
Abstract
In the past several decades, intensive research in this field has uncovered a surprising number of regulatory factors and their associated enzymatic properties to reveal the network of complexes that function in activation and repression of the transcriptional programs mediated by nuclear receptors (NR). These factors and their associated complexes have been extensively characterized both biochemically and functionally [34, 87, 94]. Several principles have emerged: (1) It is widely recognized that ligand-dependent cofactor complexes mediating repression and activation exhibit ligand-dependent exchange. (2) These complexes mediate modifications of chromatin structure consequent to their binding at regulatory elements, particularly at promoter and enhancer sites. (3) The concept about the rapid exchange of coregulatory complexes at regulatory sites has been suggested [88]. Key questions in the NR field have included: (a) What are the cofactors and exchange complexes used to mediate the ligand and signaling network-dependent switches in gene regulation programs; (b) Do long non-coding RNAs (lncRNAs) serve as regulatory “factors” for ligand-dependent gene programs, and do enhancers actually regulate transcription units encoding enhancer non-coding RNAs (eRNAs) that might have functional significance; (c) What is the relationship between DNA damage repair machinery and transcriptional machinery? (d) Do Retinoic Acid Receptors (RAR) also regulate Pol III-dependent, non-coding repeat transcriptional units in stem cells? and (e) How have new technologies such as deep sequencing altered our ability to investigate transcriptional regulatory mechanisms utilized by NRs?
Z. Liu and Q. Hu made equal contributions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Nucleotide Excision Repair
- Retinoic Acid Receptor
- Transcriptional Program
- Retinoic Acid Response Element
- Nucleotide Excision Repair Factor
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
History: Transcriptional Cofactors That Regulate RAR Transcriptional Activities
Coactivators and Corepressors
The definition of coactivators and corepressors has rapidly expanded as chromatin-associated factors that modify transcriptional programs based on their interactions with NRs. These cofactors have become recognized to include proteins, RNAs, and most recently, lipids. They often form large complexes with exchangeable components and their exchanges are regulated by covalent modifications including phosphorylation, ubiquitylation, SUMOylation, acetylation and methylation [42, 53, 86]. A partial list of this growing number of functional cofactors is provided in Table 10.1. For RAR, these cofactors can be divided into three different groups based on their function. (1) Coactivators that function with liganded RAR to activate transcription, often dependent on the presence of LXXLL motifs. RAR coactivators have a wide variety of functions, including serving as platforms for the assembly of coactivator complexes, or as enzymes that modify histone, RNA polymerase, other cofactors, or RAR itself. (2) Corepressors that function with unliganded RAR at target sites to repress gene transcription, often dismissed after RA binds to the receptor. However, in some instances, components present in corepressor complexes are retained and even required for activation functions, as exemplified by transducin-beta-like protein 1 (TBL1) and transducin beta-like 1-related protein 1 (TBLR1), with phosphorylation activating their ubiquitin ligase functions [86, 89]. (3) RA‐dependent corepressors characterized by the presence of LXXLL protein motifs that have usually been identified from coactivators. Intriguingly, this third group of repressors unconventionally uses a ligand-dependent interaction strategy to allow them to repress, rather than activate, gene expression. The mechanisms for coactivator and corepressor functions are detailed in Chaps. 2 and 3 of this volume.
ncRNAs as RAR Cofactors
While prevailing research has long focused on the function of protein coregulators, more recent data argues for the involvement of non-coding RNAs (ncRNAs) as potential “coregulators” of transcriptional regulation by many classes of transcription factors (TF), including NRs. Several contemporary technologies beginning first with the use of quantitative PCR followed by the development and use of genome‐wide deep sequencing technology and genome‐wide transcription run-on assays (Global Run‐On Sequencing or GRO-seq), have expanded our recognition of the presence of surprisingly massive transcription of genome-wide ncRNAs, which infers that the majority of the human genome is transcribed [3, 11, 38]. GRO-seq technology allows us to specifically detect only RNAs that are newly synthesized by RNA polymerase. These RNAs derive from both repeated and non-repeated genomic regions. Many thousands of relatively abundant lncRNAs have been identified [36, 37]. Many ncRNAs have also been found to be transcribed, but are present at much lower than coding genes levels of approximately 3–6 copies/cell. In some cases, the biological functions of the ncRNAs have already been established. Further studies have revealed that lncRNAs regulate the function of histone modification enzymes, and that they can often act in trans, at a distance far from the site of their transcription [104, 110, 113].
