
Abstract
Despite the encouraging advances made to date in cancer therapy, the benefits to patients are frequently offset by the development of resistance to therapeutics. Given their involvement in regulating multiple aspects of gene expression and cell signaling that dictates the behaviors of malignant cells, it is not surprising that noncoding RNAs (ncRNAs) play pivotal roles in the resistance of cancers to clinically available therapeutics. Aberrant expression of these ncRNAs, attributed to inherent defects or stress-responsive variations, mediates cellular signaling that compensates for unfavorable molecular events elicited by the therapeutics, thereby preventing the pharmaceuticals from exerting their desired effects on their cellular targets; alternatively, ncRNAs may regulate cancer therapeutic sensitivity by affecting drug accessibility to neoplastic cells and in vivo drug metabolism. In addition, dysregulation of ncRNA expression in cancer stromal cells can impair the responsiveness of neoplastic cells to appropriate therapies. In this chapter, we will describe ncRNA-related mechanisms underlying cancer resistance to routine therapeutics, hopefully providing rationales for the development of drug-sensitizing strategies targeted against or based on these ncRNAs.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Drug resistance
- Noncoding RNA
- Chemotherapy
- Radiotherapy
- Targeted therapy
- Tumor microenvironment
- Drug accessibility
- ABC transporter
10.1 Introduction
In the history of modern medicine, the wish to cure human malignancies has provided primitive and persistent impetus for the mechanistic studies on cancers. Thanks to the novel insights achieved in these studies, the clinical practice of cancer therapy has gained substantial improvement in the past decades, which is evidenced by the sustained decrease in cancer-related mortality worldwide albeit the ascending incidence of many types of malignancies in recent years [1]. However, the frequently occurring cancer resistance to the regular and innovative treatment leads to compromised efficacy of these therapeutics. As a large category of gene transcripts which fulfill a role without being translated into proteins, noncoding RNAs (ncRNAs) coordinate with proteins to regulate almost every detail of the intracellular signaling machinery. Consistently, accumulating studies have disclosed the indispensable roles of ncRNAs, particularly microRNAs (miRNAs) and long noncoding RNAs (lncRNAs), in regulation of cancer therapeutic resistance [2].
10.2 Conventional and Targeted Therapy of Cancer
Because of the lack of effective therapies, cancers occurring in diverse tissue types remain among the most life-threatening human disorders. However, chemotherapy and radiotherapy, in addition to surgery, have in many cases significantly improved the progression-free survival (PFS) of cancer patients [3, 4]. Moreover, a cohort of molecular targeted pharmaceuticals, e.g., monoclonal antibodies and small-molecule inhibitors, has been approved as first-line treatments for malignancies and has improved the outcomes of various cancers when applied alone or in combination with conventional therapies [5]. Cancer biotherapy, which is characterized by the delivery of a therapeutic gene or immunoregulatory protein or by the transfer of modified cells, is moving from a vision to clinical reality, thus providing additional options for personalized therapy of cancers [3].
10.2.1 Chemo- and Radiotherapy
Chemotherapy is a drug therapy that kills cancer cells or stops them from multiplying [6]. Most chemotherapeutic drugs are cytotoxic, in that they restrain cell division (mitosis), and are therefore more effective against fast-dividing cells such as cancer cells. These drugs block cell proliferation via various mechanisms involving DNA damage or inhibition of cellular machinery (exemplified by rearrangement of the cytoskeleton), usually culminating in a form of programmed cell death known as apoptosis [7].
Chemotherapeutic reagents currently used in clinical practice can be classified as follows:
-
1.
Alkylating agents, which bind covalently to DNA and cross-link DNA strands via their alkyl groups, causing DNA strand breaks and apoptosis. These agents work in a cell cycle-independent manner. This category of agents includes nitrogen mustards, nitrosoureas, tetrazines, aziridines, cisplatins, and their derivatives.
-
2.
Antimetabolites, further subcategorized into antifolates, fluoropyrimidines, deoxynucleoside analogs, or thiopurines, include nucleobase or nucleoside analogs that impair DNA synthesis by competitively binding to polymerases and/or cause DNA damage upon incorporation into growing DNA strands. Antimetabolites selectively inhibit carcinoma cells in S phase of the cell cycle.
-
3.
Anti-microtubule agents, e.g., vinca alkaloids, taxanes, and podophyllotoxin, are plant-derived or semisynthetic chemicals that block cell division by interfering with the function of cytoskeletal proteins, in particular, the assembly and disassembly of microtubules. They also inhibit angiogenesis in solid tumors.
-
4.
Topoisomerase inhibitors, such as irinotecan, topotecan, etoposide, doxorubicin, mitoxantrone, teniposide, novobiocin, merbarone, and aclarubicin, can affect the DNA binding or catalytic activity of two enzymes, topoisomerases I and II, which are critical for DNA unwinding required for replication.
-
5.
Cytotoxic antibiotics, such as anthracyclines (doxorubicin, daunorubicin, etc.), actinomycin, bleomycin, plicamycin, and mitomycin, are a large group of drugs with various mechanisms of action. The combination of these cytotoxic agents leads to numerous chemotherapeutic regimens that can be used against cancers of different types or different clinical stages [7].
Radiation therapy (radiotherapy) is commonly applied to cancerous tissue either alone or as part of adjuvant therapy [4]. To avoid injury to normal tissue, shaped radiation beams from several angles are aimed such that they intersect at the tumor (or the draining lymph nodes, if the tumor cells may have spread). Ionizing radiation triggers cell death by damaging DNA via release of two types of energy, photons and charged particles. Photon therapy kills cells mainly through production of free radicals, which cause severe and irreparable DNA damage, including double-stranded DNA breaks. Failure to repair damaged DNA leads to apoptotic cell death. Cancer cells reproduce more rapidly at the expense of a diminished ability to repair chromosomal abnormalities, allowing sublethal damage to accumulate. Consequently, the cells die or divide more slowly. By contrast, charged particles, e.g., protons and ions of boron, carbon, and neon, cause DNA damage in malignant cells through direct energy transfer independent of tumor oxygen supply and can be more tightly focused on the tumor due to their relatively large mass. Thanks to improved tumor targeting and attenuated cytotoxicity against healthy tissues, radiotherapy is used in clinical treatment of a growing list of malignancies [8].
10.2.2 Molecular Targeted Therapy
The past decade has witnessed the emerging and rapid expansion of molecular targeted therapy of various malignancies. In these approaches, therapeutic agents selectively bind and functionally inhibit dominant oncogenic proteins [5]. To date, two classes of oncoproteins have been targeted for therapeutic purposes:
-
1.
Cell surface or matrix proteins such as growth factors, receptors, or leukocyte differentiation antigens, which are accessible to antibodies. Examples of monoclonal antibodies approved for cancer therapy (and their respective targets) include trastuzumab/Herceptin (erbB2/HER2), cetuximab (EGFR), rituximab (CD20), and bevacizumab/Avastin (VEGF) [9].
-
2.
Cancer-promoting enzymes such as the protein tyrosine kinases (PTKs), which are key components of cellular signaling pathways that drive uncontrolled proliferation and apoptosis resistance. The activity of these PTKs can be selectively inhibited by a cohort of small-molecule compounds. Tyrosine kinase inhibitors (TKIs) already in the oncologist’s armamentarium include gefitinib, erlotinib, and Gleevec, which target EGFR; lapatinib, which targets both EGFR and HER2; and sorafenib, which inhibits the Raf/MEK/ERK pathway; and several multi-target inhibitors [5]. Whereas TKIs exert their anticancer effects solely by inhibiting kinase activity (and consequently attenuating downstream signaling), therapeutic antibodies may play inhibitory roles on tumors both by ablating the function of the target protein and by eliciting tumoricidal immune responses such as antibody-dependent cell-mediated cytotoxicity (ADCC). The latter depends strongly on the cancer microenvironment [5, 9].
The concept of molecular targeting in cancer therapy is also reminiscent of the long-standing clinical use of antihormone therapy against specific types of cancers. Because estrogens and androgens play pivotal roles in the occurrence of some categories of breast, ovarian, and prostate cancers, compounds that competitively bind steroid hormone receptors and block hormone/receptor interactions have been used for prevention and treatment of these malignancies [10, 11]. Of note are the selective estrogen receptor modulators (SERMs), e.g., tamoxifen, which are prescribed for and are effective against estrogen receptor (ER)-positive invasive breast cancers. Similarly, steroidal antiandrogens can counteract the carcinogenic effect of androgens by targeting the androgen receptor (AR) and have consequently been used for clinical treatment of androgen-dependent prostate cancers [10, 11].
10.2.3 Gene, Cell, and Immune Therapy
Cancers are characterized by genetic alterations leading to aberrant expression of oncogene or tumor suppressors, as well as by failures of immunosurveillance and eradication of transformed cells [12]. Suppression of cancers can be achieved via delivery of tumor-suppressive or cytotoxic protein-coding genes. Alternatively, autologous or allogenic cells can be propagated or modified in vitro, endowed with tumor-inhibitory capacities, and transferred to individual patients for adoptive therapy. The anticancer proteins, genes, and cells used for these purposes include those that can elicit immunological responses or attenuate immune tolerance to cancers. Although very few technical breakthroughs have been obtained to date in regard to clinical treatment, these novel therapeutics hold great promise for improving patient outcomes when applied as adjuvants or in personalized therapy [13].
10.3 General Mechanisms of Resistance to Cancer Therapeutics
Cancer resistance to clinical treatment arises from the failure of therapeutics to inhibit the malignant phenotypes of neoplastic cells. In principle, cancer cells become unresponsive to a drug for one or more of the following reasons:
-
1.
Inaccessibility of cells or molecular targets to the drug. Although most small-molecule anticancer pharmaceuticals can easily diffuse into dividing neoplastic cells where they impair the structure or biosynthesis of macromolecules and thereby trigger programmed cell death, cells also express machinery for outward transport of drug molecules. Of note in this regard are the members of ATP-binding cassette transporter (ABC transporter) superfamily of transmembrane proteins, which utilize the energy of ATP to transport a variety of substrates, including exogenous chemicals, across the membrane and out of the cell [14].
-
2.
Inability of drugs to cause lethal or suppressive molecular events. This phenomenon can be attributed to detoxification of the drug by the cell, reduced production of cytotoxic mediators such as reactive oxygen species (ROS), and dysfunction in the machinery involved in DNA repair or cell death. In such cases, a specific population of cells within a heterogeneous population can continue to survive and proliferate despite the presence of an anticancer drug [14].
-
3.
Alternative signaling that compensates for the impairment caused by the drug. Although carcinogenesis is driven mainly by key genetic variation(s), cancer cells may harbor multiple abnormalities in gene expression and intracellular signaling. More importantly, their unstable genomes may give rise to new variations that facilitate the maintenance of malignant phenotypes. Therefore, although a tumoricidal drug can successfully target a single signal pathway that drives cell survival or proliferation, activation of alternative or branched pathways may suffice to activate common downstream signaling events that deregulate oncogene and tumor-suppressor effectors [14].
