develop invasiveness. To break the tissue microenvironment, invade into the local site or even metastasize to distant organ, cancers arising from epithelial tissues need to activate a program called epithelial-mesenchymal transition (EMT) to acquire an invasive phenotype.
EMT is a complex molecular and cellular program, during which polarized epithelial cells lose their epithelial features, including cell-cell adhesion and planar and apical-basal polarity, while acquiring a mesenchymal characteristics, including enhanced motility, invasiveness, and resistance to apoptosis [10, 11]. Abundance of cellular processes and extracellular signals are engaged to initiate and regulate an EMT process, including activation of specific transcription factors, expression of specific cell surface markers, expression of specific microRNA, epithelial cell-stromal cell interaction, hypoxia, cytokines and growth factors derived from tumor environment, and so on. For instance, loss of E-cadherin, a major marker of epithelial cells, is considered essential during EMT. Transcription factors that repress E-cadherin directly or indirectly are supposed to promote EMT. For example, Snail1, Slug, ZEB1, and SIP1 (ZEB2) can bind directly to E-cadherin promoter to repress its transcription, while Twist1 indirectly represses E-cadherin [12, 13]. Moreover, Onder et al. have disclosed that loss of E-cadherin upregulates Twist1 and ZEB1 expression, and Twist1 is a crucial downstream effector on cellular function. Therefore, they proposed a feed-forward signaling loop between Twist and E-cadherin [14]. Signals from tumor environment could also have a significant effect on EMT. According to Cannito’s study, hypoxia can promote EMT via promoting Wnt/β-catenin pathway and therefore resulting Snail translocation. Furthermore, late migration and invasiveness can be sustained in a hypoxia-inducible factor-1α (HIF-1α)-dependent mechanism [15]. Reverse process of EMT is mesenchymal-epithelial transition (MET), characterized by reestablished apical-basal polarity, tight junction, and expression of cell-cell-adhesion molecules such as E-cadherin. MET is often thought to be critical in tumor growth in distant organ, which reendows tumor cells with epithelial characteristics similar to cells in primary tumor.
The evidence of CSC was first described distinctly in 1994 by Lapidot et al. as they found tumorigenic leukemic cancer stem cells and a hierarchical organization in leukemic cells [16]. Since then CSC has been gradually accepted. The CSC hypothesis raises that many human cancers, including breast cancer, colon cancer, liver cancer, glioblastoma, leukemia, pancreas cancer, melanoma, and so on, are driven by a subpopulation of cancer cells that possess stem cell properties. These cells have the ability to self-renew and differentiate into progeny without stemness, therefore driving tumor formation, maintaining the cancer cell pool, mediating metastasis, resistance to therapies, and relapse leading to therapy failure. Numerous studies have showed that, CSCs, a small subset of cancer cells within a tumor, can be identified and isolated by a distinct set of markers. For example, based on cell surface marker expression, Al-Hajj et al. have successfully distinguished the tumorigenic cells from the non-tumorigenic cells in human breast cancer and identified the CSC as CD44+CD24−/low lineage− [17]. Utilizing in vitro and in vivo experimental system, Ginestier and colleagues have found another CSC marker aldehyde dehydrogenase activity 1(ALDH1). They found that, in human breast cancers, cells with high ALDH1 activity displayed tumorigenesis capable of self-renewal and recapitulated the original heterogeneity of the parental tumor [18]. Similar observation was made by Singh in brain cancer when using cell surface marker CD133. CD133+ cell fractions are able to initiate tumor in nonobese diabetic, severe combined immunodeficient (NOD-SCID) mice brain [19]. Subsequent studies have found that CD133 highly expression cells also contain tumorigenic cells in colon cancer [20], which indicates that CSCs share something conserved between distinct cancers. What’s more, combining different CSC markers identifies a more tumorigenic population. For instance, ALDH+CD133+ cells show an increased ability to initiate tumor compared with ALDH+CD133− or ALDH+ alone [21].
Studies have found that CSCs are regulated by both cell-intrinsic and cell-extrinsic pathways which are tightly regulated in normal cells. Accumulating evidence indicates that the core signaling pathways, including Wnt, Notch, Hedgehog, PI3K/AKT, etc., which are deregulated in cancer processes and CSCs, critically regulate survival and self-renewal of CSCs. Hedgehog signaling pathway plays a pivotal role in self-renewal and differentiation of normal stem cell and are tightly regulated by the stem cell niche. Deregulation of Hedgehog signaling pathway may play an important role in carcinogenesis, and activation of Hedgehog signaling pathway has been observed in the CSCs [22, 23]. Similarly, Notch and its downstream signaling are also critical in normal tissue stem cells or progenitor cells [24], and there is substantial evidence that abnormal Notch signaling pathway associates with cancer progressions [25, 26]. Wnt signaling pathway, a pivotal regulator of cell-fate decision, has been implicated in a variety of cancers [27–29], including ovarian cancer, breast cancer, non-small cell lung cancer, etc. Beside intracellular signaling pathway mentioned above, tumor environment also has a significant impact on tumor progression and CSCs. Cytokines derived from tumor niche, such as IL-6 and IL-8, have been observed to play a vital role in cancers [30–33].
