
Abstract
Metabolic reprogramming is one of the biochemical signatures of cancer cells. Particularly, aerobic glycolysis (i.e. the process of conversion of glucose into pyruvate followed by fermentation into lactate even in the presence of oxygen) has been of immense interest due to its impact not only on cancer cells but on the tumor microenvironment as well. Conceptual advancement in understanding the oncogenic regulation of glycolysis and multifunctional properties of glycolytic enzymes underscore the relevance and significance of targeting glycolysis in cancer cells. This chapter will discuss, in the light of recent research the intricacies of glycolytic adaptation in cancer cells, and the rationale for exploiting it for therapeutic intervention.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Glycolysis
- Aerobic glycolysis
- Metabolic reprogram
- Acidosis
- Chemoresistance
- Microenvironment
- Metabolic stress
- ATP
- Hypoxia
- Apoptosis
- Monocarboxylate transporters
- Reactive oxygen species
- Warburg effect
- Embden–Meyerhof–Parnas pathway
- Antiglycolytic therapy
15.1 Introduction
Cancer cells take up glucose vividly, and this metabolic phenotype is witnessed in most if not all solid tumors. This tumor specific change in glucose consumption is so ubiquitous in cancer that it has already been exploited in the clinical diagnosis of neoplasms, using the glucose analog, 18F (fluoro)-2-deoxy glucose (FDG) by positron emission tomography (PET) imaging. A combined PET and computed tomography (CT) imaging could detect neoplasms with >90 % sensitivity and specificity (Bomanji et al. 2001; Tang et al. 2011), which depends upon the viability and/or metabolic activity of cancer cells. The common and frequent occurrence of “increased utilization of glucose” by cancer cells also indicates the necessity or preference for such a biochemical alteration. Thus, glucose metabolism in general represents a potential and sensitive therapeutic target.
Intracellularly, glucose catabolism primarily occurs via one of the two major pathways. An energy-efficient but extended pathway that involves mitochondrial respiration (also known as oxidative phosphorylation) or a short but less energy efficient pathway (glycolysis) that does not depend on mitochondria. Depending upon the intracellular requirements and available resources (e.g. oxygen, nutrients) cells direct glucose metabolism either by oxidative phosphorylation or glycolysis. In the absence of sufficient levels of molecular oxygen, glucose catabolism does not occur via mitochondrial oxidation but rather through glycolysis resulting in the conversion of pyruvate into lactate which then can be exported. This process (under less oxygenated conditions) is referred to as “anaerobic glycolysis”. Interestingly, in cancer cells glycolysis has been witnessed even in the presence of oxygen (hence referred to as “aerobic glycolysis”). The existence of an aerobic glycolytic phenotype in cancer cells has been known for almost a century since the seminal discovery by the German scientist Otto Warburg who proposed the “Warburg hypothesis” also known as the “Warburg effect” (Warburg et al. 1924). However, the causal factors and cancer-specific advantages of such altered metabolic phenotype remained obscure for several decades. Recent progress in understanding the regulation of energy metabolism has provided renewed impetus to explore the biological significance and clinical relevance of targeting tumor metabolism (Ganapathy-Kanniappan and Geschwind 2013). As a result, deregulated or altered energy metabolism has been recognized as one of the “hallmarks of cancer” (Hanahan and Weinberg 2011). Several elegant reviews have delineated a wealth of information on the biochemical processes of glycolysis and its biological significance with respect to tumor growth and poor prognosis (Gatenby and Gillies 2007; Pelicano et al. 2006). The objective of this chapter is to provide insights into the role of cellular stress in metabolic reprogramming, focusing particularly on glycolysis and its therapeutic potential as a drug target.
15.2 Tumor Glycolysis
Prior to the discussion of tumor glycolysis, in the interest of the readers, it is essential to understand its biochemical definition and the current usage in literature. In classical biochemistry, glycolysis sensu stricto refers to the conversion of glucose into pyruvate. Next, the conversion of pyruvate into lactate is known as fermentation. However, as indicated by several investigators, the process of conversion of glucose to pyruvate is common in both the lactate producing pathway as well as mitochondrial oxidation. This implies that irrespective of the mode of glucose oxidation, glycolysis will be an integral process. However, in cancer cells, as the difference between mitochondrial-dependent and independent pathways of glucose utilization is linked to either inhibition or production of lactate, respectively, the term glycolysis from a cancer perspective is indicative of the conversion of glucose into lactate. Correspondingly, the non-glycolytic, oxidative phosphorylation refers to the conversion of glucose into pyruvate which will then be metabolized via the TCA cycle in mitochondria. Similarly, the most common type of glycolysis discussed elaborately in the literature is the Embden–Meyerhof–Parnas (EMP) pathway, named after its discoverers Gustav Embden, Otto Meyerhof, and Jakub Karol Parnas. However, modified processes of glycolysis are also known (e.g. Entner–Doudoroff pathway). For brevity and clarity, and also due to the relevance to tumor metabolism, the discussion here will be limited to the EMP pathway.