The critical roles of ncRNAs in mediating these signaling events and conferring resistance to routine and molecular targeted therapies are now being characterized. In cancer cells, these ncRNAs directly regulate the intracellular signaling that determines cell responsiveness to various therapeutics. Alternatively, ncRNAs may regulate the behaviors of stromal or immune cells in the tumor microenvironment, thereby affecting drug sensitivity in a non-cell-autonomous manner [14–16].
10.4 Counteracting Roles of Therapy-Evoked and ncRNA-Related Signaling Events in Cancer Cells
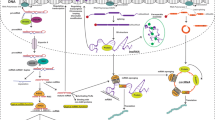
Although therapeutics trigger macromolecule damage and stresses that are detrimental to the survival of cancer cells, drug-refractory cell populations develop mechanisms to circumvent growth inhibition or death induced by cytotoxic drugs. Frequently, these signaling processes are regulated by ncRNAs [14] (see Fig. 10.1).

ncRNAs regulate intracellular signal pathways that counteract the cytotoxicity of anticancer therapeutics. Therapeutics impede cancer cell survival, proliferation, and other malignant phenotypes by suppressing intracellular signaling that leads to the expression of pro-survival and pro-proliferative genes, as well as by damaging DNA or the cytoskeleton, producing ROS, or impairing metabolism. Ultimately, these events trigger apoptotic cell death. Therapeutic-resistant cancer cells circumvent these detrimental events via constitutive activation of downstream or alternative receptor-mediated pro-survival and pro-proliferative signaling or through blockade of apoptotic signaling. All of these processes are potentially regulated by ncRNAs, including miRNAs and lncRNAs
10.4.1 Canonical Intracellular Pathways for Cell Survival and Division
Cancer arises from the aberrant activation of cellular signaling pathways that promote survival and proliferation. Upon stimulation by environmental factors and coupled in many cases to intracellular messengers, these pathways initiate a cascade of kinase activation, thereby inducing activation and nuclear translocation of transcription factors or the assembly of complexes of transacting factors. Ultimately, these events culminate in the expression of genes responsible for cell survival, cell cycle entry, migration, and other behaviors [17]. Although routine therapeutic approaches such as chemotherapy, radiation, and molecular targeted pharmaceuticals elicit different upstream events, they may converge on blockade of the same pathways to inhibit cancer progression. Accordingly, sustained activation of these pathways may underlie cancer resistance to clinical therapeutics [14].
The phosphatidylinositol-3 kinase (PI3K)/Akt pathway, in which PI3K phosphorylates inositol ring 3ʹ-OH groups in inositol phospholipids to generate the second messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3) and activate Akt, is among the most frequently activated pro-survival and pro-mitotic pathways [18]. Arcaroli et al. have found that a mutation in the PI3K catalytic subunit PIK3CA reduces its affinity to miR-520a and increases the sensitivity of colorectal cancers to Src inhibitors, suggesting that crosstalk between the Src and PI3K pathways contributes to regulation of malignant behaviors of such tumors [19]. The tumor-suppressor PTEN negatively regulates the PI3K/Akt pathway by dephosphorylating PIP3. Numerous studies have demonstrated the involvement of PTEN deregulation in therapeutic resistance of cancers. In particular, Meng et al. have screened for miRNAs that regulate the chemosensitivity of cholangiocarcinomas. They found that miR-21 and miR-200b increased sensitivity to gemcitabine and that PTEN was a direct target of miR-21 [20]. In non-small cell lung cancers (NSCLCs) and hepatocellular carcinomas (HCCs), miR-221 and miR-222, both of which are induced by Met activation of c-Jun, can target PTEN, thereby activating Akt signaling and imparting resistance to apoptosis triggered by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) or Apo 2 ligand (Apo2L) [21]. In addition, miR-214 promotes cell survival and cisplatin resistance by targeting PTEN in ovarian cancer [22]. In hepatocellular carcinomas (HCCs), miR-216a/217 targets PTEN and Smad7 to reinforce the PI3K/Akt and TGF-β signaling, respectively, thus maintaining the malignant and stem-like phenotypes of HCC cells even under TKI treatment [23]. Although widely recognized as a tumor suppressor, miR-200c activates Akt and induces chemoresistance by targeting PPP2R1B, a subunit of protein phosphatase 2A, in esophageal cancers [23]. In prostate and breast cancers, miR-95 confers resistance to radiotherapy by targeting the sphingolipid phosphatase SGPP1, an antagonist of sphingosine-1-phosphate signaling downstream of the canonical PI3K-Akt pathway [24]. The mammalian target of rapamycin (mTOR) acts downstream of PI3K/Akt to maintain the key malignant behaviors of cancer cells. MiR-199a-39, which targets mTOR and c-Met, is downregulated in various malignancies including HCC, contributing to resistance of malignant cells to chemotherapeutics such as doxorubicin [25].
The Ras/mitogen-activated protein kinase (MAPK) pathway is another fundamental pathway required for cell growth and transformation. Ras is a small GTPase that responds to upstream signaling and elicits the cascade of Raf/MEK/MAPK kinase activation, and mutation or constitutive activation of Ras has been implicated in the development of various malignancies and the acquisition of cancer resistance to different therapeutics. Weidhaas et al. have highlighted the role of the let-7 family in improving the radiosensitivity of cancer cells by targeting Ras and other oncogenes [26]. In lung carcinomas, miR-27a modulates chemosensitivity by targeting the Raf kinase inhibitory protein (RKIP) [27, 28].
Other documented intracellular signal pathways also play important roles in potentiating cell growth and conferring therapeutic resistance to cancers. In this regard, let-7 can directly repress the interleukin-6 (IL-6)-activated JAK/STAT pro-survival pathway, and its expression correlates with a relatively optimistic prognosis for esophageal squamous cell carcinoma patients receiving cisplatin treatment [29]. Phosphodiesterase 8A (PDE8A) and UV radiation resistance-associated gene (UVRAG), which negatively regulate cAMP/PKA and Notch signaling, respectively, are targets of miR-33a in glioblastoma [30]. In addition, miR-155-3p is involved in Toll-like receptor (TLR)-mediated resistance to the anti-chronic lymphocytic leukemia (CLL) drug, fludarabine [31]. Thus, ncRNAs play critical roles in regulating therapeutic susceptibility of carcinomas by fine-tuning the potency and cross talk of canonical intracellular pathways.
10.4.2 Oncogenic Ligands and Receptors
The signals that drive survival and proliferation of cells originate from the extracellular matrix. The engagement of ligands with their receptors transfers environmental signals into the cell, where intracellular pathways are activated to support cell growth. When these signals are exaggerated or uncontrollable, they lead to malignant transformation. Growth factors, ontogenesis-related ligands, steroid hormones, and their specific receptors are representative initiators of oncogenic signaling [17].
10.4.2.1 Growth Factors/Receptors
Numerous growth factors and receptors drive oncogenic signaling and malignant transformation of cells, making them potential targets for cancer therapeutics. Human epidermal growth factor receptors (HER) are well-characterized biomarkers of various cancers. These proteins form heterologous dimers in response to binding of growth factors and subsequently phosphorylate downstream substrate proteins to activate classical signal pathways [32]. HER1/EGFR-targeted TKIs are most commonly used for treatment of lung cancers, whereas the monoclonal antibody cetuximab is approved for clinical treatment of colorectal cancers and squamous cell carcinoma of the head and neck (SCCHN) [32]. Garofalo et al. have determined the mechanisms underlying TKI resistance of EGFR-positive lung cancers and identified a cohort of downstream miRNAs that repress the master regulators of cell survival and division [33]. Rai et al. have observed that overexpression of miR-7 in TKI-resistant lung cancers increases drug sensitivity in cancers harboring an EGFR mutation (T790M) by targeting EGFR, insulin receptor substrate-1 (IRS-1), and Raf-1 [34]. An analysis of the miRNA transcriptome and global network structure in colorectal carcinoma suggests that downregulation of the K-Ras-targeting miRNAs let-7b and let-7e and upregulation of miR-17* are candidate molecular markers for cetuximab resistance [35].
The humanized HER2/erbB2 antibody, trastuzumab (Herceptin), is a pioneer antitumor antibody that expedites revolutionary progress in treatment of breast cancers and, more recently, advanced gastric cancers. Nevertheless, the majority of patients with HER2-positive cancers exhibit resistance to primary trastuzumab treatment or develop acquired resistance upon repeated administration. Recent studies have revealed that both cancer cell-autonomous mechanisms, e.g., inaccessibility or decreased affinity of HER2 for the antibody or activation of alternative growth factor pathways, and modifications of the tumor microenvironment that suppress antibody-elicited immunological responses may underlie resistance to trastuzumab [36]. To decipher the role of miRNAs in mediating trastuzumab resistance of breast cancers, our laboratory screened for miRNAs differentially expressed in trastuzumab-refractory and trastuzumab-sensitive neoplastic cells. We found that miR-200c downregulation decreased trastuzumab responsiveness by alleviating suppression of transforming growth factor-β (TGF-β) signaling, whereas downregulation of miR-375 and consequently depression of its target gene, insulin-like growth factor 1 receptor (IGF1R), maintained cell growth in the context of blocked HER2 signaling [37, 38]. These studies demonstrate that miRNAs play a regulatory role in cancer resistance to molecular targeted drugs by modulating drug-targeted or alternative growth factor pathways.
Other growth factors involved in carcinogenesis include platelet-derived growth factors (PDGFs), hepatic growth factor (HGF), IGF1R, and (very rarely) bone morphogenetic proteins (BMPs). For instance, the active A receptor type 1 (ACVR1), a key receptor in BMP signaling, is targeted by miR-148 in hepatocytes. Meanwhile, downregulation of miR-148 defines a cancer stem cell-like, aggressive, and therapy-resistant subtype of hepatocellular carcinoma via the miR-148a–ACVR1–BMP–Wnt regulatory circuit [39]. Thus, failure to abolish driving or alternative growth factor signaling is a common mechanism of drug resistance regulated by ncRNAs.