In the recent decades, several intriguing studies have described a link between the EMT and the CSC. One study has found expression of CD44 is controlled by Wnt/β-catenin cascade [34]. Since CD44 is a marker of CSC, it might imply a role for EMT-related Wnt/β-catenin cascade in CSC maintenance. Mani et al. have observed a direct link between the EMT and the epithelial stem cell properties, and found that the induction of EMT via expression of either Twist or Snail in a non-tumorigenic state immortalizes human mammary epithelial cells (HMLEs) or that via exposure to TGF-β generates CD44highCD24low stem cell-like cells exhibiting not only enhanced ability to form mammospheres, a property correlated with mammary stem cells, but also EMT characteristics such as loss of E-cadherin and expression of Twist, Snail, and N-cadherin. Simultaneously, stem cells isolated from normal human mammary and breast carcinomas express the EMT markers [35]. Most recently, Liu et al. has uncovered the relationship between breast cancer stem cell (BCSC) and EMT. They showed that BCSCs could exist in at least two distinct states, namely, mesenchymal-like (EMT) state and epithelial-like (EMT) state. Moreover, BCSCs in distinct state were diverse in phenotype and function. The EMT state BCSCs, expressing a set of cell surface marker CD24−CD44+, were primarily quiescent and localized to the tumor-invasive front, whereas the MET state BCSCs, characterized as ALDH+, were proliferative and localized inside of the tumor. They proposed that the plasticity of BCSCs allowed them to undergo reversible EMT/MET transitions, which finally contributed to tumor invasiveness, metastasis, and growth at distant sites. Therefore, it’s worthy to note that it may be necessary to target alternative CSC states to achieve a better curative effect [5].
6.3 MicroRNAs Regulate Cancer Cell Plasticity and Tumor Progression
MicroRNA (miRNA) is a 19–23-nucleotide-long noncoding RNA, which functions in gene silence and posttranscriptional regulation of gene expression. The target of miRNA is usually a messenger RNA (mRNA). Via base pairing with the complementary sequences, miRNA represses the translational efficiency or destabilizes the target mRNA and can act on one or more target mRNAs. miRNA has diverse functions in cell biology including cell proliferation, differentiation, and apoptosis. Deregulated miRNAs have been proved to correlate closely with cancers [36, 37]. Depending on the mRNAs they target, miRNAs can be tumor suppressive or oncogenic. As early as 10 years ago, through utilizing the bead-based flow cytometric miRNA expression-profiling method, studies have observed a general downregulation of miRNAs in cancerous tissues compared with normal tissues. Moreover, the miRNA profiles in a way imply the developmental lineage and differentiation state of cancers. Furthermore, poorly differentiated tumors can be successfully classified by miRNA profiles [36]. Researches in breast cancer also indicate a significant deregulated miRNA expression in cancer versus normal tissues [37]. Meanwhile, through miRNA expression-profiling analysis, miRNAs, such as let-7e, miR-151-5p, miR-222, miR-21, miR-155, and miR-221, have been identified to be upregulated in cancerous tissues [38–40]. All of these suggest that we can discriminate cancerous tissues from normal tissues using miRNA profiles, which prompts a potential role for miRNA in cancer diagnosis. Indeed, subsequent studies have revealed that serum miRNA signature could be a useful biomarker for tumor progression and prediction of the outcome of several cancers [41–43].
Accumulating evidence has indicated that miRNA plays a critical role in cancer formation and development [44]. The c-Myc oncogene, which has been proved to act as both miRNA inducer and repressor, functions in inducing multiple cancer formation. On one hand, for example, the oncogenic miR-17-92 cluster is frequently found amplified in varieties of human cancers and is regulated by the MYC gene in transcriptional level [45–47]. Mu et al. have disclosed that expression of endogenous miR-17-92 is indispensable for suppressing apoptosis in Myc-induced B-cell lymphomas [48]. Via directly suppressing the expression of chromatin regulatory genes Sin3b, Hbp1, Suv420h1, and Btg1 and proapoptotic gene BIM, the MYC-regulated miR-17-92 cluster sustains autonomous proliferation and survival in MYC-induced tumors and therefore maintains the neoplastic state [49]. Besides, there is a network among MYC, miR17-92, and E2F1, a transcription factor that promotes cell cycle progression, in regulation of cell cycle, in which MYC and E2F1 positively regulate each other, while MYC-induced miR-17-92 negatively regulates E2F1 [45, 50]. Another important downstream target of miR-17-92 is the tumor suppressor PTEN. On one hand, c-Myc can mediate cell proliferation and apoptosis resistance in non-small cell lung cancer (NSCLC) cells through suppressing PTEN by miR-17-92 [51]. On the other hand, which is more often, MYC represses dozens of tumor-suppressive miRNAs including Let-7, miR-23, miR-34a, and so on [52]. Let-7, for instance, cooperates with an RNA-binding protein HuR to inhibit the expression of c-Myc in an interdependent manner. HuR represses the MYC oncogene by recruiting the let-7-loaded RNA-induced silencing complex (RISC) to the 3′-untranslated region (UTR) of c-Myc [53]. MYC can bind to the let-7 promoter, while there are let-7 binding sites in MYC 3′UTR. Hence, there exists a negative loop between let-7 and MYC. It is reported that multiple genes regulating cell cycle, proliferation and apoptosis, are responsive to the alteration of let-7. The major targets of let-7 are RAS and HMGA2 oncogenes. Johnson et al. have suggested that let-7 may function as a tumor suppressor through acting on the RAS oncogene, for there are multiple let-7 complementary sites in the RAS 3′UTR [54]. Similarly, Lee et al. have found that let-7 destabilizes HMGA2, a high-mobility group protein, via multiple target sites in 3′UTR of HMGA2 oncogene, to repress cell proliferation [55]. Deregulation of let-7 is generally found in cancer tissues, suggesting that let-7 is poorly expressed in cancer tissues compared with normal tissues [56].