The first ncRNA gene shown to function in NR signaling, steroid receptor RNA activator (SRA), was identified from a screen for NR coactivators. The SRA ncRNA was reported to be capable of serving as a cofactor for RAR as well as other NRs [15, 16]. Several lines of evidence support the view that the functional gene product of the SRA gene is an RNA and not a protein. The biological function of this gene was not affected when the translation inhibitor, cycloheximide, was added to cell assays, and even when multiple translocation stop codons were introduced into SRA, its function in NR signaling was not impacted. The latter finding underscores the surprising revelation that SRA is an RNA without protein-coding potential. Co‐fractionation experiments showed that SRA ncRNAs co‐purify with SRC‐1, a canonical coactivator of NRs. Studies have shown that SRA ncRNAs can enhance the activity of active function 1 domain (AF1) and active function 1 domain (AF2) in NRs. A pseudouridine synthase, mPus1p, can pseudouridylate SRA ncRNA, to enhance the transcriptional activation of RARγ [66, 116, 117], providing yet another layer regulation of NR‐mediated transcriptional activity.
Based on these findings, we can now conclude that RAR binds at least one lncRNA, SRA, to exert its genomic regulatory effect, as shown in Fig. 1a. The sheer number of lncRNAs identified to date strongly implies that there are likely to be multiple ncRNA “cofactors” involved in the actions of NRs in addition to SRA. We therefore expect that additional ncRNA cofactors for RAR will be identified.

Regulation of enhancers by liganded nuclear receptors and non-coding RNAs. a Proposed interaction of RAR with the lncRNA, SRA, following the binding of RAR/RXR heterodimers to RARE. b Effects of ERα in stimulating transcription of eRNAs at regulatory enhancers, which are also occupied by cohesin, resulting in target coding gene activation
Enhancers as Transcription Units: Induction of Enhancer RNAs
Investigation of enhancers has revealed that ligand- or signal-induced activation is often accompanied by transcription of rather small (1–2 kb) transcripts, referred to as enhancer RNAs (eRNAs). One of the initial examples was provided in neurons, where signals such as potassium chloride (KCl)—a neuron stimulator—induced transcription of bidirectional eRNAs [54]. Similar eRNA production activities also have been observed on enhancers bound by NRs, including estrogen receptor (ER) and androgen receptor (AR), as shown in Fig. 1b [38, 68, 109]. The function of eRNAs is still controversial, but the data from several labs suggest that after hormone/ligand signaling induces their transcription, together with cohesins and components of the mediator complex, they help to mediate enhancer/promoter looping events [38, 54, 68, 109]. Knockdown of eRNAs and enhancer like lncRNAs affects the transcription of target coding genes [56, 57, 68].
DNA Damage Repair Components also Function as RAR Cofactors
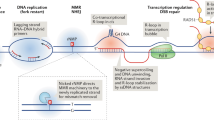
Studies of NR‐mediated gene activation have traditionally focused on the RNA Polymerase II (Pol II)‐containing RNA synthesis machinery. In response to inductive signals, a cohort of factors, coactivators and Pol II, are recruited to the promoter or enhancer regulatory regions of the activated gene, leading to initiation of RNA synthesis. Recent studies suggest that components of the DNA damage repair machinery may also be required for efficient transcription of target genes [61, 62]. In response to exogenous or endogenous factors (such as irradiation and drugs) that induce DNA damage, a number of factors, including those having endonuclease activities, are recruited to repair DNA [70]. Among these factors are the nucleotide excision repair (NER) factors, XPC, CSB, RPA, XPA, XPG, XPF and ERCC1 [81]. These proteins function together to participate in the nucleotide excision repair (NER) process.
Le May et al. [62] showed that in the absence of genotoxic stress, XPF and XPG appear to play an unexpected role. Stimulation of HeLa cells with all-trans retinoic acid (atRA) leads to recruitment of XPF and XPG, along with Pol II and the transcription factor IIF (TF IIF), to the promoter region and gene coding region of the RARβ2 gene, a atRA target gene. In contrast, no atRA‐induced recruitment of XPF or XPG is observed for the 3-Hydroxy-3-Methylglutaryl-Coenzyme A Synthase 2 (HMGCS2) gene, a peroxisome proliferator-activated receptor α (PPARα) target gene. However, in PPARα‐overexpressing HeLa cells, treatment with PPARα ligand similarly induces NER factor enrichment at HMGCS2, suggesting that in the absence of genotoxic stress, NER recruitment at gene loci depends on the activation status of the gene. Consistent with this interpretation, when endogenous NER factors are knocked down in HeLa cells, atRA-induced activation of RARβ2 is dramatically impaired, indicating the presence of these NER factors is required for effective activation of atRA target genes. A requirement for NER factors is further suggested by close examination of the epigenetic changes that occur following their depletion. Loss of these factors leads to suboptimal DNA demethylation and histone post‐transcriptional modifications, including H3K4/K9 methylation and H3K9/K14 acetylation at the promoter of RARβ2 resulting in poor expression of this gene.