10.4.2.2 Ontogenesis-Related Ligands/Receptors
Aberrant signaling through canonical pathways involved in embryonic development, e.g., the Wnt, Notch, and Hedgehog pathways, can drive the transformation of various types of cells. Meanwhile, reactivation of these pathways may underlie resistance to clinical cancer treatments [40]. Wnt signaling is activated by the binding of a Wnt-protein ligand to a Frizzled family receptor, which transfers the biological signal to the Dishevelled protein inside the cell. The canonical Wnt pathway triggers accumulation and nuclear translocation of β-catenin, coactivating TCF/LEF family of transcription factors to switch on gene expression [41]. The miRNA-mediated regulation of the Wnt pathway is involved in therapeutic resistance in a wide range of malignancies. In colorectal cancers, asymmetric cell division (ACD) and stem cell homeostasis are disrupted, thereby facilitating carcinogenesis, via a regulatory loop involving miR-146a. The transcriptional factor Snail upregulates miR-146a through the β-catenin-TCF4 complex, whereas miR-146a targets Numb to stabilize β-catenin, maintaining Wnt activity and driving symmetrical cell division. This mechanism is critically involved in the resistance of colorectal cancer to molecular targeted drugs [42]. In pancreatic ductal adenocarcinoma cells, Smad4 deficiency ablates TGF-β-triggered expression of miR-494, which in turn upregulates FoxM1, an miR-494 target, and facilitates nuclear translocation of β-catenin, leading to oncogenesis and resistance to gemcitabine chemotherapy [43].
The Notch signaling pathway is a fundamental signaling system used by neighboring cells to communicate with each other. Notch receptors are single-pass transmembrane proteins whose ligands include members of the Delta-like (DLL1, DLL3, DLL4) and Jagged (JAG1, JAG2) families. Ligand binding causes cleavage of Notch and release of the Notch intracellular domain (NICD), which undergoes nuclear translocation and associates with the CSL (CBF1/Su[H]/Lag-1) transcription factor complex, resulting in activation of the canonical Notch target genes. Notch signaling is involved in carcinogenesis and cancer drug resistance, although it plays disparate roles in various malignancies [41]. Park et al. have found that miR-34a levels are reduced in p53-deficient breast cancers, contributing to resistance to conventional chemotherapy by upregulating the miR-34a target Notch1 [44].
The Hedgehog (Hh) signaling pathway is one of the key regulators of animal development and cell lineage commitment. In the absence of Hh ligands, the cell surface transmembrane protein Patched (PTCH) suppresses the activity and expression of the receptor Smoothened (SMO). PTCH engagement by Hh (e.g., Sonic Hedgehog [SHH], the best-studied ligand) leads to the dissociation and activation of SMO, which in turn activates the GLI transcription factors to initiate downstream gene expression. The Hh pathway has been implicated in the development of various cancers, including basal cell carcinoma and medulloblastoma [41]. Recent studies have revealed that Hh signaling, which is regulated by miRNAs, is also involved in resistance to routine cancer treatment. For example, miR-9 contributes to temozolomide resistance by targeting PTCH in glioblastoma [45]. Drugs that specifically target Hedgehog signaling are being developed for treatment of these malignancies. Thus, the classical ontogenesis-related pathways, which are fine-tuned by miRNAs, also contribute to carcinogenesis and the occurrence of drug resistance.
10.4.2.3 Steroid Hormone and Receptors
Depending on the homology relationships of their specific receptors, steroid hormones are classified as glucocorticoids, mineralocorticoids, androgens, estrogens, or progestogens [46]. By binding to and prompting the nuclear translocation of a class of intracellular receptors, they transcriptionally activate a cohort of genes that participate in cell metabolism, inflammation, immunity, and development of sexual characteristics. The exaggerated signaling by overexpression of ERs and ARs plays an important role in the development of mammary and genital carcinomas. Hence, antihormone therapeutics using estrogen antagonists such as SERMs and antiandrogens like flutamide and bicalutamide have emerged as first-line treatments for breast cancer and prostate cancer, respectively [46]. However, neoplastic cells have evolved intricate signaling mechanisms to circumvent the cytotoxic effect of these antagonists, leading to acquisition of resistance to antihormone therapeutics [47].
The involvement of ncRNAs in cancer resistance to tamoxifen, the most-prescribed SERM, has been intensively investigated. Consistent with the reported suppression of ER expression by hyperactivation of MAPKs in breast cancer, Miller et al. have identified an MAPK-regulated miRNA signature that associates significantly with reduced ER expression and poor response to tamoxifen, suggesting that miRNAs can be targeted to reverse resistance to hormone therapy [48]. Maillot has determined miRNA profiles that are regulated by estrogen signaling or altered by antiestrogen therapy in breast cancers, highlighting the role of individual miRNAs in conferring antiestrogen resistance on breast cancers [49]. ER-α can be directly targeted and inhibited by miR-221/miR-222 in breast cancers, compromising the therapeutic efficacy of tamoxifen and enabling ER-α-independent growth of tamoxifen-resistant cancer cells [50]. Other miRNAs play regulatory roles in tamoxifen responsiveness of breast cancers by affecting alternate molecular machineries that govern cell cycle entry, cell survival, and metastasis [49]. Aberrant expression of a set of miRNAs and the lncRNA BCAR4 predicts poor response to tamoxifen, whose effectiveness in breast cancer relies on expression of HER2 [49, 51]. As a direct target of ER-mediated transcriptional repression, the lncRNA HOTAIR is upregulated by tamoxifen and compensatorily increases the level of ER protein, ultimately resulting in resistance of breast cancer to chemotherapy [52]. The alternative approach to blocking ER signaling is the use of inhibitors of aromatase, a rate-limiting enzyme in the conversion of androgens such as testosterone and androstenedione into estrogens. However, breast cancer resistance to aromatase inhibitors (e.g., letrozole) arises concurrently with overexpression of miR-128a and miR-181a or downregulation of miR-125b and let-7c. Letrozole treatment also increases expression of let-7f, which downregulates aromatase, thereby desensitizing breast cancer cells to subsequent letrozole treatment [53]. In terms of cancer resistance to antiandrogen therapy, miR-616 induces androgen-independent growth of prostate cancer cells by suppressing expression of tissue factor pathway inhibitor 2 (TFPI-2), thereby contributing to drug resistance of prostate cancers [54]. In addition, two lncRNAs, PRNCR1 (also known as PCAT8) and PCGEM1, can bind and cooperate with ARs to transcriptionally activate target genes independently of ligand engagement, resulting in prostate cancer resistance to castration or antiandrogen therapy [55]. As a miRNA that mediates androgen-dependent growth of prostate cancer cells, miR-21 is also sufficient to induce castration resistance of prostate cancers [56]. Taken together, these observations show that ncRNAs play diverse roles in conferring or counteracting resistance to antihormone therapy of cancers by modulating sex steroid pathways or coordinated signaling involved in cancer progression.
10.4.3 Key Transcriptional Factors
Oncogenic and differentiation-determining transcriptional factors may promote cancer progression and drug resistance following activation by upstream signals or acquisition of constitutive activity upon mutation [57]. The oncoprotein c-Myc is overexpressed in various malignancies and is correlated with poor outcomes of routine clinical therapies. In non-Hodgkin B-cell lymphoma, stromal adhesion promotes cell survival and imparts resistance to cytotoxic drugs like mitoxantrone via an amplification loop in which c-Myc induces epigenetic silencing of miR-548m and subsequently increases the expression of the miR-548m targets c-Myc and HDAC6 [58]. Numerous other transcriptional factors that expedite drug resistance are also regulated by miRNAs. For instance, glioma cells acquire chemoresistance as a result of inhibitor of differentiation 4 (ID4) depression of miR-9-mediated suppression of Sox2 [59]. Downregulation of transcriptional factors that drive differentiation also underlies cancer resistance to clinical therapeutics, as exemplified by forkhead box O3a (FOXO3a), which is targeted and silenced by miR-153, thus attenuating platinum-induced apoptosis of colorectal cancers [60]. Therefore, transcription factors, which can both be regulated by miRNAs and dictate the expression of specific miRNAs, may play distinct roles in therapeutic resistance, depending on the repertoires of their transcriptional targets.
10.4.4 Cell Cycle Progression
Cell proliferation requires continuous entry into and progression of the cell cycle, which is divided into different phases with checkpoints controlled by numerous factors [61]. Although anticancer therapeutics may trigger cell cycle arrest through intracellular signaling, refractory subsets of malignant cells can develop miRNA-mediated regulatory mechanisms that facilitate cell cycle progression. Pouliot et al. have found that miR-155 and miR-15 improve the sensitivity of epidermoid carcinoma cells to cisplatin by targeting and repressing the cell cycle kinases WEE1 and CHK1 [62]. Salerno et al. have found in a mouse model of chronic lymphocytic leukemia (CLL) that exogenous miR-15a and miR-16-1, which target cyclin D1, improve the responses of cells to nutlin, a mouse double minute 2 (MDM2) antagonist, and genistein, a tyrosine kinase inhibitor [63]. MiR-122 sensitizes HCC to doxorubicin by modulating cyclin G1, thereby influencing p53 protein stability and transcriptional activity [64]. Thus, ncRNAs may contribute to the etiology of cancer drug resistance by governing cell cycle progression in the context of various clinical treatments.
10.4.5 Apoptotic Machinery
Both ontogenesis of the organism and maintenance of tissue homeostasis involve the removal of senescent or aberrant cells through programmed cell death [65]. In contrast to necrosis, which occurs under stressful conditions like tissue injury, apoptosis represents the most common pattern of physiological cell death. Inadequate apoptosis underlies carcinogenesis in multiple tissues, and desensitization of cells to chemotherapy- or radiotherapy-triggered apoptosis accounts for therapeutic resistance of a variety of clinical cancers [65]. The apoptotic machinery consists of two major pathways:
-
1.
In the extrinsic pathway, extracellular ligands such as Fas ligand, tumor necrosis factor-α (TNF-α), and TRAIL bind to and trigger the oligomerization and activation of death receptors such as Fas, TNFR, and death receptor 4 (DR4). Signaling from these receptors leads in turn to the sequential processing and activation of initiator and effector caspases and ultimately to widespread degradation proteins and the collapse of the entire cell.
-
2.
The intrinsic pathway, which senses intracellular stress signals like DNA damage, causes permeabilization of the mitochondria and release of cytochrome c into the cytoplasm, thereby initiating activation of the caspase cascade via caspase-9. Bcl-2 family members fine-tune apoptotic signaling via pore formation on the mitochondrial membrane and reciprocal interactions to determine the fate of individual cells. In addition, negative regulators of apoptosis such as the inhibitors of apoptosis (IAP) and FLICE-inhibitory protein (FLIP) impede caspase activation in the context of various apoptotic signals [65]. The aforementioned apoptosis executioners and regulators, which play critical roles in determining responses to cytotoxic therapeutics, can be targeted by ncRNAs in various types of malignancies. In particular, the sensitivity of osteosarcoma cells to FasL is regulated by miR-20a, which targets the death receptor Fas [66]. In cholangiocarcinoma, miR-25 is upregulated by Hedgehog signaling, which desensitizes neoplastic cells to TRAIL-induced apoptosis by targeting DR4 [67]. In addition, miR-21 silencing also exerts synergistic cytotoxicity with TRAIL in gliomas [68]. Conversely, miR-212 increases TRAIL sensitivity in non-small cell lung cancer by targeting the antiapoptotic protein PED/PEA-15 [69].