Since aberrant activation of EMT triggers malignant tumor progression, a large amount of evidence has proved an miRNA network in regulating EMT process. Much work has observed an EMT in cancer cells in response to transforming growth factor (TGF)-β. TGF-β seems to play a dominant role in EMT in advanced cancer via directly activating transcription factors ZEB, Snail, and Twist [10]. Studies have identified a number of miRNAs that possibly take part in TGF-β-induced EMT pathway, including miR-200, miR-21, miR-31, and so on. It is reported that expression level of miR-21 and miR-31 is significantly increased in response to TGF-β stimulation. MiR-21 and miR-31 synergize with TGF-β to enhance the EMT by targeting TIAM1, a guanidine exchange factor of the Rac GTPase [57].
The miR-200 family contains miR-200a, miR-200b, miR-200c, miR-141, and miR-429. There is growing evidence suggesting that miR-200 participates in tumor metastasis via regulating EMT. All five members of miR-200 family have been found to markedly decrease in cells that have undergone EMT induced by TGF-β and in invasive breast cancer cell lines, which is reported to depend on the SMAD signaling pathway [58, 59]. The major targets of miR-200 are ZEB1 and SIP1, the E-cadherin transcription repressors [58]. Through directly repressing mRNA of ZEB1 and SIP1, miR-200 maintains the expression of E-cadherin and the epithelial morphology. Meanwhile, in mesenchymal cells, ZEB1 and SIP1 act as repressors of miR-200 through binding to a conserved site at the miR-200 promoter region [60], hence forming a reciprocal negative feedback loop between miR-200 and ZEB1/SIP1. Brabletz et al. have also found that the miR-200-ZEB1 feedback loop controls the Notch signaling in cancer cells, especially in poorly differentiated invasive tumor cells [61]. The Notch signaling pathway component, such as Jagged 1, Maml2, and Maml3, is also one of the miR-200 targets. Via inhibiting miR-200, ZEB1 upregulates the Notch signaling, contributing to cancer cells properties. Similarly, Yang et al. have discovered a miR-200-dependent pathway in the Notch-induced EMT [62]. The Notch ligand Jagged 2 has been found to upregulate the expression of GATA-binding factors, which in turn suppress the miR-200, thus promoting the EMT and metastasis. Furthermore, study in lung cancer has demonstrated that ZEB1 shows altered expression level in erlotinib-sensitive cancer cells and that ectopic expression of miR-200c can alter the drug sensitivity [63], suggesting that miR-200-ZEB1 feedback loop might be a potent target for cancer therapy.
6.4 MicroRNAs Play Important Roles in Regulating Cancer Stem Cells
The tumor consists of heterogeneous cells, in which cancer stem cells (CSCs), with the ability to self-renew and differentiate, are thought to be the driving force of the tumor development, therapy resistance, and recurrence. The CSCs have been found in a wide spectrum of cancer types, among which breast cancer stem cell (BCSC) is the first described CSC in solid tumor and also the best characterized CSC so far. Numerous extracellular factors and intracellular elements have been uncovered to regulate the CSCs properties, among which microRNAs have been validated to play a key role in regulating CSC properties [64]. Through comparing miRNA profiles, several miRNA clusters, such as miR-200c-141, miR-200b-200a-429, and miR-183-96-182, have been found to differentially express between BCSCs and non-tumorigenic bulk tumor cells [65]. According to Liu et al., the BCSC subpopulation is likewise heterogeneous containing distinct groups characterized by different markers, such as ALDH+ and CD24−CD44+. They have pointed out that BCSCs may exist in at least two alternative states (EMT and MET) on the basis of the CSC markers they express. The mesenchymal-like (EMT) BCSC represents the CD24−CD44+ subpopulation and is characterized as primarily quiescence and invasive marginal location. The ALDH+ subpopulation is described as epithelial-like (MET) BCSCs, which are proliferative and located centrally. Moreover, they have proposed that BCSCs may transition between the EMT and MET states to achieve tumor invasion, dissemination, and growth at distant organs [5]. Interconversion of EMT state and MET state is regulated by the microRNA network in that miR-9, miR-100, miR-221, and miR-155 induce the EMT state, while miR-34c, miR-200, miR-205, and miR-93 induce the MET state [66, 67].