Uncontrolled proliferation and insensitivity to growth inhibitory signals result in the production of enormous biomass of cancer cells. Consequently, it is inevitable for a multicellular, three-dimensional tumor to be anatomically displaced from the primary source of blood supply. In this situation, cancer cells induce the formation of new blood vessels (neo-angiogenesis) to establish an alternative vascular network with existing vessels. However, due to incomplete or aberrant circuitry of capillaries cancer cells still remain under fluctuating levels of oxygen and/ or nutrients supply. Hence a metabolic switch from mitochondrial respiration to glycolysis under hypoxia and/ or mitochondrial dysfunction (Hu et al. 2012; Lu et al. 2012) is an adaptive mechanism necessitated to maintain uninterrupted growth of cancer cells. Nonetheless, a metabolic alteration to aerobic glycolysis under normoxic condition despite the presence of functionally competent mitochondria is intriguing. Furthermore, glycolysis is known to produce fewer adenosine triphosphate (ATP; the principal form of energy) molecules than oxidative phosphorylation per every molecule of glucose. Arguably, aerobic glycolysis will indeed increase the intracellular demand for ATP, a condition that will add metabolic stress. Yet, cancer cells of different tissue origin consistently exhibit an aerobic glycolytic phenotype. Until recently, understanding the biological rationale and cellular advantages of such metabolic shift remained a challenge. Teleological evidences demonstrate that a metabolic switch to glycolysis could provide selective advantage to cancer cells despite the low-yield in ATP (de Souza et al. 2011). For example, in order to minimize the difference in the production of total number of ATP due to the metabolic switch from mitochondrial respiration to glycolysis, cancer cells facilitate a higher rate of glycolysis. A higher glycolytic rate in turn elevates the rate of glucose oxidation into lactate as has been witnessed in cancer cells. This increase in the glycolytic rate thus maintains a faster rate of ATP production (Pfeiffer et al. 2001). In addition, such an elevated glycolytic rate has also been proposed to confer selective advantage under competition (between cancerous and non-cancerous (healthy) cells) for shared energy sources (Zhou et al. 2012). Some investigators opined that in cancer cells the bulk of the ATP pool is primarily required for cell maintenance rather than proliferation suggesting a minimal decrease in total number of ATP (due to the glycolytic switch) is not detrimental to cancer cells (Gatenby and Gillies 2004; Lunt and Vander Heiden 2011). Next, it is increasingly evident that glycolytic intermediates serve as precursors for the biosynthesis of macromolecules (e.g. NADPH and ribose-5-phosphate) which in turn are critical for cell growth (Deberardinis et al. 2008). In addition, the generation of NADPH via glycolysis facilitates the maintenance of adequate levels of the antioxidant, reduced glutathione (GSH). GSH is indispensable not only for maintaining the redox balance but also to thwart the anticancer effects of some antineoplastic agents as well (Backos et al. 2012; Traverso et al. 2013). Thus aerobic glycolysis has been known to provide chemoresistance and resistance to radiotherapy (Pitroda et al. 2009). While aerobic glycolysis facilitates the pentose phosphate pathway (PPP) which in turn is critical for macromolecular biosynthesis, the PPP by itself has been known to render resistance to therapy as well (Riganti et al. 2012). Thus the cancer specific advantages of glycolysis could underly the preferential metabolic switch from mitochondrial oxidation to aerobic glycolysis.
15.3 Cellular Stress and Metabolic Reprogramming
In the words of Chi Dang, “metabolism generates oxygen radicals, which contribute to oncogenic mutations. Activated oncogenes and loss of tumor suppressors in turn alter metabolism” (Dang 2012). Cellular metabolic processes release several reactive molecules like hydrogen peroxide (H2O2), oxyradicals, hydroxyl (•OH) radicals etc., that are collectively known as reactive oxygen species (ROS). These ROS have been known to promote deleterious effects hence maintaining the cellular ROS level within threshold is critical for the maintenance of genomic integrity and cell survival. (Ray et al. 2012). Similarly, excessive accumulation of protons (H+) reduce the intracellular pH resulting in the disruption of normal physiology leading to cell death. Nevertheless, cells have evolved inherent mechanisms to respond to such undesirable changes in the levels of H+, ROS etc. For instance, intracellular levels of H+ are constantly maintained in homeostasis by proton exchange transporters by which protons are pumped out to the exterior. Similarly, the cellular response to mitigate the deleterious effects of oxidants (ROS) includes utilization of antioxidants (e.g. glutathione) which upon neutralization of the ROS becomes reduced.