The cancer response to cytotoxic therapeutics also involves intrinsic apoptotic signaling, which is likewise regulated by ncRNAs. A natural product, oridonin, increases the sensitivity of leukemia to chemotherapy by downregulating miR-17 and miR-20a and thus restoring expression of their common target, the S variant of BIM, resulting in promotion of mitochondrial apoptotic signaling [70]. STAT3 signaling maintains the expression of miR-17 and suppresses its target BIM, thereby conferring MEK inhibitor resistance on lung cancers, suggesting the cooperative antitumor potential of STAT3 and MEK inhibitors [71]. Signaling from chemokine receptor CXCR4 in acute myeloid leukemia (AML) cells downregulates let-7a through the transcription factor Yin Yang 1, resulting in chemoresistance due to increased expression of let-7a targets such as Bcl-xL [72]. Lam et al. have identified miRNA modulators of colorectal cancer responsiveness to the Bcl-2 inhibitor ABT-263 (navitoclax) and found that a majority of these miRNAs sensitize neoplastic cells by downregulating the pro-survival Bcl-2 family member Mcl-1 [73]. Hepatitis C virus increases HCC sensitivity to sorafenib via miR-193b targeting of Mcl-1, thereby promoting apoptosis of HCC cells [74]. These findings suggest that modulation of apoptotic signaling by ncRNAs causes altered responses to clinical therapeutics.
10.4.6 Genotoxic Stress
Although cells have evolved machinery for comprehensive genome surveillance and DNA repair, deficiencies in these machineries (or, alternatively, severe DNA injury) may prevent restoration of genomic homeostasis [75]. From the standpoint of the tumor cell, DNA abnormalities are a double-edged sword. On one hand, genomic DNA instability and mutation are the key drivers of carcinogenesis: activation of oncogenes or dysfunction of tumor suppressors elicits uncontrolled mitosis and apoptosis resistance, explaining the intimate relationship between DNA repair defects and tumorigenesis. On the other hand, irreparable DNA damage triggers cell death to maintain the purity of the genetic material, providing the rationale for radiation therapy and the large proportion of chemotherapeutic drugs that kill cells by extensively damaging the DNA [75]. The types of DNA damage include undesired modification or mismatch of bases, single-strand damage, and double-strand break (DSB). Sensors of DNA damage establish checkpoints prior to the initiation of DNA repair. Once activated by damaged DNA, these checkpoints halt the cell cycle and give the cell time to repair the damage. Checkpoint activation is controlled by two master kinases, ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3 related (ATR), which phosphorylate downstream targets in a signal transduction cascade and eventually induce cell cycle arrest [76]. In addition, checkpoint mediator proteins including BRCA1, MDC1, and 53BP1 are required for transmission of the checkpoint activation signal to downstream proteins. miRNAs are involved in these processes. MiR-205 inhibits DNA damage repair by targeting zinc finger E-box-binding homeobox 1 (ZEB1) and the ubiquitin-conjugating enzyme Ubc13, thus acting as a tumor radiosensitizer by targeting the DNA repair machinery [77]. Notably, however, radiotherapy downregulates miR-205 through ATM and ZEB1 in breast cancer. In NSCLCs, miR-181a and miR-630 promote and reduce cisplatin-triggered cell death, respectively, in the former case via regulation of the intrinsic apoptotic pathway and in the latter case via miR-630-mediated blockade of early manifestations of the DNA damage response such as activation of ATM [78].
As a type of severe DNA damage, DSBs can be repaired via three mechanisms: nonhomologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), and homologous recombination (HR) [76]. Poly ADP ribose polymerase 1 (PARP1) plays crucial roles in DNA repair by preventing the development of DSBs from single-strand breaks and by participating in the MMEJ repair of DSBs. Because MMEJ is an error-prone repair pathway, PARP1 overexpression has been detected in various malignancies and therapy-resistant cancer cells. The lncRNA PCAT1 sensitizes prostate cancers to genotoxic drugs, e.g., inhibitors of PARP1, by posttranscriptionally repressing the DSB repair protein BRCA2 [79]. Although miR-223 supports the aggressive phenotype of esophageal adenocarcinomas, it also improves the response of malignant cells to genotoxic drugs by directly targeting PARP1 [80]. RAD51 is critically involved in HR of DNA during DSB repair. DNA repair in malignant cells is also attenuated by miR-96, which targets RAD51 and the trans-lesion synthesis DNA polymerase REV1, increasing the sensitivity of cancers to the interstrand cross-linking drug cisplatin and PARP1 inhibitors [81].
The tumor-suppressor p53 responds to diverse cellular stresses to regulate expression of target genes, thereby inducing cell cycle arrest, apoptosis, senescence, or metabolic changes. Most importantly, p53 serves as a guardian of the genome by coupling DNA damage to the cellular DNA repair machinery or to apoptotic cell death when repair fails [82]. In p53-deficient cancers, the functional balance and cross talk between p73, which mediates chemosensitivity, and p63, which promotes cell survival, proliferation, and cell survival, are crucial for cancer progression. This phenomenon is at least partially mediated by miRNAs, such as miR-193a-5p, which targets p73, and is itself regulated by both p63 and p73. Chemotherapy causes p63/p73-dependent induction of this miRNA, thereby inducing chemoresistance due to miRNA-mediated feedback inhibition of p73 [83]. In liver tumor-initiating cells, miR-130b maintains cell growth, self-renewal, and chemotherapy resistance by targeting tumor protein 53-induced nuclear protein 1 (TP53INP1) [84]. Cisplatin-induced apoptosis of testicular cancer cells is counteracted by cytoplasmic p21WAF1/CIP1, a p53 target that accumulates due to reduced Oct4 transactivation of miR-106b and miR-17-5p [85]. Collectively, ncRNAs are strongly implicated in the regulation of susceptibility to cancer therapeutics that elicit genotoxic stress.
10.4.7 Oxidative Stress
Cells produce ROS during the course of normal metabolism and eliminate them via various mechanisms [86]. Oxidative stress arises from a dynamic imbalance between the systemic manifestation of ROS and a biological system’s ability to detoxify these reactive intermediates [86]. Although severe oxidative stress is cytotoxic, oxidative stress underlies carcinogenesis, and the insusceptibility of carcinoma cells to oxidative stress leads to drug resistance [87]. The physiological ROS-scavenging systems include intracellular antioxidants such as glutathione and a variety of antioxidant enzymes such as superoxide dismutase (SOD). Meanwhile, the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) is a master regulator of the body’s antioxidant response: Nrf2 is activated by different sensors of redox status and constitutively degraded by the key regulator Kelch-like ECH-associated protein 1 (KEAP1). Activated Nrf2 binds to the antioxidant response element (ARE) and switches on expression of a wide range of detoxification or stress-response genes [87]. These machineries, which are implicated in cell detoxification and therapeutic resistance, are also targeted by ncRNAs. Drayton et al. have found that miR-27a improves the responsiveness of bladder cancers to cisplatin by targeting the cystine/glutamate exchanger SLC7A11, thereby disrupting glutathione biosynthesis [88]. In addition, histone deacetylase inhibition can overcome lung cancer resistance to polyamines by upregulating miR-200c, which in turn upregulates Nrf2-mediated transcription of the polyamine catabolic enzyme spermidine/spermine N(1)-acetyltransferase (SSAT) by directly targeting KEAP1 [89].
10.4.8 Malignant Phenotypes: Cancer Stem Cells (CSCs), Autophagy, and Epithelial–Mesenchymal Transition (EMT)
According to the CSC theory, cancers are initiated and maintained by a small subset of stem-like or cancer-initiating cells that are capable of self-renewal and differentiation into other populations of the tumor mass [41]. CSCs are also the primary cause of distal metastasis and therapeutic resistance. The properties and behaviors of this specific subset of cancer cells are regulated by miRNAs, a topic that is beyond the scope of this chapter [41]. In addition, expression of the lncRNA X-inactive specific transcript (XIST) is a biomarker that predicts the response of breast cancer to HDAC inhibitors, although the underlying mechanisms remain to be elucidated [90].
Cancer cells undergo the EMT to acquire the migratory and invasive properties required for metastasis. In addition, the EMT plays a vital role in acquisition of resistance to anticancer therapeutics [91]. As a master regulator of the EMT, TGF-β signaling plays essential roles in regulating malignant phenotypes, e.g., drug resistance, of various cancers. MiR-200 family members, especially miR-200c, are downregulated in various cancers that are refractory to chemotherapy or treatment with TKIs or monoclonal antibodies [92, 93]. This phenomenon is intimately related to miR-200c regulation of TGF-β signaling and the EMT via targeting of the transcription factors ZEB1 and ZNF217 and mesenchymal genes such as FN1, NTRK2, and QKI [94–97]. MiR-200 also inhibits EGFR-independent cell growth by targeting MIG6, thereby conferring resistance to EGFR-targeted therapeutics [92, 94]. In lung adenocarcinomas, the miR-134/487b/655 cluster regulates TGF-β-induced EMT and drug resistance to gefitinib by targeting MAGI2, a scaffold protein required for PTEN stabilization [98]. MiR-34a sensitizes head and neck cancers to EGFR TKIs by targeting the protein tyrosine kinase receptor Axl and repressing the EMT [99]. MiR-30c antagonizes breast cancer chemoresistance by targeting the EMT-related cytoskeleton proteins actin-binding protein twinfilin 1 (TWF1) and vimentin. In addition to mediating the EMT, TWF1 also desensitizes cancer cells to chemotherapy by promoting IL-11 production [100].
Autophagy is an intracellular process of macromolecule and organelle recycling or turnover. Targeted cytoplasmic constituents are isolated within a double-membraned vesicle known as an autophagosome, which subsequently fuses with a lysosome, where the cargo is degraded [101]. Autophagy enables cells to survive stress from the external environment, such as nutrient deprivation, and also allows them to withstand internal stresses like accumulation of damaged organelles and invasion by pathogens. Moreover, autophagy can cause programmed cell death, depending on the cell type and the context of intracellular signaling [101]. Autophagy maintains cellular homeostasis and prevents early transformation of cells by eliminating superfluous or damaged proteins, enhancing host defense against pathogens and circumventing precancerous chronic tissue damage; by contrast, after the onset of cancer, autophagy facilitates tumor progression, including the development of therapeutic resistance [101]. In this regard, miR-23b sensitizes pancreatic cancers to radiotherapy by targeting ATG12 and blocking radiation-initiated cell-protective autophagy [102]. However, it remains unclear to what extent the various ncRNAs responsible for the regulation of autophagy are involved in therapeutic resistance of cancers.