More recently, several intriguing studies have described the role of miRNAs in modulating the cell fate of CSC. Through evaluating expression level in different-stage breast cancer samples, miR-9 has been found to overexpress in late-stage tumors with aggressive phenotypes and associate with a CD24−CD44+ phenotype and EMT [68]. Via repressing the CSC regulatory gene SMADCA5, SMADCD1, and BMPR2, miR-100 has been found to directly regulate self-renewal and differentiation of BCSCs and reduce the ALDH+ population [69]. Depending on the differentiation states, miRNA seems to have different impact on the CSCs. In the less differentiated breast cancer cells, miR-93 can induce MET accompanied by decreased expression of TGF-β and numerous stem cell regulatory genes, such as JAK1, STAT3, AKT3, SOX4, EZH1, and HMGA2, thus reducing the CSC population. However, the CSC subpopulation is increased as miR-93 expresses in a more differentiated breast cancer cells [70]. Another miRNA, let-7, has been reported as well to regulate multiple properties of BCSCs through its target gene H-RAS and HMGA2. It is interesting to point out that, via repressing H-RAS, let-7 reduces CSC self-renewal while having no effect on its differentiation. Via repressing HMGA2, let-7 enhances CSC differentiation while having no effect on its self-renewal [71]. In addition, let-7 repression seems to promote the BCSC expansion by the Wnt/β-catenin pathway through transactivation of a negative let-7 biogenesis regulator Lin28 [72]. Similar with let-7, miR-30 has also been found to regulate the self-renewal and apoptosis of BCSCs via its target Ubc9 and ITGB3, respectively [73]. More recently, a signaling axis involving Snail, miR-146a, and Numb has been identified in regulating symmetric and asymmetric cell division in colorectal cancer stem cells [74]. All these demonstrate that miRNA could be both repressor and promoter in regulating CSC properties, implying a more accurate miRNA-targeted therapy according to distinct tumor differentiation states.
Decreased expression of miR-200 family members has been proved to be important in tumor metastasis, apoptosis resistance, and drug resistance. The major targets of miR-200 are ZEB1 and SIP1. Of note here, stem cell factors, such as Sox2 and Klf4, are also candidate targets of miR-200 family members, suggesting a link between miR-200 and stem cells. Recently, molecular links between miR-200 and CSCs have drawn particular attention. Lim’s study has found that expression of miR-200 gradually loses in a non-stem human mammary epithelial (HMLE) cells during its transition to a stem-like phenotype and that the restoration of its expression can promote MET and decrease the stem cell properties. Similar phenomenon has been observed in the stem cell fractions of metastatic breast cancer. Furthermore, their research has uncovered an epigenetic modification mechanism of miR-200. According to Lim, miR-200 is repressed through polycomb-group-mediated histone modifications and DNA methylation [75]. Shimono et al. has disclosed that miR-200c is downregulated in human BCSCs and its expression can inhibit the growth of breast cancer cell and breast tumor formation driven by BCSCs in vivo [65], illustrating an indispensable role of miR-200c in BCSCs. Besides, it has been reported that the polycomb repressor Bmi1, a key regulator of cancer stem cell self-renewal, is repressed by miR-200 [65, 76]. Research in melanoma has also revealed that miR-200c overexpression leads to diminished expression of Bmi1, as well as melanoma tumor growth and metastasis inhibition [77]. In addition, the miR-200-ZEB1 reciprocal negative feedback loop not only promotes tumor cells dissemination but also regulates tumor-initiating cells in pancreatic and colorectal cancer cells. Through repressing the stemness-inhibiting miRNA, such as miR-200 and miR20-203, ZEB1 connects EMT activation with stemness maintenance [76]. These results suggest that miR-200 and miR-200-ZEB1 negative loop may be critical targets for CSC-targeted therapies.
6.5 Long Noncoding RNAs (LncRNAs) in Cancer Biology and Regulating Cancer Cell Plasticity
With the advance of high-resolution microarray and genome-wide sequencing technology, a large amount of novel transcripts have been found. It is reported that about 70 % of the genome is actively transcribed [78]. Of note here, noncoding RNA has drawn particular attention. Long noncoding RNA (lncRNA) is a new class of noncoding RNAs, with length ranged from 200 bp to 100 kbp, representing a large population of the noncoding RNA. Although there are thousands of lncRNA, it is the least well understood, and the vast majority of it is functionally uncharacterized [79]. Still, recent studies have gradually discovered one of lncRNA’s roles as the driver for tumor-suppressive or oncogenic function in multiple cancer types, including breast cancer, prostate cancer, pancreatic cancer, hepatocellular cancer, and so on [80, 81]. Expression of lncRNA is often altered and deregulated in tumors [82].
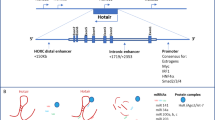
The HOX antisense intergenic RNA (HOTAIR), one of the best well-known lncRNAs, is a 2.2 kbp lncRNA molecule located on chromosome 12q13.13. HOTAIR is reported to be upregulated in many prevalent human cancers. There is substantial evidence that HOTAIR takes part in carcinogenesis, metastatic dissemination, and drug resistance. Expression of HOTAIR is an important prognostic marker of many cancers [83, 84]. Li et al. have discovered that high expression level of HOTAIR in laryngeal squamous cell carcinoma promotes methylation of the tumor suppressor PTEN, resulting to tumorigenesis [85]. While suppressed expression of HOTAIR could inhibit tumorigenesis and tumor proliferation [86], elevated expression level of HOTAIR was found in primary breast tumor and metastases, with its expression level in primary tumor as a powerful predictor of poor prognosis. HOTAIR increased the tumor invasiveness and metastasis in a polycomb repressive complex 2 (PRC2)-dependent manner [87]. Besides, the depletion of HOTAIR was found to associate with increased expression of E-cadherin and decreased expression of vimentin [86]. In clinical specimens of gastric cancer and colon cancer, HOTAIR inhibition was found to reverse the EMT process [88, 89]. All of these suggest that HOTAIR may act as an EMT modulator. In addition, recent research has uncovered that HOTAIR contributes to cisplatin resistance through downregulation of p21 expression [90]. Furthermore, substantial evidence indicates that HOTAIR may take part in the CSC regulation. It was found to be expressed at a much higher level in the colon CSC subpopulation (CD133+CD44+) compared with the other non-stem cancer cells. And knockdown of HOTAIR by siRNA correlated with a decreased colony forming capacity of colon and breast cancer cells [91]. These results suggest that HOTAIR may be an important regulator of cancer cell plasticity and a valuable predictor of tumor progression. HOTAIR inhibition may be a potential option for cancer prevention and CSC-targeted therapies.