When RARβ2 is activated, its promoter and terminator regions form a looping structure in a CCCTC-binding factor (CTCF)-dependent manner that promotes optimal expression of the gene. Le May et al. [61] found that, upon atRA stimulation, XPG and XPF were required for the proper assembly of the transcriptional machinery at both the promoter and terminator of RARβ2, and that CTCF recruitment preceded the docking of the transcriptional machinery. Using quantitative chromatin conformation capture (3C) assays, they observed an atRA‐triggered chromatin looping between the promoter and terminator of RARβ2. However, when endogenous XPG or XPF was depleted, the looping was significantly suppressed, resulting in a dramatically decreased expression of RARβ2. They further proved that the participation of XPG and XPF in chromatin looping required the endonuclease activity of XPG and XPF. The catalytic activity induced DNA nicks or breaks and DNA demethylation, two events essential for efficient recruitment of CTCF and consequent chromatin looping. Thus, they revealed an essential role for XPG/XPF in atRA‐triggered chromatin reorganization [61]. It will be of particular interest to find how general this strategy proves to be for the broader regulatory transcriptional program.
RAR Modifications also Affect its Interaction with Cofactors
RAR normally forms a heterodimeric structure with RXR, and ligand binding triggers canonical RAR/RXR signaling. A subsequent corepressor‐coactivator switch is essential for RAR/RXR‐regulated gene transcription [86]. Studies have shown that modification of RAR itself has a profound impact on its signaling activity in terms of heterodimerization, cofactor binding and transcriptional activity.
Rochette‐Egly et al. [93] found that phosphorylation of RARα1 by protein kinase A (PKA) was required for RA‐induced parietal endodermal differentiation. In RARα1 null F9 cells, RA‐induced parietal endoderm differentiation was abolished but RARγ‐regulated primitive endoderm differentiation was not impacted. Rescuing with wild‐type RARα1 restored parietal endoderm differentiation in the RARα1 gene-knockout cells. However, RARα1 with mutation at the PKA phosphorylation site could not efficiently rescue parietal endoderm differentiation in the mutant cells.
Phosphorylation of a different RAR isotype, RARγ2, was also shown to be critical for its function. Upon ligand binding, RARγ2 is normally degraded, a step that is required for its transactivation function. Gianni et al. [33] found that the AF1 and AF2 activation domains of RARγ2 were involved in promoting the turnover of the receptor, and in particular, that the p38MAPK pathway phosphorylated the AF1 domain, thus facilitating the recognition and degradation of RARγ2 by proteasomes [33]. When the phosphorylation of AF1 was blocked, the RARγ2‐mediated transactivation was dramatically impaired, supporting an important role of phosphorylation modification in NR signaling.
Other types of receptor modification with biological significance have also been investigated. With mass spectrometric analysis, Huq et al. [50] identified a trimethylation modification at Lys347 in the ligand binding domain (LBD) of murine RARα. This event is critical in promoting the dimerization of RARα and RXRα and the binding of cofactors to RARα, including CBP/p300 and RIP140. The ligand‐dependent recruitment of these cofactors is essential for the transactivation activity of RARα. Interestingly, although trimethylation of Lys347 occurs within the LBD, the ligand binding kinetics is not affected. In another study, Huq et al. [49] identified two monomethylated residues, Lys109 and Lys171, in RARα using an accurate and sensitive liquid chromatography-electrospray ionization/multi-stage mass spectrometry technique [LC‐ESI‐MS/MS] that can detect covalent modifications of proteins. These two new methylated residues were located within the DNA binding domain (DBD) and hinge regions of the receptor. Similar to the trimethylation of Lys347 in the RARα LBD, the monomethylation of Lys109 and Lys171 was found to facilitate the heterodimerization of RARα and RXRα. It also participates in the recruitment of cofactors to liganded RARα and promotes their transactivation activity. These studies have unveiled an important role of non‐histone methylation events in NR‐regulated transcription networks.