10.5 Drug Accessibility Regulated by ncRNA in Cancer Treatment
The cytotoxicity of anticancer drugs can be diminished by limiting the access of pharmaceutical molecules to malignant cells. This can occur when cancer cells develop mechanisms to pump out drugs via transporter proteins on the cell membrane or when cells manage to shield target proteins from drug engagement. Both paradigms are regulated by ncRNAs [14] (see Fig. 10.2). The detailed miRNA-mediated regulation of ABC transporters has been reviewed elsewhere [103].

ncRNAs regulate therapeutic accessibility and pharmacokinetics of drugs, as well as the cancer microenvironment. Cancer cells develop autonomous resistance to therapeutics or anticancer drugs by reducing therapeutic/drug accessibility. In addition, the systemic absorbance and biological transformation of anticancer drugs, as well as the microenvironment, which may be educated by cancer cells or modulated by the therapeutics, influence therapeutic outcomes. A wide range of genes involved in these machineries are candidate targets of ncRNAs, which include (I) intracellular mechanisms that downregulate drug-targeted proteins, (II) mutations that lower target protein affinity to the drug, and (III) molecules that shield drug-binding sites on the target protein
10.5.1 Drug Export
Eukaryotes express a class of transporter proteins on the cell membrane that pump out xenobiotics, toxins, and drugs from inside the cell. The efflux of cytotoxic drugs decreases intracellular drug concentrations and represents a common mechanism by which neoplastic cells acquire resistance to anticancer drugs [104].
10.5.1.1 ATP-Binding Cassette (ABC) Transporters
ABC transporters, a group of active transporter proteins ubiquitously expressed by mammalian cells, hydrolyze ATP to ADP and use the energy to drive the efflux of intracellular substrates against a concentration gradient. The 48 members of the ABC transporter family identified to date have been divided into seven subfamilies: ABCA through ABCG. ABC transporter proteins are composed of two nucleotide-binding domains (NBDs) and two transmembrane domains (TMDs) [105]. The classification is based on the sequence of the NBDs, also known as ABC domains, which are mainly involved in hydrolyzing ATP, binding physiological and xenobiotic substrates, and extruding them out of the cell. The majority of ABC transporters are full or complete transporters, although some (e.g., the ABCG subfamily) are half transporters that contain only one NBD and TMD per protein [105]. Two NBDs are required for normal transporter activity, consistent with the observation that ABCB1 (P-gp or MDR1) hydrolyzes two ATPs in a stepwise process during drug trafficking. The hydrolysis of the first ATP structurally modifies the TMDs by flipping the inner leaf to the outer side of the cell membrane, resulting in efflux of the drug from the cell. The hydrolysis of the second ATP restores the structure of the transporter to its original high-affinity state [105].
ABC transporters are responsible for outward transportation of xenobiotics and numerous agents including amino acids, cholesterol and its derivatives, carbohydrates, vitamins, peptides, lipids, certain important proteins, hydrophobic drugs, and antibiotics [105]. Given their capability to potentiate efflux of anticancer agents, ABC transporters play a pivotal role in conferring resistance to chemotherapeutic and molecular targeted drugs on neoplastic cells. However, depending on their individual structures, different members of the ABC transporters are involved in the efflux of different tumoricidal drugs. ABC drug transporters increase the efflux of their substrates (e.g., anticancer agents), thereby reducing the intracellular concentration of drugs and resulting in an MDR phenotype [105]. Meanwhile, the expression of ABC transporters is regulated in neoplastic cells through multiple mechanisms, including posttranscriptional silencing by ncRNAs. Borel et al. have identified 13 miRNAs that regulate the ABC transporter family in HCCs. Deregulation of these miRNAs contributed to significant upregulation of drug efflux pumps and MDR of HCCs [106]. Jaiswal et al. have found that multidrug resistance (MDR) can be transferred intercellularly by delivery of the transcripts and regulatory miRNAs of drug efflux proteins, including ABC transporters, via microparticles derived from membrane budding, thereby “retemplating” the transcriptional landscape of recipient cells from MDR donor cells to drug-sensitive recipient cells [107].
10.5.1.1.1 ABCB1
ABCB1/P-glycoprotein (P-gp/MDR1) is a 160–170 kDa protein encoded by the MDR1 gene. As an apical membrane transporter localized in cells of the kidney, placenta, liver, adrenal glands, intestine, and blood-brain barrier, ABCB1 transports xenobiotics and cellular toxicants not only out of the cell but also into the urine and bile, thereby facilitating their excretion from the body. ABCB1 overexpression confers resistance to a variety of anticancer compounds like vinblastine (VLB), vincristine (VCR), paclitaxel (PTX), and colchicine (COL). ABCB1 also imparts TKI resistance to carcinoma cells [108]. Kovalchuk et al. have found that miR-451 antagonizes chemoresistance of the breast cancer cell line, MCF-7, by directly targeting ABCB1 [109]. The H19 mRNA, which is encoded by the imprinted H19 gene and is thought to function as an RNA component of the ribonucleoprotein, is expressed at significantly higher levels in breast, lung, or hepatocellular cancer cells refractory to chemotherapeutic drugs like doxorubicin. H19 is implicated in ABCB1 expression through the control of promoter methylation [110, 111].
10.5.1.1.2 ABCCs
The ABCC/multidrug resistance protein (MRP) family can be further subdivided into three groups: long ABCCs such as ABCC1 (MRP1), ABCC2 (MRP2), ABCC3 (MRP3), ABCC6 (MRP6), and ABCC10 (MRP7); short ABCCs such as ABCC4 (MRP4), ABCC5 (MRP5), ABCC11 (MRP8), and ABCC12 (MRP9); and ABCC7 to ABCC9, which are components of ion channels rather than transporters. These ABCCs are critical mediators of drug resistance arising in various types of carcinomas [108]. In particular, ABCC1 overexpression correlates with doxorubicin resistance of leukemia and lung cancer, whereas ABCC10 expression confers resistance to various anticancer drugs including docetaxel, PTX, VCR, VLB, cytarabine, gemcitabine, 2ʹ,3ʹ-dideoxycytidine, 9-(2-phosphonyl-methoxyethyl) adenine (PMEA), and epothilone B. Both ABCC1 and ABCC2 increase the efflux of TKIs such as imatinib and sorafenib, whereas imatinib exposure causes further upregulation of ABCC1, thus conferring TKI resistance on various malignancies [108]. All of these ABCCs have been verified as targets of miRNAs [103].
10.5.1.1.3 ABCG2
ABCG2 is also known as breast cancer resistance protein (BCRP), mitoxantrone resistance protein (MXR), or ABC transporter expressed in placenta (ABCP) [108]. As a half transporter with one TMD and one NBD, it must homodimerize or oligomerize with other transporters to exhibit transporter activity and mediate MDR. ABCG2 is expressed in the placenta, small intestines, colon, liver, and blood vessels, where it protects cells or tissues against toxins and xenobiotics. ABCG2 also transports organic anion conjugates, nucleoside analogs, organic dyes, TKIs, anthracyclines, and topoisomerase I inhibitors and is responsible for cancer resistance to mitoxantrone (MX) and doxorubicin (DX). In addition, mutations of ABCG2 may result in significant conformational changes and alter the drug-binding and efflux capacity of the transporter [108]. By demonstrating that two miRNAs, miR-519c and miR-520h, target ABCG2, To et al. have demonstrated that the acquisition of MX resistance in various cancers can be attributed to the shortening of the ABCG2 3ʹ UTR, resulting in loss of miRNA binding sites or sequestering of the miRNA by highly expressed ABCG2 mRNA [112].
10.5.1.2 Nucleoside Transporter (NT) Proteins
NTs are integral membrane proteins involved in the salvage of natural nucleobases and nucleosides involved in nucleic acid synthesis [113]. They belong to solute carrier families 28 and 29 (SLC28 and SLC29), which encode human concentrative NTs (hCNTs) and equilibrative NT proteins (hENTs), respectively. Localized on the apical membrane of polarized epithelia, these NTs are required for uptake of numerous nucleoside and nucleobase analogs currently used for treatment of cancers and viral infections and are therefore determinants of drug action. hCNTs prompt the influx of nucleoside drugs coupled to the influx of sodium ions [113]. Different members of the SLC28 gene family exhibit preferences for pyrimidine or purine nucleosides and their derivatives as substrates, as exemplified by hCNT1, a high-affinity pyrimidine nucleoside transporter involved in intracellular delivery of chemotherapeutics such as gemcitabine. Similarly, hENT family members are responsible for transport of natural nucleosides and nucleoside-derived drugs. SLC22, which encodes human organic cation transporters (hOCTs) and organic anion transporters (hOATs), plays a predominant role in the uptake of nucleoside-derived drugs with specific structural variations, e.g., lack of 3ʹ-OH [113]. Among the growing number of ncRNAs known to modulate the expression of NTs, several miRNAs including miR-122, miR-214, miR-339-3p, and miR-650 target hCNT1/SLC28A1, suggesting that these ncRNAs are involved in acquisition of chemoresistance by pancreatic cancers [114].
10.5.2 Blockade of Drug–Target Interactions
The therapeutic efficacy of anticancer drugs relies on efficient drug–target interactions. Consequently, cancer cells have developed various mechanisms to suppress drug binding to target proteins [115, 116]. For instance, a well-documented mutation (T790M) in the kinase domain of EGFR dramatically decreases the receptor’s affinity for TKIs, thereby imparting resistance to these drugs [115]. Acquisition of resistance to trastuzumab occurs in a subset of HER2-positive breast cancer cells expressing mucin 1 or mucin 4. In addition to promoting cell invasion and enhancing HER2–HER3 signaling, these O-glycosylated transmembrane proteins interfere with trastuzumab targeting by masking the antibody-binding epitope of HER2 [116]. These situations can be ameliorated by miRNA-mediated suppression of the mucin proteins [117, 118]. Meanwhile, although miRNAs targeting HER family oncogenes can impair the onset of malignancies, they can also facilitate growth factor-independent cancer progression and resistance of advanced tumors to therapies targeting these cancer drivers [119]. In addition, Boni et al. have found that miR-192 and miR-215 directly repress thymidylate synthase (TYMS), thereby imparting resistance to TYMS-targeted chemotherapeutic agents such as 5-fluorouracil (5-FU) in gastrointestinal cancers [120].
10.6 Drug Pharmacokinetics Controlled by ncRNAs
The tumor-inhibitory potency of a chemical drug is determined by drug pharmacokinetics and metabolism, which together control the time the drug is retained in tumor tissue [121]. It is worth noting that the aforementioned ABC transporters and nucleoside transporter proteins play vital roles in regulating the pharmacokinetics of tumoricidal drugs. Moreover, these transporters are not expressed exclusively by malignant cells, but are ubiquitously present in the intestine, kidney, liver, and blood-brain barrier, which determine the absorption, in vivo distribution, and renal or hepatic processing of drugs [108, 121] (see Fig. 10.2).