Another lncRNA associated tightly with tumorigenesis is antisense noncoding RNA in the INK4 locus (ANRIL). Similar with HOTAIR, through binding to and recruiting PCR2, ANRIL represses the expression of p15INK4B locus, which encodes a tumor suppressor p15INK4B and has a pivotal role in cell cycle inhibition, senescence, and stress-induced apoptosis [92]. Besides, Nie et al. have uncovered another mechanism of ANRIL repression of p15 via silencing of KLF2 and P21 transcription [93]. These observations suggest that one of lncRNA’s mechanisms in mediating tumorigenesis may be through silencing of tumor suppressor genes.
The metastasis-associated lung adenocarcinoma transcript 1 (lncRNA MALAT1) is widely expressed in normal organs, such as lung and pancreas, and upregulated in several cancer types [94–96]. Through comparing metastatic and nonmetastatic early stage non-small cell lung cancer (NSCLC) tumors, MALAT1 was first demonstrated to be significantly associated with high metastatic potential and poor patient prognosis [97]. Consequently, a number of studies began to investigate the correlation of MALAT1 and metastasis. MALAT1 was then proved to be a regulator of numerous metastasis-associated gene expressions in lung cancer [96]. Through siRNA-mediated silencing of MALAT1 in bladder cancer cells, Ying et al. have found a decreased level in EMT-associated transcription factors, such as ZEB1, SIP1, and Slug, and an increased level of E-cadherin. They further demonstrated that MALAT1 promoted EMT in a Wnt signaling pathway activation-dependent manner [98]. MALAT1 was found to function in regulating the TGF-β-induced EMT [99]. In addition, it could promote tumor growth and metastasis in osteosarcoma by activating the PI3K-AKT pathway [100]. These results indicate that MALAT1 acts as a novel EMT regulator and may be a potential therapeutic target for cancer metastasis.
Besides oncogenic lncRNA, such as HOTAIR, ANRIL, and MALAT1, there are some lncRNAs acting as tumor suppressors. For instance, lincRNA-p21, a 3.1 kbp transcript located in the proximity of the cell cycle regulator gene Cdkn1a, via physically associating with heterogeneous nuclear ribonucleoprotein K (hnRNP-K), mediates transcription repression in the p53 pathway to regulate hundreds of p53 downstream target genes and triggers apoptosis [101]. Another tumor-suppressive lncRNA is GAS5, growth arrest-specific 5, which is abundant in cells with arrested growth owing to nutrients lacking, influences the cell survival and metabolism by modulating the transcriptional activity of the glucocorticoid receptor [102]. Significant reduction of GAS5 level has been observed in breast cancer tissues relative to corresponding adjacent normal tissues [103], partially explaining cancer cell survival in nutrient-lacking environment. Collectively, these studies show that tumor-suppressive lncRNA may play critical roles in cancer biology, but the underlying mechanisms still require much more exploration.
Since both miRNA and lncRNA are critical regulators of cancer, the interaction of miRNA and lncRNA in regulating cancer properties has drawn much attention. Recently, increasing studies have described the interaction between miRNA and lncRNA. LncRNA and miRNA may act as either decoy or decay reciprocally. The well-known lncRNA HOTAIR, on one hand, was found to suppress the tumor suppressor miR-7, by modulating the expression of HoxD10, and therefore sustain the expression of C-myc, Twist, and miR-9, hence maintaining the EMT process and the CSC pool of breast cancer [104]. On the other hand, expression of HOTAIR was repressed by miR-34a via directly binding to HOTAIR mRNA sequence in prostate cancer cells [105]. Liu et al. have also found a reciprocal suppressive relation between the p53-regulated tumor suppressor loc285194 and the tumor promoter miR-211. The tumor growth inhibition mediated by loc285194 was in part due to loc285194-specific suppression of miR-211 [106]. Besides, lncRNA and miRNA might synergize with each other. Studies in embryo development have discovered that miR-675 is embedded in the lncRNA H19’s first exon. By controlling the release of miR-675, H19 limited the growth of placenta before birth [107]. However, whether H19 and miR-675 function in the same way in tumor, we need more exploration.