Development of the Field: Newly Developed Technologies Have Expanded Our Understanding of the RAR-Mediated Transcriptional Program
RAR Genome‐Wide Binding Data Suggest More Complex RAR Transcriptional Programs at Both Promoter and Enhancer Sites
On a global level, a deeper understanding of NR transcriptional regulatory programs has been licensed by the rapid development of global genomic technologies based on next generation deep sequencing methodology. For example, Chromatin Immunoprecipitation (ChIP)-sequencing has allowed genome wide identification of potential interaction sites for DNA binding TFs and cofactors. Several such studies in different cell lines have identified RAR genome-wide binding sites [21, 48, 73, 95], and these data have altered our viewpoint of the most cogent regulatory elements for RAR action. GRO-seq analyses have also allowed us to visualize transcription events genome-wide and to delineate regions with transcription on both strands of DNA, differences in elongation, and promoter pausing events in transcriptional regulation [17, 71]. The new technology also permits determination of the location of lncRNAs in the genome. One of the most potent methods is Chromatin Isolation by RNA purification-sequencing (ChIRP-seq) [14], which permits investigation of the genomic regions interacting with lncRNAs and eRNAs. While the full impact of these new technologies has not yet been fully realized, one powerful aspect of these global technologies is that they have begun to reveal that different cohorts of regulated transcription units can use distinct molecular mechanisms in regulating different aspects of the full transcriptional program.
Before the availability of deep‐sequencing technology, the identification of RAR‐binding sites had focused only on the promoter and proximal promoter regions of RA targets [19, 24, 72, 76, 102]. Global genomic data analyses obtained with the newer ChIP-sequencing technology revealed a different picture. The data from the two groups conducting global genomic studies [48, 95] indicated that only a relatively small portion of RAR‐binding sites were actually at proximal promoter regions; rather most RAR‐binding sites were found in intronic or distal promoter intergenic regions. These results suggest that the regulation of RA targets likely involves the action of RAR receptors on regulatory elements that included enhancers marked with specific histone modifier binding marks, such as H3K4me1, H3K4me2, CBP/p300 and H4K16Ac [1, 12, 39, 41, 107, 111], as well as other potential distal regulatory sequences. To date, comprehensive functional studies of RAR‐bound enhancers are still lacking, but we expect to see intensive investigation on this subject in the future. This is because enhancers participate in critical aspects of transcriptional regulation [40], alter chromatin interactions, and contribute to putative looping activities with promoters to deliver activating factors, such as components of the MLL complex [110].
RAR Binding is Dynamically Regulated During Differentiation
Although RAR can bind constitutively to target sites, several recent publications report a ligand‐dependent shift in RAR binding sites during RA induced differentiation and the different RAR binding patterns in mouse embryonic fibroblasts and mouse embryonic stem cells (ESCs) [21, 73]. These data suggest that RAR binding is dynamically regulated by ligand treatment or cell differentiation status.
Using a pan‐RAR antibody for ChIP-sequencing during RA induced differentiation, the David Gifford group found that RA treatment could cause widespread changes in RAR genome‐wide binding during RA‐induced neuronal differentiation [73]. Based on their RAR binding data, they concluded that only a small subset of RAR binding sites were constitutively bound, with two other sets of RAR binding sites present only in the absence or presence of RA. When they compared RAR binding sites occupied by unliganded and liganded receptors with the well‐characterized TF regulatory network in mouse ES cells, they found the binding information of ES cell TFs and other TF regulatory proteins can accurately predict both constitutive and ligand-induced RAR binding. The binding of core ES cell regulators is highly correlated with unliganded RAR binding sites, and slightly less correlated with liganded RAR binding sites.
RAR ChIP‐chip assays performed in both mouse embryonic fibroblasts and ES cell also revealed different RAR binding patterns in these two cell lines [21]. Because their ChIP‐chip experiments were performed using extended promoter array (−5 to +2 kb of promoters) and we now realize that most RARs bind at intergenic enhancer regions [48, 95], they only found 354 binding peaks in MEFs and 462 peaks in ES cells [21]. They found only 58 common RAR binding peaks for both cell lines [21], suggesting that RARs have cell‐type specific functions through binding to the different regulatory regions controlling different subsets of gene targets. It will be important in future studies to clarify whether the chromatin environment or other tissue‐specific TFs, such as FoxA1, as reported by the Kevin White group in MCF7 cells [48], determines whether RAR binds to a specific locus.