Upon exerting a cytotoxic role in the desired tissue, a drug may undergo biotransformation prior to excretion. Consequently, the key enzymes responsible for inactivation of anticancer compounds dictate the half-life and persistence of drugs [121]. In particular, miR-27a and miR-27b sensitize malignant cells to 5-FU by targeting and repressing dihydropyrimidine dehydrogenase (DPD), a key uracil catabolic enzyme responsible for conversion of 5-FU to the inactive metabolite 5-dihydrofluorouracil [122]. Persson et al. have found that RNAs in the vault particle, a conserved organelle, are implicated in multidrug resistance of malignant cells. One of these so-called small vault RNAs (svRNAs), svRNAb, negatively regulates the expression of CYP3A4, which encodes a cytochrome P450 enzyme crucially involved in the metabolism of many chemotherapeutic compounds and almost 60 % of all marketed drugs [123].
10.7 Drug-Refractory Cancer Microenvironment Modulated by Noncoding RNAs
Numerous cutting-edge studies highlight the role of the microenvironment on the development, progression, and therapeutic responsiveness of cancers [124]. In theory, the tumor-suppressive efficacy of therapeutics represents the combined outcome of direct cytotoxicity to neoplastic cells and the modulation of the tumor microenvironment by the drug [125] (see Fig. 10.2). The microenvironment includes the extracellular matrix (ECM), stromal fibroblasts, immune cells, and blood vessels supplying solid tumors, all of which affect cancer progression via direct cell–cell contact or secretion of diverse factors [124, 125]. Cells in the microenvironment are extremely important for the tumor-inhibitory action of monoclonal antibodies, which in addition to their cancer cell-autonomous mechanisms elicit antitumor immunity [125]. This is exemplified by miR-27a/miR-27b, which efficiently induces the transformation of normal fibroblasts into cancer-associated fibroblasts (CAF), as evidenced by induction of α-smooth muscle actin (α-SMA) expression and TGF-β production, thereby conferring cisplatin resistance of esophageal cancers [126]. In addition, attenuated miR-142-3p suppression of the ecto-nucleoside triphosphate diphosphohydrolase CD39 leads to a reduction of ATP levels in regulatory T (Treg) cells relative to those in conventional T cells, explaining the vulnerability of Tregs to low-dose cyclophosphamide. This observation has implications for overcoming immune tolerance to carcinomas receiving chemotherapy [127].
10.8 Complicated Solo Performance: Combined Versus Unknown Targets
Regulation of gene expression by ncRNAs is characterized by the ability of individual ncRNAs, e.g., miRNAs, to simultaneously target multiple mRNAs. Conversely, a given transcript can be concurrently inhibited by several miRNAs [128]. In this regard, a growing number of miRNAs have been determined to target various genes that synergistically regulate sensitivity to therapeutics. For example, miR-128 downregulation accounts for drug resistance of breast cancer-initiating cells, because it directly targets both the stem cell transcriptional factor Bmi-1 and the ABC transporter ABCC5 [129]. Giovannetti et al. have demonstrated that miR-21-mediated gemcitabine resistance in pancreatic ductal adenocarcinoma (PDAC) can be attributed to the modulation of apoptosis, Akt phosphorylation, and expression of genes involved in invasive behavior [130]. Using a genome-wide screening approach, Ziliak et al. have identified an SNP (rs1649942) that significantly affects platinum sensitivity. They attributed this effect to changes in the miRNA profile and specifically to altered expression of miR-193b, which targeted a set of platinum-associated genes including CRIM1, IFIT2, OAS1, KCNMA1, and GRAMD1B [131]. MiR-301 mediates various malignant phenotypes of breast cancers, including tamoxifen resistance, through multiple targets including FOXF2, BBC3, PTEN, and COL2A1 [132]. Alternatively, a single ncRNA involved in therapeutic resistance may regulate several pathways by targeting a multifunctional gene. In this regard, Eto et al. have found that miR-223 is highly expressed in trastuzumab-resistant gastric cancers. MiR-223 directly targeted F-box and WD repeat domain-containing 7 (FBXW7), the substrate recognition component of an evolutionarily conserved SCF (complex of SKP1, CUL1, and F-box protein)-type ubiquitin ligase complex, thereby attenuating FBXW7-dependent degradation of oncoproteins including cyclin E, c-Myc, Notch, c-Jun, mTOR, and Mcl-1 [133]. These studies suggest that ncRNAs may play a more important role than protein-coding genes in determining the therapeutic responsiveness of cancers due to their ability to target multiple functional genes.
The extensive roles of ncRNAs in the therapeutic resistance of cancers are far from completely elucidated. In addition to the numerous undefined targets of miRNAs that demarcate therapy-refractory cell subsets, many lncRNAs are believed to determine therapeutic responses via mechanisms yet to be characterized [14, 134]. For example, expression of the inactive XIST, a spliced noncoding polyadenylated RNA and the only transcript expressed exclusively from the inactive X chromosome, correlates with high sensitivity to Taxol in ovarian cancers. However, its mode of action remains poorly understood [135]. Future breakthroughs in deciphering the characteristics of ncRNAs characters will provide novel functional annotations for these RNA species in the context of therapeutic resistance of cancers.
10.9 Strategies for Overcoming ncRNA-Mediated Therapeutic Resistance
The critical involvement of ncRNAs in regulating the therapeutic resistance of various cancers warrants the development of strategies based on or targeting ncRNAs in order to reverse refractory phenotypes of carcinomas [15, 16]. First, ncRNAs can be directly manipulated to improve the sensitivity of cancers to specific therapeutics. ncRNAs or their antisense inhibitors (in particular, miRNAs and antagomirs) can be synthesized and introduced into cultured cells for therapeutic purposes or delivered in vivo through nonviral carriers such as liposomes or positively charged agents that encapsulate the RNAs in nanoparticles. Moreover, cancer-targeted delivery of small RNAs can be achieved via generation of an RNA delivery system using antibodies or ligands that recognize tumor-specific antigens or receptors [136]. ncRNAs such as miRNAs and their inhibitors can also be expressed from eukaryotic expression cassettes and then expressed ectopically in malignant cells via viral or nonviral delivery of the cassettes [136]. Second, ncRNA-regulated pathways can be targeted, providing important guidance for selection and optimization of combined medication or therapy [137]. Finally, ncRNAs can be used as biomarkers for drug responsiveness and for the relapse or prognosis of cancers after treatment targeting the drug-resistant cell populations [15, 16]. These strategies will be beneficial to the development of adjuvant therapy and will potentially increase the efficacy of routine cancer treatment.
10.10 Future Perspectives
In light of the immense diversity of anticancer therapeutics per se and the paradigms by which they eliminate malignant cells, cancer cells need to evolve widely varied mechanisms to survive cytotoxic attacks. Recent studies have underscored the critical involvement of ncRNAs in regulating the therapeutic susceptibilities of different malignancies. Nevertheless, the full regulatory network underlying therapeutic resistance of cancers (e.g., the ways in which therapeutics exert selective pressure for or even fuel the development of the molecular machineries of therapeutic resistance, presumably via ncRNAs), the hierarchy of regulators (including multiple ncRNAs) involved in drug resistance, and the roles of ncRNAs in mediating cross talk between various drug resistance pathways remain to be fully understood. Except for the regulation of drug transport or metabolism, the roles most ncRNAs play in therapeutic resistance are shared by those they conduct in regulating other malignant phenotypes of carcinomas. Therefore, future investigations should seek to demarcate these roles of ncRNAs for each type of malignancy. In addition, in the context of personalized medicine, it is desirable to determine the individual variations and underlying genetic discrepancies that govern the importance of particular ncRNAs in determining the therapeutic responses of different patient populations. Finally, in contrast to the substantial participation and definitive role of miRNAs in regulating therapeutic sensitivity by posttranscriptionally silencing target genes, the contribution of most lncRNAs to drug resistance of cancers remains elusive. Moreover, the few lncRNAs so far shown to regulate the therapeutic response of cancers represent an incomplete repertoire of functional patterns. Despite the challenges scientists have encountered in this area, future studies will help to illustrate the roles of ncRNAs as key nodes of the regulatory network and precisely define the landscape of molecules or signaling events involved in cancer therapeutic responses, ultimately yielding beneficial outcomes by facilitating the development of ncRNA-based interventions against therapeutic resistance of cancers.
References
Harris RE. Global epidemiology of cancer. Burlington: Jones and Bartlett Publishers; 2015.
Fabbri M. Non-coding RNAs and cancer. New York: Springer; 2014.
Ramakrishnan R, Gabrilovich DI. Novel mechanism of synergistic effects of conventional chemotherapy and immune therapy of cancer. Cancer Immunol Immunother CII. 2013;62:405–10.
Schaue D, McBride WH. Opportunities and challenges of radiotherapy for treating cancer. Nat Rev Clin Oncol. 2015;12:527–40.
Dong B, Zhu YM. Molecular-targeted therapy for cancer. Chin J Cancer. 2010;29:340–5.
Pinedo HM, Giaccone G. Chemotherapy. Lancet. 1997;349 Suppl 2:SII7–9.
Perry MC. The chemotherapy source book. Philadelphia: Lippincott Williams & Wilkins; 2007.
Chapman JD, Nahum AE. Radiotherapy treatment planning: linear-quadratic radiobiology. Boca Raton: CRC Press; 2015.
Sliwkowski MX, Mellman I. Antibody therapeutics in cancer. Science. 2013;341:1192–8.
Bachelot A, Chabbert-Buffet N, Salenave S, et al. Anti-androgen treatments. Annales D’endocrinologie. 2010;71:19–24.
Jordan VC. The science of selective estrogen receptor modulators: concept to clinical practice. Clin Cancer Res. 2006;12:5010–3.
Becker Y. Molecular immunological approaches to biotherapy of human cancers—a review, hypothesis and implications. Anticancer Res. 2006;26:1113–34.
Young A, Rowett L, Kerr D. Cancer biotherapy: an introductory guide. Oxford/New York: Oxford University Press; 2006.
Housman G, Byler S, Heerboth S, et al. Drug resistance in cancer: an overview. Cancers. 2014;6:1769–92.
Malek E, Jagannathan S, Driscoll JJ. Correlation of long non-coding RNA expression with metastasis, drug resistance and clinical outcome in cancer. Oncotarget. 2014;5:8027–38.
Zheng T, Wang J, Chen X, Liu L. Role of microRNA in anticancer drug resistance. Int J Cancer. 2010;126:2–10.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7–24.
Arcaroli JJ, Quackenbush KS, Powell RW, et al. Common PIK3CA mutants and a novel 3ʹ UTR mutation are associated with increased sensitivity to saracatinib. Clin Cancer Res. 2012;18:2704–14.
Meng F, Henson R, Lang M, et al. Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology. 2006;130:2113–29.
Garofalo M, Di Leva G, Romano G, et al. MiR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 down-regulation. Cancer Cell. 2009;16:498–509.
Yang H, Kong W, He L, et al. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008;68:425–33.