6.6 Conclusions and Perspectives
Many human tumors consist of heterogeneous components, among which cancer stem cells possess the ability to self-renew and differentiate into the non-tumorigenic progeny, therefore driving tumorigenesis, proliferation, metastatic dissemination, and drug resistance. The cancer cell plasticity enables tumor to transition between distinct morphologies and proliferate at primary or distant sites. EMT is the most important molecular mechanism of cancer cell plasticity, either in non-tumorigenic cells or in cancer stem cells. Accumulating evidence indicates that noncoding RNA plays a vital role in regulating cancer and cancer stem cell biology. Alteration in miRNA expression alone has been found to cause neoplasm [108, 109]. Herein, we have discussed that noncoding RNAs, mainly miRNA and lncRNA, may act as oncogenic or tumor suppressors in cancer formation and progression and cancer stem cell biology. There definitely exists an ncRNA network in controlling cancer cell plasticity and CSC transition through regulating EMT-associated genes and relevant signaling pathways. Interaction of miRNA and lncRNA seems to play an important role in cancer and CSC properties, prompting a potential therapeutic method by targeting both miRNAs and lncRNAs, correlative oncogenes, and signaling pathways. However, as there are large amounts of ncRNAs, many underlying mechanisms of their interactions and physiological and pathological roles are still undiscovered.
References
Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52.
Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–74.
Dick JE. Breast cancer stem cells revealed. Proc Natl Acad Sci. 2003;100(7):3547–9.
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90.
Liu S, Cong Y, Wang D, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014;2(1):78–91.
Gupta PB, Fillmore CM, Jiang G, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146(4):633–44.
Sharma SV, Lee DY, Li B, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141(1):69–80.
Roesch A, Fukunaga-Kalabis M, Schmidt EC, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141(4):583–94.
Perou CM. Molecular stratification of triple-negative breast cancers. Oncologist. 2011;16 Suppl 1:61–70.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420.
Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9(4):265–73.
Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14(6):818–29.
Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7(6):415–28.
Onder TT, Gupta PB, Mani SA, et al. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68(10):3645–54.
Cannito S, Novo E, Compagnone A, et al. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis. 2008;29(12):2267–78.
Caceres-Cortes J, Mindeni M, Patersoni B, Caligiuri MA. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:17.
Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci. 2003;100(7):3983–8.
Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555–67.
Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401.
Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445(7123):111–5.
Huang EH, Hynes MJ, Zhang T, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009;69(8):3382–9.
Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66(12):6063–71.
di Magliano MP, Hebrok M. Hedgehog signalling in cancer formation and maintenance. Nat Rev Cancer. 2003;3(12):903–11.
Dontu G, Jackson KW, McNicholas E, et al. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004;6(6):R605–15.
Radtke F, Raj K. The role of Notch in tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer. 2003;3(10):756–67.
Roy M, Pear WS, Aster JC. The multifaceted role of Notch in cancer. Curr Opin Genet Dev. 2007;17(1):52–9.
Arend RC, Londoño-Joshi AI, Straughn JM, Buchsbaum DJ. The Wnt/β-catenin pathway in ovarian cancer: a review. Gynecol Oncol. 2013;131(3):772–9.
Loh YN, Hedditch EL, Baker LA, et al. The Wnt signalling pathway is up-regulated in an in vitro model of acquired tamoxifen resistant breast cancer. BMC Cancer. 2013;13(1):174.
Bravo DT, Yang Y-L, Kuchenbecker K, et al. Frizzled-8 receptor is activated by the Wnt-2 ligand in non-small cell lung cancer. BMC Cancer. 2013;13(1):316.
Korkaya H, G-i K, Davis A, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell. 2012;47(4):570–84.
Inoue K, Slaton JW, Eve BY, et al. Interleukin 8 expression regulates tumorigenicity and metastases in androgen-independent prostate cancer. Clin Cancer Res. 2000;6(5):2104–19.
Ginestier C, Liu S, Diebel ME, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. 2010;120(2):485.
Liu S, Ginestier C, Ou SJ, et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. 2011;71(2):614–24.
Wielenga VJ, Smits R, Korinek V, et al. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am J Pathol. 1999;154(2):515–23.
Mani SA, Guo W, Liao M-J, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–15.
Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–8.
Iorio MV, Ferracin M, Liu C-G, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65(16):7065–70.
Yu J, Li A, Hong S-M, et al. MicroRNA alterations of pancreatic intraepithelial neoplasias. Clin Cancer Res. 2012;18(4):981–92.
Wang J, Sen S. MicroRNA functional network in pancreatic cancer: from biology to biomarkers of disease. J Biosci. 2011;36(3):481–91.
Yu F, Jiao Y, Zhu Y, et al. MicroRNA 34c gene down-regulation via DNA methylation promotes self-renewal and epithelial-mesenchymal transition in breast tumor-initiating cells. J Biol Chem. 2012;287(1):465–73.
Keller A, Leidinger P, Gislefoss R, et al. Stable serum miRNA profiles as potential tool for non-invasive lung cancer diagnosis. RNA Biol. 2011;8(3):506–16.
Hu Z, Chen X, Zhao Y, et al. Serum MicroRNA signatures identified in a genome-wide serum MicroRNA expression profiling predict survival of non-small-cell lung cancer. J Clin Oncol. 2010;28(10):1721–6.
Schultz NA, Dehlendorff C, Jensen BV, et al. MicroRNA biomarkers in whole blood for detection of pancreatic cancer. JAMA. 2014;311(4):392–404.
Malek NP. Another myc in the wall: MicroRNA‐101 controls important functions in liver cancer formation. Hepatology. 2014;59(5):1676–7.
Diosdado B, Van De Wiel M, et al. MiR-17-92 cluster is associated with 13q gain and c-myc expression during colorectal adenoma to adenocarcinoma progression. Br J Cancer. 2009;101(4):707–14.