In cancer cells in order to meet the constant demand for energy due to rapid proliferation and exponential growth, the rate of glucose utilization is elevated resulting in markedly high levels of glucose uptake. While the energy produced by high rate of glucose catabolism is necessary for cancer cell maintenance and growth, the by-products such as ROS released from mitochondrial oxidation will also raise to toxic levels. Though the redox balance is maintained by reduced-glutathione, an active antioxidant that neutralize enormous levels of ROS, a chronic elevation in the level of ROS will necessitate continuous replenishment of GSH. In this context, by diverting glucose catabolism away from mitochondrial respiration cancer cells can reduce the total ROS produced, decrease the protons (H+) released and more importantly conserve or minimize the utilization of available reducing equivalents (NADH) or antioxidants (GSH). Interestingly, glycolysis facilitates these desirable biochemical phenotypes besides providing several biological advantages, as discussed elsewhere. Although intracellular stress affects the overall physiology of cells, emerging data indicate that in cancer cells they do play a role in the promotion of altered tumor metabolism (Fig. 15.1). Among various stress molecules we will discuss ROS, intracellular-acidosis, and signaling mechanisms known to contribute to the metabolic switch to glycolysis.

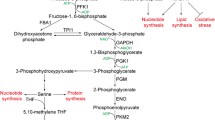
Schematic showing the link between cellular stress and glycolysis. Stress stimuli trigger the stress signaling pathway such as p38/MAPK resulting in the activation of MAPK-M2 by phosphorylation. MAPK-M2 directly accelerates glycolysis by modulating PFKFB3 activity (indicated by brown arrows). Next, ROS, a major intracellular stress inducer also affects glycolysis by diverting glycolysis towards PPP (indicated by purple arrows). ROS affects the sulphydryl group of PKM2 resulting in its inactivation. Also, ROS promotes the stabilization of HIF1α which in turn activates several glycolytic enzymes resulting in an increase in the rate of glycolysis. Acidosis, another frequent and common intracellular stress, blocks LDH activity to redirect glucose oxidation via PPP (indicated by red arrows). Also, under acidosis p53 is up regulated which in turn promotes PPP by activating GPD. Enzymes are indicated in square boxes in bold and italicized letters. Grey arrows indicate the enzymes involved in glycolysis, black arrows indicate the sequential steps in glycolysis. Purple color refers to ROS mediated effects, red color refers to acidosis mediated effects and brown color refers to signaling mechanism mediated effects. MAPK Mitogen activated protein kinase, PFKFB3 -phosphofructokinase-2-kinase/fructose-2,6-bisphosphatase 3, HIF1α hypoxia inducible factor 1alpha (α), PPP pentose phosphate pathway, ROS reactive oxygen species, HKII hexokinase II, PGI phosphoglucose isomerase, GPD glucose -6-phosphate dehydrogenase, PFK phosphofructokinase, ALD aldolase, GAPDH glyceraldehyde-3-phosphate dehydrogenase, TPI triose phosphate isomerase, PGK phosphoglycerate kinase, PGM phosphoglycerate mutase, ENO enolase, PKM2 pyruvate kinase M2 isoform, LDH lactate dehydrogenase, PPP pentose phosphate pathway, TCA cycle tricarboxylic acid cycle
15.3.1 ROS
It is widely known that excessive accumulation of ROS beyond cellular tolerance is cytotoxic. High rate of metabolism leads to an elevation in the level of cellular ROS creating an imbalance in the ratio of antioxidants and prooxidants. However, recent investigations have revealed that a minimal elevation in ROS prior to the chronic/injurious level could provide cue to cancer cells for the necessity of metabolic alteration. In order to escape ROS mediated injury and mitigate further cellular stress alternative pathways such as glycolysis and PPP are facilitated (Sosa et al. 2013).
One of the adaptive mechanisms recently identified is that during high levels of ROS, the enzyme, purvate kinase-M2 isoform (PKM2) is inactivated by the modification of its suphydryl group by ROS. PK-M2 catalysis the conversion of phosphoenol pyruvate into pyruvate for further oxidation into either acetyl coA or lactate. Thus inactivation of PK-M2 affects the glycolytic step of pyruvate formation diverting it towards the PPP (Anastasiou et al. 2011). The advantage of this altered metabolism is that PPP generates NADPH which can rejuvenate oxidized glutathione into its reduced form to act as an active antioxidant. This way, glucose is diverted away from mitochondrial oxidation which while reducing the level of ROS produced, simultaneously supports the replenishment of glutathione to neutralize the cellular ROS level (Dang 2012). It is noteworthy that the PKM2 has also been indicated as a gatekeeper of cell growth and survival (Harris et al. 2012).