Hamano R, Miyata H, Yamasaki M, et al. Overexpression of miR-200c induces chemoresistance in esophageal cancers mediated through activation of the Akt signaling pathway. Clin Cancer Res. 2011;17:3029–38.
Huang X, Taeb S, Jahangiri S, et al. miRNA-95 mediates radioresistance in tumors by targeting the sphingolipid phosphatase SGPP1. Cancer Res. 2013;73:6972–86.
Fornari F, Milazzo M, Chieco P, et al. MiR-199a-3p regulates mTOR and c-Met to influence the doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res. 2010;70:5184–93.
Weidhaas JB, Babar I, Nallur SM, et al. MicroRNAs as potential agents to alter resistance to cytotoxic anticancer therapy. Cancer Res. 2007;67:11111–6.
Deng Y, Bai H, Hu H. Rs11671784 G/A variation in miR-27a decreases chemo-sensitivity of bladder cancer by decreasing miR-27a and increasing the target RUNX-1 expression. Biochem Biophys Res Commun. 2015;458:321–7.
Li J, Wang Y, Song Y, et al. MiR-27a regulates cisplatin resistance and metastasis by targeting RKIP in human lung adenocarcinoma cells. Mol Cancer. 2014;13:193.
Sugimura K, Miyata H, Tanaka K, et al. Let-7 expression is a significant determinant of response to chemotherapy through the regulation of IL-6/STAT3 pathway in esophageal squamous cell carcinoma. Clin Cancer Res. 2012;18:5144–53.
Wang H, Sun T, Hu J, et al. MiR-33a promotes glioma-initiating cell self-renewal via PKA and NOTCH pathways. J Clin Invest. 2014;124:4489–502.
Fonte E, Apollonio B, Scarfo L, et al. In vitro sensitivity of CLL cells to fludarabine may be modulated by the stimulation of Toll-like receptors. Clin Cancer Res. 2013;19:367–79.
Mok TS, Lee K, Leung L. Targeting epidermal growth factor receptor in the management of lung cancer. Semin Oncol. 2014;41:101–9.
Garofalo M, Romano G, Leva D, et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat Med. 2012;18:74–82.
Rai K, Takigawa N, Ito S, et al. Liposomal delivery of MicroRNA-7-expressing plasmid overcomes epidermal growth factor receptor tyrosine kinase inhibitor-resistance in lung cancer cells. Mol Cancer Ther. 2011;10:1720–7.
Ragusa M, Majorana A, Statello L, et al. Specific alterations of microRNA transcriptome and global network structure in colorectal carcinoma after cetuximab treatment. Mol Cancer Ther. 2010;9:3396–409.
Spector NL, Blackwell KL. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2009;27:5838–47.
Bai WD, Ye XM, Zhang MY, et al. MiR-200c suppresses TGF-beta signaling and counteracts trastuzumab resistance and metastasis by targeting ZNF217 and ZEB1 in breast cancer. Int J Cancer. 2014;135:1356–68.
Ye XM, Zhu HY, Bai WD, et al. Epigenetic silencing of miR-375 induces trastuzumab resistance in HER2-positive breast cancer by targeting IGF1R. BMC Cancer. 2014;14:134.
Li L, Liu Y, Guo Y, et al. Regulatory MiR-148a-ACVR1/BMP circuit defines a cancer stem cell-like aggressive subtype of hepatocellular carcinoma. Hepatology. 2015;61:574–84.
Walker CL, Ho SM. Developmental reprogramming of cancer susceptibility. Nat Rev Cancer. 2012;12:479–86.
Takebe N, Miele L, Harris PJ, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12:445–64.
Hwang WL, Jiang JK, Yang SH, et al. MicroRNA-146a directs the symmetric division of Snail-dominant colorectal cancer stem cells. Nat Cell Biol. 2014;16:268–80.
Li L, Li Z, Kong X, et al. Down-regulation of microRNA-494 via loss of SMAD4 increases FOXM1 and beta-catenin signaling in pancreatic ductal adenocarcinoma cells. Gastroenterology. 2014;147:485–97 e418.
Park EY, Chang E, Lee EJ, et al. Targeting of miR34a-NOTCH1 axis reduced breast cancer stemness and chemoresistance. Cancer Res. 2014;74:7573–82.
Munoz JL, Rodriguez-Cruz V, Ramkissoon SH, et al. Temozolomide resistance in glioblastoma occurs by miRNA-9-targeted PTCH1, independent of sonic hedgehog level. Oncotarget. 2015;6:1190–201.
Ahmad N, Kumar R. Steroid hormone receptors in cancer development: a target for cancer therapeutics. Cancer Lett. 2011;300:1–9.
Jordan VC, O’Malley BW. Selective estrogen-receptor modulators and antihormonal resistance in breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2007;25:5815–24.
Miller PC, Clarke J, Koru-Sengul T, et al. A novel MAPK-microRNA signature is predictive of hormone-therapy resistance and poor outcome in ER-positive breast cancer. Clin Cancer Res. 2015;21:373–85.
Maillot G, Lacroix-Triki M, Pierredon S, et al. Widespread estrogen-dependent repression of micrornas involved in breast tumor cell growth. Cancer Res. 2009;69:8332–40.
Rao X, Di Leva G, Li M, et al. MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene. 2011;30:1082–97.
Godinho M, Meijer D, Setyono-Han B, et al. Characterization of BCAR4, a novel oncogene causing endocrine resistance in human breast cancer cells. J Cell Physiol. 2011;226:1741–9.
Xue X, Yang YA, Zhang A, et al. LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene. 2015. doi:10.1038/onc.2015.340.
Muluhngwi P, Klinge CM. Roles for miRNAs in endocrine resistance in breast cancer. Endocr Relat Cancer. 2015;22:R279–300.
Ma S, Chan YP, Kwan PS, et al. MicroRNA-616 induces androgen-independent growth of prostate cancer cells by suppressing expression of tissue factor pathway inhibitor TFPI-2. Cancer Res. 2011;71:583–92.
Yang L, Lin C, Jin C, et al. LncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature. 2013;500:598–602.
Ribas J, Ni X, Haffner M, et al. MiR-21: an androgen receptor-regulated microRNA that promotes hormone-dependent and hormone-independent prostate cancer growth. Cancer Res. 2009;69:7165–9.
Rahman N. Realizing the promise of cancer predisposition genes. Nature. 2014;505:302–8.
Lwin T, Zhao X, Cheng F, et al. A microenvironment-mediated c-Myc/miR-548m/HDAC6 amplification loop in non-Hodgkin B cell lymphomas. J Clin Invest. 2013;123:4612–26.
Jeon HM, Sohn YW, Oh SY, et al. ID4 imparts chemoresistance and cancer stemness to glioma cells by derepressing miR-9*-mediated suppression of SOX2. Cancer Res. 2011;71:3410–21.
Zhang L, Pickard K, Jenei V, et al. MiR-153 supports colorectal cancer progression via pleiotropic effects that enhance invasion and chemotherapeutic resistance. Cancer Res. 2013;73:6435–47.
Williams GH, Stoeber K. The cell cycle and cancer. J Pathol. 2012;226:352–64.
Pouliot LM, Chen YC, Bai J, et al. Cisplatin sensitivity mediated by WEE1 and CHK1 is mediated by miR-155 and the miR-15 family. Cancer Res. 2012;72:5945–55.
Salerno E, Scaglione BJ, Coffman FD, et al. Correcting miR-15a/16 genetic defect in New Zealand Black mouse model of CLL enhances drug sensitivity. Mol Cancer Ther. 2009;8:2684–92.
Fornari F, Gramantieri L, Giovannini C, et al. MiR-122/cyclin G1 interaction modulates p53 activity and affects doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res. 2009;69:5761–7.
Mohammad RM, Muqbil I, Lowe L, et al. Broad targeting of resistance to apoptosis in cancer. Seminars in Cancer Biology. 2015;35(Supplement):S78–103.
Huang G, Nishimoto K, Zhou Z, et al. MiR-20a encoded by the miR-17-92 cluster increases the metastatic potential of osteosarcoma cells by regulating Fas expression. Cancer Res. 2012;72:908–16.
Razumilava N, Bronk SF, Smoot RL, et al. MiR-25 targets TNF-related apoptosis inducing ligand (TRAIL) death receptor-4 and promotes apoptosis resistance in cholangiocarcinoma. Hepatology. 2012;55:465–75.
Corsten MF, Miranda R, Kasmieh R, et al. MicroRNA-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered S-TRAIL in human gliomas. Cancer Res. 2007;67:8994–9000.
Incoronato M, Garofalo M, Urso L, et al. MiR-212 increases tumor necrosis factor-related apoptosis-inducing ligand sensitivity in non-small cell lung cancer by targeting the antiapoptotic protein PED. Cancer Res. 2010;70:3638–46.
Weng H, Huang H, Dong B, et al. Inhibition of miR-17 and miR-20a by oridonin triggers apoptosis and reverses chemoresistance by derepressing BIM-S. Cancer Res. 2014;74:4409–19.
Dai B, Meng J, Peyton M, et al. STAT3 mediates resistance to MEK inhibitor through microRNA miR-17. Cancer Res. 2011;71:3658–68.
Chen Y, Jacamo R, Konopleva M, et al. CXCR4 down-regulation of let-7a drives chemoresistance in acute myeloid leukemia. J Clin Invest. 2013;123:2395–407.
Lam LT, Lu X, Zhang H, et al. A microRNA screen to identify modulators of sensitivity to BCL2 inhibitor ABT-263 (navitoclax). Mol Cancer Ther. 2010;9:2943–50.
Braconi C, Valeri N, Gasparini P, et al. Hepatitis C virus proteins modulate microRNA expression and chemosensitivity in malignant hepatocytes. Clin Cancer Res. 2010;16:957–66.
Swift LH, Golsteyn RM. Genotoxic anti-cancer agents and their relationship to DNA damage, mitosis, and checkpoint adaptation in proliferating cancer cells. Int J Mol Sci. 2014;15:3403–31.
Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12:801–17.
Zhang P, Wang L, Rodriguez-Aguayo C, et al. MiR-205 acts as a tumour radiosensitizer by targeting ZEB1 and Ubc13. Nat Commun. 2014;5:5671.
Galluzzi L, Morselli E, Vitale I, et al. MiR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res. 2010;70:1793–803.
Prensner JR, Chen W, Iyer MK, et al. PCAT-1, a long non-coding RNA, regulates BRCA2 and controls homologous recombination in cancer. Cancer Res. 2014;74:1651–60.
Streppel MM, Pai S, Campbell NR, et al. MicroRNA 223 is up-regulated in the multistep progression of Barrett’s esophagus and modulates sensitivity to chemotherapy by targeting PARP1. Clin Cancer Res. 2013;19:4067–78.