Xiao C, Srinivasan L, Calado DP, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9(4):405–14.
Hayashita Y, Osada H, Tatematsu Y, et al. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005;65(21):9628–32.
Mu P, Han Y-C, Betel D, et al. Genetic dissection of the miR-17∼ 92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev. 2009;23(24):2806–11.
Li Y, Choi PS, Casey SC, et al. MYC through miR-17-92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell. 2014;26(2):262–72.
O’Donnell KA, Wentzel EA, Zeller KI, et al. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435(7043):839–43.
Krysan K, Kusko R, Grogan T, et al. PGE2-driven expression of c-Myc and oncomiR-17-92 contributes to apoptosis resistance in NSCLC. Mol Cancer Res. 2014;12(5):765–74.
Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22–35.
Kim HH, Kuwano Y, Srikantan S, et al. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009;23(15):1743–8.
Johnson SM, Grosshans H, Shingara J, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120(5):635–47.
Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21(9):1025–30.
Takamizawa J, Konishi H, Yanagisawa K, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64(11):3753–6.
Cottonham CL, Kaneko S, Xu L. miR-21 and miR-31 converge on TIAM1 to regulate migration and invasion of colon carcinoma cells. J Biol Chem. 2010;285(46):35293–302.
Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601.
Xiong M, Jiang L, Zhou Y, et al. The miR-200 family regulates TGF-β1-induced renal tubular epithelial to mesenchymal transition through Smad pathway by targeting ZEB1 and ZEB2 expression. AM J Physiol-Ren Physiol. 2012;302(3):F369–79.
Bracken CP, Gregory PA, Kolesnikoff N, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68(19):7846–54.
Brabletz S, Bajdak K, Meidhof S, et al. The ZEB1/miR‐200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011;30(4):770–82.
Yang Y, Ahn Y-H, Gibbons DL, et al. The Notch ligand Jagged2 promotes lung adenocarcinoma metastasis through a miR-200-dependent pathway in mice. J Clin Invest. 2011;121(4):1373.
Bryant J, Britson J, Balko J, et al. A microRNA gene expression signature predicts response to erlotinib in epithelial cancer cell lines and targets EMT. Br J Cancer. 2012;106(1):148–56.
Liu C, Kelnar K, Liu B, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17(2):211–5.
Shimono Y, Zabala M, Cho RW, et al. Down-regulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138(3):592–603.
Yu S, Liu Y, Wang J, Guo Z, Zhang Q, Yu F, et al. Circulating microRNA profiles as potential biomarkers for diagnosis of papillary thyroid carcinoma. J Clin Endocrinol Metab. 2012;97(6):2084–92.
Liu S, Clouthier SG, Wicha MS. Role of microRNAs in the regulation of breast cancer stem cells. J Mammary Gland Biol Neoplasia. 2012;17(1):15–21.
Gwak JM, Kim HJ, Kim EJ, et al. MicroRNA-9 is associated with epithelial-mesenchymal transition, breast cancer stem cell phenotype, and tumor progression in breast cancer. Breast Cancer Res Treat. 2014;147(1):39–49.
Deng L, Shang L, Bai S, et al. MicroRNA100 inhibits self-renewal of breast cancer stem-like cells and breast tumor development. Cancer Res. 2014;74(22):6648–60.
Liu S, Patel SH, Ginestier C, et al. MicroRNA93 regulates proliferation and differentiation of normal and malignant breast stem cells. PLoS Genet. 2012;8(6):e1002751.
Yu F, Yao H, Zhu P, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131(6):1109–23.
Cai W-Y, Wei T-Z, Luo Q-C, et al. The Wnt-β-catenin pathway represses let-7 microRNA expression through transactivation of Lin28 to augment breast cancer stem cell expansion. J Cell Sci. 2013;126(13):2877–89.
Yu F, Deng H, Yao H, et al. Mir-30 reduction maintains self-renewal and inhibits apoptosis in breast tumor-initiating cells. Oncogene. 2010;29(29):4194–204.
Lerner RG, Petritsch C. A microRNA-operated switch of asymmetric-to-symmetric cancer stem cell divisions. Nat Cell Biol. 2014;16(3):212–4.
Lim Y-Y, Wright JA, Attema JL, et al. Epigenetic modulation of the miR-200 family is associated with transition to a breast cancer stem-cell-like state. J Cell Sci. 2013;126(10):2256–66.
Wellner U, Schubert J, Burk UC, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11(12):1487–95.
Liu S, Tetzlaff MT, Cui R, Xu X. miR-200c inhibits melanoma progression and drug resistance through down-regulation of BMI-1. Am J Pathol. 2012;181(5):1823–35.
Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–8.
Guttman M, Amit I, Garber M, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458(7235):223–7.
Cheetham S, Gruhl F, Mattick J, Dinger M. Long non-coding RNAs and the genetics of cancer. Br J Cancer. 2013;108(12):2419–25.
Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011;1(5):391–407.
Calin GA, C-g L, Ferracin M, et al. Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell. 2007;12(3):215–29.
Nie Y, Liu X, Qu S, et al. Long non‐coding RNA HOTAIR is an independent prognostic marker for nasopharyngeal carcinoma progression and survival. Cancer Sci. 2013;104(4):458–64.