An intracellular increase in ROS levels also has been known to stabilize HIF-1alpha, a key protein which transactivates several genes of glycolysis (Semenza et al. 1994). Among the glycolytic enzymes that are up regulated, the activation of PDK results in the rewiring of the metabolic circuitry of glucose catabolism. PDK phosphorylates PDH resulting in its inhibition, directing the pyruvate to be converted to lactate by LDH. Similarly, the activation of PFKFB4 results in the degradation of 2,6-FBP (an activator of PFK1 that catalyzes the conversion of fructose 1 phosphate into fructose 1,6 bisphosphate, a rate limiting step of glycolysis) (Yalcin et al. 2009). Such inhibition of PFKFB4 has been known to redirect glucose into PPP (Ros and Schulze 2013). However, PFKFB3 is also activated by HIF-1alpha, and PFKFB3 drives glucose into glycolysis. Depending upon the cellular requirement particular isoforms of PFKFB (3 or 4) can play a critical role in the adopting the mode of glucose catabolism. Thus ROS influences energy metabolism by facilitating glucose utilization by non-mitochondrial pathways (glycolysis/PPP) enabling cancer cells to evade chronic intracellular stress.
15.3.2 Acidosis
Cellular acidosis in general can be defined as a decrease in intracellular pH that can affect normal cell physiology, eventually causing cell death. Lactic acid (lactate) generated by glycolysis may contribute to intracellular acidosis yet it is not an indispensable factor. A change in the level of intracellular H+ concentration is sufficient to cause acidosis. In an elegant report, by experimental manipulation of intracellular lactate levels and intracellular H+ concentration, Jen-Tsan Chi’s group has demonstrated that acidosis can promote metabolic reprogramming (Lamonte et al. 2013). Under intracellular acidification, cancer cells favored the diversion of glycolysis into PPP (both oxidative and non-oxidative) and enhanced glutamine-metabolism to meet biosynthetic (PPP) and bioenergetic (glutaminolysis) demands. It is also evident that acidosis also governs the oxidative and nonoxidative PPP depending upon the cellular requirements. Under nonoxidative PPP conditions (R5P-pentose phosphate), any excess or accumulation could be redirected or reversed back to glycolysis by the reversible reactions of transketolase and transaldolase.
Elevated levels of free protons (H+) are often shuttled to the extracellular tumor microenvironment to maintain intracellular pH (pHi) at physiologic levels. Increasing amounts of H+ being pumped into the extracellular space creates an acidic microenvironment, which is known to select for cells with enhanced metastatic potential as well as provide resistance to chemotherapy (Bailey et al. 2012; Moellering et al. 2008; Schlappack et al. 1991). Thus cellular response to intracellular pH enables metabolic reprogramming to evade cellular acidification-related cytotoxicity.
15.3.3 Signal Transduction
Stress responsive signal transduction mechanisms have been known to play a significant role in the regulation of several processes including cell cycle checkpoints, apoptosis etc. Mitogen activated protein kinase (MAPK) pathways (e.g. p38) account for many of such changes in cellular processes. However, evidence for any direct regulation of glycolysis by a signal transduction molecule remained elusive. Recently it has been demonstrated that MAPK-M2 (MAPK-activated protein kinase 2) activates PFKFB3, a key promoter of glycolysis (Novellasdemunt et al. 2013). Thus metabolic reprogramming to glycolysis is achieved through signaling pathways as well. Similarly, STAT1-dependent expressional regulation of glycolysis suggests a potential role for STAT1 as a transcriptional modulator of genes responsible for glycolysis (Pitroda et al. 2009). Thus increasing data indicate that a coordinated network facilitates the metabolic switch from oxidative phosphorylation to glycolysis. Taken together intracellular stress inducers including ROS, pH and others could influence redirection of glucose metabolism away from mitochondria but towards glycolysis and PPP.
15.4 Targeting Glycolysis
15.4.1 Rationale
Apart from providing the energy source, the intermediates (substrates/products) of glucose metabolism (glycolysis) are used for anabolic reactions as well. For example, glucose-6-phosphate is used for synthesis of ribose -5 phosphate for further use in nucleic acid synthesis, similarly, dihydroxyacetone for lipid synthesis. Multiple lines of evidence show that the enhanced glucose uptake witnessed in tumor cells is to meet manifold requirements, and not just the energy demand. Thus, the enhanced glucose uptake is not just a favorite biochemical change, rather an indispensable metabolic transformation that is critical for the rapid, uncontrolled proliferation of tumor cells. In principle, targeting glucose metabolism essentially involves targeting more than one pathway that is interlinked with increased-glucose utilization.