New RAR Binding Motifs
Many studies have provided evidence that RAR/RXR heterodimers bind asymmetrically to retinoic acid response element (RARE) [74]. RAR genome‐wide binding data give us a more comprehensive view of RAR binding patterns, and the data suggest that RAR binding is also enriched at other motifs [21, 48, 73]. The Kevin White group conducted an in-depth analysis for all possible hormone response element (HRE) motifs in RAR‐binding sites and found that in addition to some well-known experimentally validated RAREs, such as direct repeat (DR)5 and DR2, there were some HREs not previously known as RAREs [48], such as everted repeat (ER)2. This leads to a very intriguing question—Do different RARE motifs confer different transcriptional regulation activities on RARs? Recently, the Pierre Chambon group found several special glucocorticoid receptor (GR) binding motifs (inverted repeat (IR)0, IR1 and IR2) function as negative response elements to mediate repression by agonist‐liganded GR [100]. Further studies of these newly identified RAREs are expected to elucidate their relevance to RAR function under different conditions.
Trans‐binding of RAR and Transcriptional Regulation
Some nuclear receptors, such as GR and PPARγ, have been reported to regulate gene expression through binding to other DNA‐binding TFs, even if they do not bind directly to their DNA binding elements, which we termed “trans‐binding” effect [58, 83, 84]. This was exemplified by the unexpected discovery that in response to ligand stimulation, PPARγ was recruited in trans to mediate transrepression in macrophages [83]. Ligand-dependent SUMOylation of PPARγ, or other nuclear receptors, permits their recruitment in trans to specific regulatory regions, repressing coding gene transcription (Shown in Fig. 2a) [32, 83, 106].

Models of ligand-dependent transrepression by nuclear receptors. a Transrepression by liganded PPARγ. Shown is a model of liganded PPARγ that is SUMOylated (Su) with recruited nuclear corepressors. Here, the complex is shown inhibiting NK-κB gene activation. b Transrepression by liganded RAR. In this case, the SUMOylated RAR complex brings nuclear receptor corepressors (NCoR) to AP1 and represses AP1 target gene activation
Recently, substantial data indicate that RAR also exhibits trans-binding activities by interacting with other signaling pathways, including estrogen/ERα signaling, Wnt signaling and activating protein 1 (AP1) transcription factor complex [25, 48, 55, 69, 80]. One well‐established example is that RA-bound RAR represses the transcriptional activation of AP1 transcription factor complex, which consists of Fos and Jun [55, 69, 80]. Using various selective retinoids for RAR, researchers were able to dissociate its inhibition ability on AP1 from its classical RARE‐binding transcriptional regulation activity [10, 28, 92], suggesting that RAR interferes with AP1 activity by a different functional mechanism from its regular DNA‐binding function. Several models have been proposed to explain the transrepression of AP1 by RAR, including the fact that RAR directly interacts with Jun‐Fos at their binding targets through trans‐binding effect as shown in Fig. 2b [2, 20, 23, 96, 97, 103, 112, 118], even though we still do not have direct evidence to confirm that RAR can bind to AP1 in trans and repress its activation.
Current State of the Field: Deeper Understanding of RAR-Mediated Transcriptional Regulation
RAR Regulates Both Pol II and Pol III Transcriptional Programs
It is well documented that liganded RAR induces the expression of a cohort of the RNA Pol II‐transcribed protein‐coding genes [75], exemplified by the Hox genes, which are critically involved in development. Many of these genes harbor typical RAREs featuring a direct repeat cassette, DR2 or DR5, at their promoter or enhancer regions for efficient activation.
Bioinformatics analysis of over one million copies of human Alu repeats revealed that some of these repetitive elements contain canonical motifs for many TFs and NRs, such as NF‐κB, RAR, ER and TR [90]. In particular, around one tenth of the human Alu repeats contain the DR2 cassette for RAR recognition and binding within the B box [59]. Given that the A/B boxes constitute an internal promoter for Pol III, it is possible that RA might trigger RAR to bind to the embedded DR2 cassette and thus drive Pol III‐dependent transcription of this class of Alu repeats.