Wang Y, Huang JW, Calses P, et al. MiR-96 down-regulates REV1 and RAD51 to promote cellular sensitivity to cisplatin and PARP inhibition. Cancer Res. 2012;72:4037–46.
Duffy MJ, Synnott NC, McGowan PM, et al. P53 as a target for the treatment of cancer. Cancer Treat Rev. 2014;40:1153–60.
Ory B, Ramsey MR, Wilson C, et al. A microRNA-dependent program controls p53-independent survival and chemosensitivity in human and murine squamous cell carcinoma. J Clin Invest. 2011;121:809–20.
Ma S, Tang KH, Chan YP, et al. MiR-130b Promotes CD133(+) liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear protein 1. Cell Stem Cell. 2010;7:694–707.
Koster R, di Pietro A, Timmer-Bosscha H, et al. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J Clin Invest. 2010;120:3594–605.
Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer. 2014;14:709–21.
Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603–16.
Drayton RM, Dudziec E, Peter S, et al. Reduced expression of miRNA-27a modulates cisplatin resistance in bladder cancer by targeting the cystine/glutamate exchanger SLC7A11. Clin Cancer Res. 2014;20:1990–2000.
Murray-Stewart T, Hanigan CL, Woster PM, et al. Histone deacetylase inhibition overcomes drug resistance through a miRNA-dependent mechanism. Mol Cancer Ther. 2013;12:2088–99.
Salvador MA, Wicinski J, Cabaud O, et al. The histone deacetylase inhibitor abexinostat induces cancer stem cells differentiation in breast cancer with low Xist expression. Clin Cancer Res. 2013;19:6520–31.
Shang Y, Cai X, Fan D. Roles of epithelial-mesenchymal transition in cancer drug resistance. Curr Cancer Drug Targets. 2013;13:915–29.
Izumchenko E, Chang X, Michailidi C, et al. The TGFbeta-miR200-MIG6 pathway orchestrates the EMT-associated kinase switch that induces resistance to EGFR inhibitors. Cancer Res. 2014;74:3995–4005.
Mirzoeva OK, Collisson EA, Schaefer PM, et al. Subtype-specific MEK-PI3 kinase feedback as a therapeutic target in pancreatic adenocarcinoma. Mol Cancer Ther. 2013;12:2213–25.
Adam L, Zhong M, Choi W, et al. MiR-200 expression regulates epithelial-to-mesenchymal transition in bladder cancer cells and reverses resistance to epidermal growth factor receptor therapy. Clin Cancer Res. 2009;15:5060–72.
Ali S, Ahmad A, Banerjee S, et al. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 2010;70:3606–17.
Cochrane DR, Spoelstra NS, Howe EN, et al. MicroRNA-200c mitigates invasiveness and restores sensitivity to microtubule-targeting chemotherapeutic agents. Mol Cancer Ther. 2009;8:1055–66.
Li Y, VandenBoom TG, Kong D, et al. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69:6704–12.
Kitamura K, Seike M, Okano T, et al. MiR-134/487b/655 cluster regulates TGF-beta-induced epithelial-mesenchymal transition and drug resistance to gefitinib by targeting MAGI2 in lung adenocarcinoma cells. Mol Cancer Ther. 2014;13:444–53.
Giles KM, Kalinowski FC, Candy PA, et al. Axl mediates acquired resistance of head and neck cancer cells to the epidermal growth factor receptor inhibitor erlotinib. Mol Cancer Ther. 2013;12:2541–58.
Bockhorn J, Dalton R, Nwachukwu C, et al. MicroRNA-30c inhibits human breast tumour chemotherapy resistance by regulating TWF1 and IL-11. Nat Commun. 2013;4:1393.
Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. The EMBO J. 2015;34:856–80.
Wang P, Zhang J, Zhang L, et al. MicroRNA 23b regulates autophagy associated with radioresistance of pancreatic cancer cells. Gastroenterology. 2013;145:1133–43 e1112.
Haenisch S, Werk AN, Cascorbi I. MicroRNAs and their relevance to ABC transporters. Br J Clin Pharmacol. 2014;77:587–96.
Wu CP, Hsieh CH, Wu YS. The emergence of drug transporter-mediated multidrug resistance to cancer chemotherapy. Mol Pharm. 2011;8:1996–2011.
Choi YH, Yu AM. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr Pharm Des. 2014;20:793–807.
Borel F, Han R, Visser A, et al. Adenosine triphosphate-binding cassette transporter genes up-regulation in untreated hepatocellular carcinoma is mediated by cellular microRNAs. Hepatology. 2012;55:821–32.
Jaiswal R, Gong J, Sambasivam S, et al. Microparticle-associated nucleic acids mediate trait dominance in cancer. FASEB J. 2012;26:420–9.
Anreddy N, Gupta P, Kathawala RJ, et al. Tyrosine kinase inhibitors as reversal agents for ABC transporter mediated drug resistance. Molecules. 2014;19:13848–77.
Kovalchuk O, Filkowski J, Meservy J, et al. Involvement of microRNA-451 in resistance of the MCF-7 breast cancer cells to chemotherapeutic drug doxorubicin. Mol Cancer Ther. 2008;7:2152–9.
Doyle LA, Yang W, Rishi AK, et al. H19 gene overexpression in atypical multidrug-resistant cells associated with expression of a 95-kilodalton membrane glycoprotein. Cancer Res. 1996;56:2904–7.
Tsang WP, Kwok TT. Riboregulator H19 induction of MDR1-associated drug resistance in human hepatocellular carcinoma cells. Oncogene. 2007;26:4877–81.
To KK, Robey RW, Knutsen T, et al. Escape from hsa-miR-519c enables drug-resistant cells to maintain high expression of ABCG2. Mol Cancer Ther. 2009;8:2959–68.
Molina-Arcas M, Casado FJ, Pastor-Anglada M. Nucleoside transporter proteins. Curr Vasc Pharmacol. 2009;7:426–34.
Bhutia YD, Hung SW, Patel B, et al. CNT1 expression influences proliferation and chemosensitivity in drug-resistant pancreatic cancer cells. Cancer Res. 2011;71:1825–35.
Dahabreh IJ, Linardou H, Siannis F, et al. Somatic EGFR mutation and gene copy gain as predictive biomarkers for response to tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2010;16:291–303.
Nahta R, Yu D, Hung MC, et al. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3:269–80.
Lahdaoui F, Delpu Y, Vincent A, et al. MiR-219-1-3p is a negative regulator of the mucin MUC4 expression and is a tumor suppressor in pancreatic cancer. Oncogene. 2015;34:780–8.
Sachdeva M, Mo YY. MicroRNA-145 suppresses cell invasion and metastasis by directly targeting mucin 1. Cancer Res. 2010;70:378–87.
Gomez GG, Wykosky J, Zanca C, et al. Therapeutic resistance in cancer: microRNA regulation of EGFR signaling networks. Cancer Biol Med. 2013;10:192–205.
Boni V, Bitarte N, Cristobal I, et al. MiR-192/miR-215 influence 5-fluorouracil resistance through cell cycle-mediated mechanisms complementary to its post-transcriptional thymidylate synthase regulation. Mol Cancer Ther. 2010;9:2265–75.
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26.
Offer SM, Butterfield GL, Jerde CR, et al. MicroRNAs miR-27a and miR-27b directly regulate liver dihydropyrimidine dehydrogenase expression through two conserved binding sites. Mol Cancer Ther. 2014;13:742–51.
Persson H, Kvist A, Vallon-Christersson J, et al. The non-coding RNA of the multidrug resistance-linked vault particle encodes multiple regulatory small RNAs. Nat Cell Biol. 2009;11:1268–71.
Blonska M, Agarwal NK, Vega F. Shaping of the tumor microenvironment: stromal cells and vessels. Semin Cancer Biol. 2015;34:3–13.
Kohlhapp FJ, Mitra AK, Lengyel E, Peter ME. MicroRNAs as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene. 2015;34:5857–68.
Tanaka K, Miyata H, Sugimura K, et al. MiR-27 is associated with chemoresistance in esophageal cancer through transformation of normal fibroblasts to cancer-associated fibroblasts. Carcinogenesis. 2015;36:894–903.
Zhao J, Cao Y, Lei Z, et al. Selective depletion of CD4 + CD25 + Foxp3+ regulatory T cells by low-dose cyclophosphamide is explained by reduced intracellular ATP levels. Cancer Res. 2010;70:4850–8.
Kong YW, Ferland-McCollough D, Jackson TJ, et al. MicroRNAs in cancer management. Lancet Oncol. 2012;13:e249–58.
Zhu Y, Yu F, Jiao Y, et al. Reduced miR-128 in breast tumor-initiating cells induces chemotherapeutic resistance via Bmi-1 and ABCC5. Clin Cancer Res. 2011;17:7105–15.
Giovannetti E, Funel N, Peters GJ, et al. MicroRNA-21 in pancreatic cancer: correlation with clinical outcome and pharmacologic aspects underlying its role in the modulation of gemcitabine activity. Cancer Res. 2010;70:4528–38.
Ziliak D, Gamazon ER, Lacroix B, et al. Genetic variation that predicts platinum sensitivity reveals the role of miR-193b* in chemotherapeutic susceptibility. Mol Cancer Ther. 2012;11:2054–61.
Shi W, Gerster K, Alajez NM, et al. MicroRNA-301 mediates proliferation and invasion in human breast cancer. Cancer Res. 2011;71:2926–37.
Eto K, Iwatsuki M, Watanabe M, et al. The sensitivity of gastric cancer to trastuzumab is regulated by the miR-223/FBXW7 pathway. Int J Cancer. 2015;136:1537–45.
Liu Q, Paroo Z. Biochemical principles of small RNA pathways. Annu Rev Biochem. 2010;79:295–319.
Huang KC, Rao PH, Lau CC, et al. Relationship of XIST expression and responses of ovarian cancer to chemotherapy. Mol Cancer Ther. 2002;1:769–76.
Kaboli PJ, Rahmat A, Ismail P, Ling KH. MicroRNA-based therapy and breast cancer: a comprehensive review of novel therapeutic strategies from diagnosis to treatment. Pharmacol Res. 2015;97:104–21.
Wang Z, Li Y, Ahmad A, et al. Targeting Notch signaling pathway to overcome drug resistance for cancer therapy. Biochim Biophys Acta. 2010;1806:258–67.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media Singapore
About this chapter
Cite this chapter
Jia, L., Yang, A. (2016). Noncoding RNAs in Therapeutic Resistance of Cancer. In: Song, E. (eds) The Long and Short Non-coding RNAs in Cancer Biology. Advances in Experimental Medicine and Biology, vol 927. Springer, Singapore. https://doi.org/10.1007/978-981-10-1498-7_10
Download citation
DOI: https://doi.org/10.1007/978-981-10-1498-7_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-1496-3
Online ISBN: 978-981-10-1498-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)