Thus it is evident that aerobic glycolysis in conjunction with the PPP provide multiple benefits to cancer cells such as promoting tumor progression and providing resistance to therapy. Hence, this key signature of cancer cells, tumor metabolism, particularly the tumor glycolysis, provides an ideal target for therapeutic intervention.
Emerging data also substantiate several non-glycolytic functions of glycolytic enzymes and the metabolic intermediates of glycolysis (Fig. 15.2). Many enzymes of the glycolytic pathway such as hexokinase II (HKII), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), pyruvate kinase (PK)-M2 isoform and lactate dehydrogenase (LDH) participate in a number of subcellular functions including gene regulation and histone modifications (Kim and Dang 2005). Similar to the enzymes of glycolysis, some of the metabolic intermediates of glycolysis are also involved in non-glycolytic functions. Fructose-1, 6 bisphosphate by maintaining cytochrome C in an inactive state plays an anti-apoptotic role in cancer (Diaz-Ruiz et al. 2008). While pyruvate, another metabolic intermediate of glycolysis, is known to promote chemoresistance by the upregulation of p-glycoprotein (Wartenberg et al. 2010) its transporters (monocarboxylate transporters, MCTs) regulate CD147, a matrix metalloproteinase inducer (Izumi et al. 2011; Pertega-Gomes et al. 2011). Taken together, these findings demonstrate that glycolytic enzymes and metabolic intermediates play a key role beyond glycolysis, impacting cancer cell growth.

Non-glycolytic functions of glycolytic enzymes and metabolic intermediates. In the innermost circle, thick arrows represent enzymes and thin arrows indicate intermediate metabolites. The short arrows pointing towards the outer circle represent the non-glycolytic functions of corresponding enzymes/metabolites. HKII hexokinase II, FBP fructose 1,6-bisphosphate, PFKFB3 6-phosphofructokinase-2-kinase/fructose-2,6-bisphosphatase 3, GAPDH glyceraldehyde-3-phosphate dehydrogenase, PKM2 pyruvate kinase M2, LDH lactate dehydrogenase, MCT monocarboxylate transporters (Reproduced with permission from Molecular Cancer (2013))
It is noteworthy that a higher lactate level has been known to correlate with aggressive phenotype including tumor recurrence and the metastatic potential resulting in poor prognosis (Walenta et al. 2000). Since elevated lactate levels indicate the preponderance of glycolysis, antiglycolytic agents could be very effective in targeting such metabolic-phenotype in tumors. It is increasingly evident that lactate export mitigates intracellular acidification while its import into normoxic cancer cells provides a substrate source for TCA cycle (mitochondrial oxidation) and energy production. Thus a “metabolic symbiosis” prevails within a tumor due to the metabolic heterogeneity, viz. a central hypoxic and glycolytic population of cells, and a peripheral oxygenated tumor cells (Sonveaux et al. 2008). The existence of such a depending upon tumor vasculature and “give and take lactate” mechanism (Semenza 2008) will benefit both lactate-exporting and lactate-importing cells.
15.4.2 Therapeutic Opportunities
Oncogenic driver mutations have been known to culminate in altered signal transduction pathways enabling tumor cells to reprogram their metabolic circuitry to adapt to the microenvironment. For example, it has been demonstrated that enhanced nutrient uptake is an effect of oncogenic RAS mutations (Yun et al. 2009). The tumor-specific shift in metabolism has been shown to be inevitable for uncontrolled proliferation and invasion of almost all solid tumors, hence the tumor metabolism is aptly described as “Cancer’s Achilles’ Heel” (Kroemer and Pouyssegur 2008). Thus, mounting evidence points to the notion that “enhanced or increased glucose uptake” of cancer cells could be a potential therapeutic target. Several reviews have emphasized and elegantly demonstrated the potential molecular targets of glycolysis that can be exploited for anticancer therapy. Figure 15.3 illustrates the biochemical steps that are blocked by currently explored inhibitors that are either under preclinical or clinical evaluation.