To further study how the RA/RAR signal regulates Pol III‐mediated Alu repeat transcription, we have taken advantage of the RA‐induced stem cell differentiation model. In human embryonic carcinoma “stem” cells, Ntera2/D1, and in human embryonic stem cells (H9), it has been found that atRA treatment dramatically enhanced the level of DR2 Alu transcripts [46]. By knocking down Pol III or blocking TF3C, it was confirmed that the atRA-induced DR2 Alu transcription was Pol III-dependent. It was also found that RAR, together with NCoR, binds to these DR2 Alu repeats in the absence of ligand, and that the corepressor complexes were dismissed upon RA treatment. The non‐coding DR2 Alu transcripts were transported into the cytoplasm and became colocalized with the P bodies, the cytoplasmic machinery that contains Dicer and Argonaute (Ago) proteins and acts as RNA‐processing hubs. It was also observed that the DR2 Alu transcripts were processed in the P bodies into a heterogeneously‐sized population of RA-induced small (~30–65 nt) RNAs (riRNAs), initially requiring an unexpected, Dicer-dependent step.
To explore the biological function of riRNAs, bioinformatics analysis was performed to determine if riRNAs, like microRNAs, could potentially target complementary sequence in the 3′UTRs of a subunit of ES cell‐expressed mRNAs, including those critical for stem cell maintenance, such as NANOG and TDGF‐1. It was found that the treatment with atRA decreased the transcript levels of these genes in Ntera2 cells, and that the overexpression of DR2 Alu or riRNAs dramatically down‐regulated these targets. And instead of initiating mRNA processing from the 3′ terminus as is the case for microRNA‐mediated post‐transcriptional regulation, riRNAs and associated Argonaute3 (AGO3) protein recruit decapping proteins, DCP1A and DCP2, to execute exonuclease cleavage from the 5′ terminus of targeted mRNAs. Thus, a new functional mechanism for RAR has been uncovered, in which the RAR and Pol III dependent DR2 Alu transcriptional events in stem cells functionally complement the Pol II‐dependent neuronal transcriptional program (Fig. 3). This regulatory event provides a mechanism that helps to clear stem cell commitment transcripts, as RA induces the coding gene transcripts required for differentiation, and facilitates exit from the stem cell state. It is likely that other subsets of ALU repeats exert biological functions in many more differentiated cell types, and may have roles in cancer and aging.

Schematic diagram showing a proposed mechanism for Pol III transcriptional activation of a subclass of Alu repeats, referred to as DR2 Alu repeats, by the actions of liganded RAR in embryonic stem cells (ESC). Our recent studies have revealed that RAR has two transcription programs. One is the conventional RNA Pol II-driven program. The other is dependent on Pol III, in which liganded RAR, together with associated coactivators (CoA), drives the transcription of human DR2 Alu repeats. The resultant non-coding Alu RNAs are transported into the cytoplasm where they are processed into a new type of small RNAs, riRNAs, in a DICER-dependent manner. riRNAs require AGO3 to efficiently bind to complementary sequences in the 3′UTRs of many key stem cell mRNAs, which leads to recruitment of decapping complexes containing DCP1 and DCP2, resulting in the degradation of mRNAs by exonuclease XRN1
It has also been found that the RA‐inducible DR2 Alu repeats appear to be located close to (<10 kb) active Pol II transcription units, suggesting that there might be a critical architectural chromatin “domain” adjacent to active Pol II‐transcribed coding gene loci required for the effective RA induction of Alu repeats by Pol III.
Cross-talk Between the RAR-Mediated Transcriptional Program and the Estrogen/Estrogen Receptor Pathway
The function of RAR-mediated transcriptional regulation in breast cancer, especially in estrogen receptor positive (ER+) breast cancer, has been the focus of several groups. The Jason Carroll and Kevin White laboratory groups set out to identify RAR genomic targets using ChIP and microarray gene expression analysis in an MCF7 breast cancer cell line [48, 95]. Both studies shed light on how RAR regulates gene expression together with another dominant hormone signaling estrogen in breast cancer cells. However, although both groups found a large number of common binding sites for RAR and ERα, they came to totally different conclusions. One paper [95] suggests that RARα functions cooperatively with ERα to regulate the loading of coactivators at ERα enhancers [95], while the other paper [48] proposed that RARs and ERα actually compete to bind to common regulatory elements, thus mediating the genomic antagonism between RA and estrogen signaling in breast cancer [48].