Lv X-B, Lian G-Y, Wang H-R, et al. Long non-coding RNA HOTAIR is a prognostic marker for esophageal squamous cell carcinoma progression and survival. PLoS One. 2013;8(5):e63516. doi:10.1371/journal.pone.0063516.
Li D, Feng J, Wu T, et al. Long intergenic non-coding RNA HOTAIR is overexpressed and regulates PTEN methylation in laryngeal squamous cell carcinoma. Am J Pathol. 2013;182(1):64–70.
Wu Y, Liu J, Zheng Y, et al. Suppressed expression of long non-coding RNA HOTAIR inhibits proliferation and tumourigenicity of renal carcinoma cells. Tumor Biol. 2014;35(12):11887–94.
Gupta RA, Shah N, Wang KC, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464(7291):1071–6.
Wu Z-H, Wang X-L, Tang H-M, et al. Long non-coding RNA HOTAIR is a powerful predictor of metastasis and poor prognosis and is associated with epithelial-mesenchymal transition in colon cancer. Oncol Rep. 2014;32(1):395–402.
Xu Z-Y, Yu Q-M, Du Y-A, et al. Knockdown of long non-coding RNA HOTAIR suppresses tumor invasion and reverses epithelial-mesenchymal transition in gastric cancer. Int J Biol Sci. 2013;9(6):587.
Liu Z, Sun M, Lu K, et al. The long non-coding RNA HOTAIR contributes to cisplatin resistance of human lung adenocarcinoma cells via downregulation of p21 (WAF1/CIP1) expression. PLoS One. 2013;8(10):e77293.
Alves CP, Fonseca AS, Muys BR, et al. The lincRNA Hotair is required for epithelial-to-mesenchymal transition and stemness maintenance of cancer cells lines. Stem Cells. 2013;31(12):2827–32.
Kotake Y, Nakagawa T, Kitagawa K, et al. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15INK4B tumor suppressor gene. Oncogene. 2011;30(16):1956–62.
F-q N, Sun M, Yang J-s, et al. Long non-coding RNA ANRIL promotes non-small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Mol Cancer Ther. 2015;14(1):268–77.
Schmidt LH, Spieker T, Koschmieder S, et al. The long non-coding MALAT-1 RNA indicates a poor prognosis in non-small cell lung cancer and induces migration and tumor growth. J Thorac Oncol. 2011;6(12):1984–92.
Jiao F, Hu H, Yuan C, et al. Elevated expression level of long non-coding RNA MALAT-1 facilitates cell growth, migration and invasion in pancreatic cancer. Oncol Rep. 2014;32(6):2485–92.
Gutschner T, Hämmerle M, Eißmann M, et al. The non-coding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013;73(3):1180–9.
Ji P, Diederichs S, Wang W, et al. MALAT-1, a novel non-coding RNA, and thymosin β4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22(39):8031–41.
Ying L, Chen Q, Wang Y, et al. Up-regulated MALAT-1 contributes to bladder cancer cell migration by inducing epithelial-to-mesenchymal transition. Mol Biosyst. 2012;8(9):2289–94.
Fan Y, Shen B, Tan M, et al. TGF-β-induced up-regulation of malat1 promotes bladder cancer metastasis by associating with suz12. Clin Cancer Res. 2014;20(6):1531–41.
Dong Y, Liang G, Yuan B, et al. MALAT1 promotes the proliferation and metastasis of osteosarcoma cells by activating the PI3K/Akt pathway. Tumor Biol. 2014;36(3):1477–86.
Huarte M, Guttman M, Feldser D, et al. A large intergenic non-coding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142(3):409–19.
Kino T, Hurt DE, Ichijo T, et al. Non-coding RNA Gas5 is a growth arrest and starvation-associated repressor of the glucocorticoid receptor. Sci Signal. 2010;3(107):ra8.
Mourtada-Maarabouni M, Pickard M, Hedge V, et al. GAS5, a non-protein-coding RNA, controls apoptosis and is down-regulated in breast cancer. Oncogene. 2009;28(2):195–208.
Zhang H, Cai K, Wang J, et al. MiR‐7, inhibited indirectly by LincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of breast cancer stem cells by down-regulating the STAT3 pathway. Stem Cells. 2014;32(11):2858–68.
Chiyomaru T, Yamamura S, Fukuhara S, et al. Genistein inhibits prostate cancer cell growth by targeting miR-34a and oncogenic HOTAIR. PLoS One. 2013;8(8):e70372.
Liu Q, Huang J, Zhou N, et al. LncRNA loc285194 is a p53-regulated tumor suppressor. Nucleic Acids Res. 2013;41(9):4976–87.
Keniry A, Oxley D, Monnier P, et al. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat Cell Biol. 2012;14(7):659–65.
Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 2010;467(7311):86–90.
Klein U, Lia M, Crespo M, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17(1):28–40.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media Singapore
About this chapter
Cite this chapter
Xu, J., Liu, S. (2016). Noncoding RNAs in Cancer Cell Plasticity. In: Song, E. (eds) The Long and Short Non-coding RNAs in Cancer Biology. Advances in Experimental Medicine and Biology, vol 927. Springer, Singapore. https://doi.org/10.1007/978-981-10-1498-7_6
Download citation
DOI: https://doi.org/10.1007/978-981-10-1498-7_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-1496-3
Online ISBN: 978-981-10-1498-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)