Diagram showing the two phases of glycolysis and the molecular targets currently exploited for potential therapeutic drug strategies. Energy molecules such as ATP and NADH are highlighted in yellow, black arrows indicate consumption while red arrows indicate the energy release. The enzymes involved in respective reactions are abbreviated and encircled, where as the block symbol shows the targets exploited for drug development in preclinical investigations. NADH nicotinamide adenine dinucleotide reduced form, GLUT glucose transporters, HKII hexokinase II, PGI phosphoglucose isomerase, PFK phosphofructokinase, FBA fructose-bisphosphate aldolase, GAPDH glyceraldehyde-3-phosphate dehydrogenase, TPI triose phosphate isomerase, PGK phosphoglycerate kinase, PGM phosphoglycerate mutase, PK pyruvate kinase, LDH lactate dehydrogenase, MCT monocarboxylate transporter (Reproduced with permission from Molecular Cancer (2013))
15.4.3 Targeting Tumor and the Microenvironment
Several lines of evidence indicate that the impact of metabolic reprogramming to glycolysis is not confined to cancer cells but extends to the stroma/fibroblasts in the tumor microenvironment which favors tumor progression by sustained fuel or energy supply. These microenvironment-glycolytic reprogramming are also orchestrated by oncogenes. It is important to recall that oncogenetic activation (RAS, NF-kB etc.) and tumor suppressor loss have also been shown to facilitate the metabolic reprogramming of tumor microenvironment. Thus, the cancer cell’s metabolic reprogramming to glycolysis also directs the metabolic reprogramming of cancer associated fibroblasts (Lisanti et al. 2013). It has been known that tumor microenvironment acts as a barrier and renders defense against therapeutic agents. Targeting tumor glycolysis will therefore affect the tumor microenvironment which could disrupt microenvironment-related protumorigenic properties.
15.4.3.1 Combination with Chemotherapy/Radiation Therapy
Clinical outcome of chemotherapeutics thus far clearly demonstrate that a monotherapy may not be as effective as combination therapy. If under a personalized medicine approach, combinatorial strategy is required antiglycolytic agents might be a better candidate to promote potent anticancer effects. Thus antiglycolytic agents may provide an additional line of attack via combination therapy. Such approaches have already been evaluated in preclinical models to overcome drug resistance in cancer (Dwarakanath and Jain 2009; Maschek et al. 2004). As discussed earlier, since glycolysis also plays a pivotal role in resistance to therapy its inhibition potentially could sensitize tumor cells for any chemotherapy. Therefore, the combinatorial therapeutic approach with antiglycolytic agents could be a vital strategy against resistant phenotypes.
Under radiation therapy, cancer cells induce aerobic glycolysis through reactive oxygen species. Reports have identified the Warburg effect to be implicated in resistance to cytotoxic stress, including ionizing radiation as well as chemotherapy. Therefore, treatment methods which block or reduce glycolytic metabolism after radiotherapy may increase tumor cell sensitivity to radiation and chemotherapeutic killing (Zhong et al. 2013).
15.4.3.2 Antiglycolytic Strategy and Induction of Immune Response
The ability to evade immune surveillance is one of the hallmarks of cancer, and it has been well established that tumor cells escape immune detection through immunosuppressive networks. One of the factors that challenge the functional efficiency of antitumor-immune cells is the maintenance of a relatively low pH in the tumor micro environment. Tumors achieve this by regulating lactic acid secretion via modification of glucose/glutamine metabolism. Cancer-generated lactic acid could thus be viewed as a critical, immunosuppressive metabolite in the tumour microenvironment rather than a ‘metabolic waste product’ (Choi et al. 2013). Thus antiglycolytic therapy should preferably reduce or prevent lactate accumulation which in turn could promote or elicit the host immune response. The inhibition of glycolysis followed by alteration in the microenvironmental lactate levels could expose cancer cells as vulnerable to host immune surveillance, providing an opportunity for immunotherapy (Beneteau et al. 2012).
15.5 Summary
In summary, substantial evidences establish the scientific rationale for targeting glycolysis in cancer cells. Aerobic glycolysis indeed is an integral component of the altered metabolism of cancer, a hallmark that has received renewed interest in the recent decades. Several candidate drugs have been developed and evaluated mostly at the preclinical level with mixed success. Selective candidates (e.g. 2-deoxyglucose) have entered clinical trials, yet their translation remains to be witnessed. Mechanistically, the therapeutic potential of antiglycolytic strategy also includes the activation of proapoptotic pathways that are deregulated in cancer cells. Aerobic glycolysis suppresses p53 activity in cancer cells to provide selective protection from apoptosis upon loss of growth signal or inhibition of BCR-Abl (Mason et al. 2010). Thus inhibition of glycolysis could eliminate the antiapoptotic status of p53 resulting in the induction of tumor cell death. Similarly, it is also suggested that inhibition of glycolysis could sensitize cancer cells to AMPK activator-dependent induction of apoptosis (Pradelli et al. 2010). Therefore, molecular targeting of glycolysis could promote a myriad of effects including sensitization to chemotherapy and radiation therapy, and activation of apoptotic mechanisms in addition to the primary effect of disruption of energy metabolism. Up till now the major impediment for the successful clinical translation of any potent antiglycolytic agents is the manifestation of undesirable systemic toxicities which emanate from non-specific targeting. Future investigations to design and develop antiglycolytic agents that are selective in targeting cancer cells could greatly improve the therapeutic opportunities in the fight against cancer. To conclude, as the link between altered energy metabolism and cancer is increasingly evident targeting the metabolic reprogramming such as aerobic glycolysis could be an effective strategy for successful cancer therapy.