The Carroll group found that estrogen induced RARα expression and that RARα was required for estrogen‐induced growth of the MCF7 breast cancer cells [95]. They further performed RARα ChIP‐sequencing in the MCF7 line. Their data indicate that RARα exhibits substantial co-occupancy with ERα at the genome‐wide level, and that knockdown of ERα expression reduced RARα’s binding at approximately half of these co‐bound sites, suggesting a functional interaction between ERα and RARα. On the other hand, knockdown of RARα did not affect ERα binding, but did alter coactivator binding, such as p300, and affected histone H3 acetylation and RNA Pol II loading at the promoter regions of ERα targets. Hence, these authors hypothesized that besides its classic role as a heterodimeric partner of RXR proteins that respond to natural ligands such as RA, RARα can function cooperatively as an ERα‐associated protein for maintaining ERα‐cofactor interaction during estrogen‐mediated gene transcription. Thus, the addition of RA ligand can competitively trigger the classic RARα role and inhibit estrogen target genes by affecting estrogen‐ERα function. These data explain how RARα ligand can be used for an effective treatment, as well as provide a rationale for why both RARα agonists and antagonists inhibit breast cancer in animal models and preclinical trials.
The White group used GFP tag technology to map RARα and RARγ in MCF7 cell by ChIP‐chip. They found that both RARα and RARγ binding is highly coincident with ERα [48]. Their gene expression data suggested: (1) Liganded RAR can both activate and repress different gene targets, while traditional view proposed that repressive function is mediated solely by unliganded RAR; and (2) The co‐occupied RAR/ERα binding sites mediated the antagonistic actions between RA and estrogen on gene regulation. In contrast to the Carroll group’s finding, their data suggested that instead of simultaneously binding to common sites, RARα and ERα compete to bind these sites. They also reported that FoxA1 and GATA3 TFs were recruited at RAR/ERα bound enhancers. Surprisingly, knockdown of FoxA1 affects the binding of RARs at these common binding sites, suggesting that RARs also function cooperatively with FoxA1 to gain access to their binding sites on enhancers or promoters.
The different conclusions of these studies in the breast cancer cell line raise many questions and we expect that additional studies will emerge to further characterize the functional interaction between RAR and ERα. Indeed, although the exact nature of the interaction between RA/RAR and estrogen/ERα in breast cancer cell line MCF7 is still obscure, some pilot studies, such as the genome‐wide profiling of RAR and ERα bound sites, suggest a possible broad trans‐binding between RAR or ERα. It will be instructive to further explore an appropriate working model to validate the RAR/ERα interaction in breast cancer cells, and to develop novel therapies for RA-resistant tumors.
Relevance: RAR-Mediated Transcriptional Regulation and Disease
Understanding the basic principles of gene regulation by NRs, exemplified by RAR, has particular importance for designing strategies that can ultimately alter transcriptional programs in development, homeostasis, disease, aging and DNA damage repair. For example, the realization of the critical roles of enhancers in NR transcriptional programs provides motivation for investigation of new strategies to block function of cell-type specific enhancers, perhaps by novel mutation or anti-eRNA approaches. Retinoids, through binding to its NRs (RAR), are physiological regulators of embryonic development, tissue homeostasis and cell differentiation, as well as mediating apoptosis and proliferation [7]. Because of their inhibitory effects on breast cancer cell lines and suppression of carcinogenesis in experimental animal models, retinoids occupy a prominent position among the chemopreventive agents that have been examined in preclinical studies and clinical trials [114, 115]. However, the clinical trials of retinoids in patients with advanced breast cancer were not as successful as initially expected. Thus, it is of prime importance to study the molecular mechanism of RAR-mediated transcriptional regulation in cancers, and the roles played by the three types of retinoic acid receptors in various cell types, to permit more effective strategies for harnessing the potential anti-cancer effects of retinoids. Even understanding at a molecular level why binding of retinoic acid receptors to some enhancers activate their target coding genes, while binding to other enhancers results in repression of their target coding genes, will provide new approaches to fine tuning these events in both health and disease.
This period of intensive investigation has undoubtedly pointed to a surprisingly large series of cofactors as critical components in the RAR signaling program under both the physiological and pathological conditions. In particular, the functional study of corepressors promises to enhance our understanding of the inefficiency of therapeutic application of RA in different cancer diseases. One example of such a cofactor is the human tumor antigen PRAME [18, 26, 27, 85]. Studies show that PRAME functions as a dominant repressor of RAR signaling by binding to RAR in the presence of RA and preventing ligand induced receptor activation through recruitment of Polycomb proteins [26]. Thus PRAME inhibits RA‐induced differentiation, growth arrest, and apoptosis. Knockdown of PRAME expression by RNA interference in RA‐resistant human melanoma restores RAR signaling and reinstates the sensitivity of tumor cells to the anti-proliferative effects of RA both in vitro and in vivo.