References
Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS et al (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334:1278–1283
Backos DS, Franklin CC, Reigan P (2012) The role of glutathione in brain tumor drug resistance. Biochem Pharmacol 83:1005–1012
Bailey KM, Wojtkowiak JW, Hashim AI, Gillies RJ (2012) Targeting the metabolic microenvironment of tumors. Adv Pharmacol 65:63–107
Beneteau M, Zunino B, Jacquin MA, Meynet O, Chiche J, Pradelli LA, Marchetti S, Cornille A, Carles M, Ricci JE (2012) Combination of glycolysis inhibition with chemotherapy results in an antitumor immune response. Proc Natl Acad Sci U S A 109:20071–20076
Bomanji JB, Costa DC, Ell PJ (2001) Clinical role of positron emission tomography in oncology. Lancet Oncol 2:157–164
Choi SY, Collins CC, Gout PW, Wang Y (2013) Cancer-generated lactic acid: a regulatory, immunosuppressive metabolite? J Pathol 230:350–355
Dang CV (2012) Links between metabolism and cancer. Genes Dev 26:877–890
de Souza AC, Justo GZ, de Araujo DR, Cavagis AD (2011) Defining the molecular basis of tumor metabolism: a continuing challenge since Warburg’s discovery. Cell Physiol Biochem 28:771–792
Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB (2008) Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev 18:54–61
Diaz-Ruiz R, Averet N, Araiza D, Pinson B, Uribe-Carvajal S, Devin A, Rigoulet M (2008) Mitochondrial oxidative phosphorylation is regulated by fructose 1,6-bisphosphate. A possible role in Crabtree effect induction? J Biol Chem 283:26948–26955
Dwarakanath B, Jain V (2009) Targeting glucose metabolism with 2-deoxy-D-glucose for improving cancer therapy. Future Oncol 5:581–585
Ganapathy-Kanniappan S, Geschwind JF (2013) Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer 12:152, 4598-12-152
Gatenby RA, Gillies RJ (2007) Glycolysis in cancer: a potential target for therapy. Int J Biochem Cell Biol 39:1358–1366
Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4:891–899
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Harris I, McCracken S, Mak TW (2012) PKM2: a gatekeeper between growth and survival. Cell Res 22:447–449
Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, Ogasawara M, Trachootham D, Feng L, Pelicano H et al (2012) K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res 22:399–412
Izumi H, Takahashi M, Uramoto H, Nakayama Y, Oyama T, Wang KY, Sasaguri Y, Nishizawa S, Kohno K (2011) Monocarboxylate transporters 1 and 4 are involved in the invasion activity of human lung cancer cells. Cancer Sci 102:1007–1013
Kim JW, Dang CV (2005) Multifaceted roles of glycolytic enzymes. Trends Biochem Sci 30:142–150
Kroemer G, Pouyssegur J (2008) Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 13:472–482
Lamonte G, Tang X, Chen JL, Wu J, Ding CK, Keenan MM, Sangokoya C, Kung HN, Ilkayeva O, Boros LG et al (2013) Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab 1:23, 3002-1-23
Lisanti MP, Martinez-Outschoorn UE, Sotgia F (2013) Oncogenes induce the cancer-associated fibroblast phenotype: metabolic symbiosis and “fibroblast addiction” are new therapeutic targets for drug discovery. Cell Cycle 12:2723–2732
Lu W, Hu Y, Chen G, Chen Z, Zhang H, Wang F, Feng L, Pelicano H, Wang H, Keating MJ et al (2012) Novel role of NOX in supporting aerobic glycolysis in cancer cells with mitochondrial dysfunction and as a potential target for cancer therapy. PLoS Biol 10:e1001326
Lunt SY, Vander Heiden MG (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27:441–464
Maschek G, Savaraj N, Priebe W, Braunschweiger P, Hamilton K, Tidmarsh GF, De Young LR, Lampidis TJ (2004) 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res 64:31–34
Mason EF, Zhao Y, Goraksha-Hicks P, Coloff JL, Gannon H, Jones SN, Rathmell JC (2010) Aerobic glycolysis suppresses p53 activity to provide selective protection from apoptosis upon loss of growth signals or inhibition of BCR-Abl. Cancer Res 70:8066–8076
Moellering RE, Black KC, Krishnamurty C, Baggett BK, Stafford P, Rain M, Gatenby RA, Gillies RJ (2008) Acid treatment of melanoma cells selects for invasive phenotypes. Clin Exp Metastasis 25:411–425
Novellasdemunt L, Bultot L, Manzano A, Ventura F, Rosa JL, Vertommen D, Rider MH, Navarro-Sabate A, Bartrons R (2013) PFKFB3 activation in cancer cells by the p38/MK2 pathway in response to stress stimuli. Biochem J 452:531–543
Pelicano H, Martin DS, Xu RH, Huang P (2006) Glycolysis inhibition for anticancer treatment. Oncogene 25:4633–4646
Pertega-Gomes N, Vizcaino JR, Miranda-Goncalves V, Pinheiro C, Silva J, Pereira H, Monteiro P, Henrique RM, Reis RM, Lopes C et al (2011) Monocarboxylate transporter 4 (MCT4) and CD147 overexpression is associated with poor prognosis in prostate cancer. BMC Cancer 11:312, 2407-11-312
Pfeiffer T, Schuster S, Bonhoeffer S (2001) Cooperation and competition in the evolution of ATP-producing pathways. Science 292:504–507
Pitroda SP, Wakim BT, Sood RF, Beveridge MG, Beckett MA, MacDermed DM, Weichselbaum RR, Khodarev NN (2009) STAT1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect. BMC Med 7:68, 7015-7-68
Pradelli LA, Beneteau M, Chauvin C, Jacquin MA, Marchetti S, Munoz-Pinedo C, Auberger P, Pende M, Ricci JE (2010) Glycolysis inhibition sensitizes tumor cells to death receptors-induced apoptosis by AMP kinase activation leading to Mcl-1 block in translation. Oncogene 29:1641–1652
Ray PD, Huang BW, Tsuji Y (2012) Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24:981–990
Riganti C, Gazzano E, Polimeni M, Aldieri E, Ghigo D (2012) The pentose phosphate pathway: an antioxidant defense and a crossroad in tumor cell fate. Free Radic Biol Med 53:421–436
Ros S, Schulze A (2013) Glycolysis back in the limelight: systemic targeting of HK2 blocks tumor growth. Cancer Disc 3:1105–1107
Schlappack OK, Zimmermann A, Hill RP (1991) Glucose starvation and acidosis: effect on experimental metastatic potential, DNA content and MTX resistance of murine tumour cells. Br J Cancer 64:663–670
Semenza GL (2008) Tumor metabolism: cancer cells give and take lactate. J Clin Invest 118:3835–3837
Semenza GL, Roth PH, Fang HM, Wang GL (1994) Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem 269:23757–23763
Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF et al (2008) Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 118:3930–3942
Sosa V, Moline T, Somoza R, Paciucci R, Kondoh H, LLeonart ME (2013) Oxidative stress and cancer: an overview. Ageing Res Rev 12:376–390
Tang S, Huang G, Liu J, Liu T, Treven L, Song S, Zhang C, Pan L, Zhang T (2011) Usefulness of 18F-FDG PET, combined FDG-PET/CT and EUS in diagnosing primary pancreatic carcinoma: a meta-analysis. Eur J Radiol 78:142–150
Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, Marinari UM, Domenicotti C (2013) Role of glutathione in cancer progression and chemoresistance. Oxidative Med Cell Longev 2013:972913
Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfor K, Rofstad EK, Mueller-Klieser W (2000) High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res 60:916–921
Warburg O, Posener K, Negelein E (1924) Über den Stoffwechsel der Tumoren. Biochem Z 152:319–344
Wartenberg M, Richter M, Datchev A, Gunther S, Milosevic N, Bekhite MM, Figulla HR, Aran JM, Petriz J, Sauer H (2010) Glycolytic pyruvate regulates P-Glycoprotein expression in multicellular tumor spheroids via modulation of the intracellular redox state. J Cell Biochem 109:434–446
Yalcin A, Telang S, Clem B, Chesney J (2009) Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp Mol Pathol 86:174–179
Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S et al (2009) Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325:1555–1559
Zhong J, Rajaram N, Brizel DM, Frees AE, Ramanujam N, Batinic-Haberle I, Dewhirst MW (2013) Radiation induces aerobic glycolysis through reactive oxygen species. Radiother Oncol 106:390–396
Zhou Y, Tozzi F, Chen J, Fan F, Xia L, Wang J, Gao G, Zhang A, Xia X, Brasher H et al (2012) Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res 72:304–314
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Ganapathy-Kanniappan, S. (2015). Targeting Glycolytic Adaptations of Cancer Cells: From Molecular Mechanisms to Therapeutic Opportunities. In: Wondrak, G. (eds) Stress Response Pathways in Cancer. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-9421-3_15
Download citation
DOI: https://doi.org/10.1007/978-94-017-9421-3_15
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-9420-6
Online ISBN: 978-94-017-9421-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)