Future Directions: Future Questions on RAR-Mediated Transcriptional Regulation
With the current genome-wide profiling and interactome characterization, we can expect to see an ever-growing body of cofactors for the RAR program, including additional enzymes, ncRNAs, and other non-conventional RAR corepressors/coactivators. It will also be important to learn more about how DNA damage repair components, in concert with known coactivators at RAR enhancer and promoter sites, function in control of regulated transcription, looping and gene activation.
By harnessing the power of contemporary sequencing technologies, we are rapidly accumulating knowledge and gaining insight into how RAR mediates transcriptional regulation at a genome‐wide level. We expect to see in-depth studies on RAR function in different development, and disease models. These insights will, of course, answer many critical questions concerning normal development and pathological conditions in human, including:
-
1.
How does liganded RAR function for both activation and repression as reported by recent genome‐wide studies in breast cancer cell lines?
-
2.
Does RAR globally use different types of RARE information to determine its function and to recruit different cofactors?
-
3.
Does RAR act globally through trans‐binding with other TFs by protein‐protein interaction, and does the outcome require new functions of its DNA binding domain?
The era of molecular biology has brought us to a deep understanding of the biological roles and mechanisms of retinoic acid receptor function. In the near future, the era of global genomics will rapidly and significantly extend our knowledge for a clearer understanding of both the uniform and the distinct ways in which different cohorts of RA‐regulated transcription units are transcribed.
Abbreviations
- 3′UTR:
-
3′ untranslated region
- AF1:
-
Active function 1 domain
- AF2:
-
Active function 2 domain
- AP1:
-
Activating protein 1
- AR:
-
Androgen receptor
- atRA:
-
All-trans retinoic acid
- ChIP:
-
Chromatin immunoprecipitation
- CSB:
-
Cockayne syndrome B protein
- CTCF:
-
CCCTC-binding factor
- DBD:
-
DNA binding domain
- DCP1A:
-
mRNA-decapping enzyme 1A
- DCP2:
-
mRNA-decapping enzyme 2
- DR:
-
Direct repeat
- ER:
-
Estrogen receptor
- ER:
-
Everted repeat
- ERCC1:
-
Excision repair cross-complementing protein 1
- eRNA:
-
Enhancer RNA
- ESCs:
-
Embryonic stem cells
- GR:
-
Glucocorticoid receptor
- HMGCS2:
-
3-Hydroxy-3-Methylglutaryl-Coenzyme A Synthase 2
- HRE:
-
Hormone response element
- IR:
-
Inverted repeat
- LBD:
-
Ligand-binding domain
- LC‐ESI‐MS:
-
Liquid chromatography-electrospray ionization–mass spectrometry
- lncRNA:
-
Long non-coding RNA
- MAPK:
-
Mitogen-activated protein kinase
- NCoR:
-
Nuclear receptor corepressor
- ncRNA:
-
Non‐coding RNA
- NER:
-
Nucleotide excision repair
- NR:
-
Nuclear receptor
- PKA:
-
Protein kinase A
- Pol II:
-
RNA polymerase II
- Pol III:
-
RNA polymerase III
- PPAR:
-
Peroxisome proliferator-activated receptor
- RA:
-
Retinoic acid
- RAR:
-
Retinoic acid receptor
- RARE:
-
Retinoic acid response element
- RPA:
-
Replication protein A
- RXR:
-
Retinoid X receptor
- SRA:
-
Steroid receptor RNA activator
- STAT3:
-
Signal transducer and activator of transcription 3
- TBL1:
-
Transducin-beta-like protein 1
- TBLR1:
-
Transducin beta-like 1-related protein 1
- TF3C:
-
General transcription factor 3C
- TF IIF:
-
Transcription factor IIF
- TR:
-
Thyroid hormone receptor
- XPA:
-
Xeroderma pigmentosum, complementation group A
- XPC:
-
Xeroderma pigmentosum, complementation group C
- XPF:
-
Xeroderma pigmentosum, complementation group F
- XPG:
-
Xeroderma pigmentosum, complementation group G
References
Belikov S, Holmqvist PH, Astrand C, Wrange O (2012) FoxA1 and glucocorticoid receptor crosstalk via histone H4K16 acetylation at a hormone regulated enhancer. Exp Cell Res 318:61–74
Benkoussa M, Brand C, Delmotte MH, Formstecher P, Lefebvre P (2002) Retinoic acid receptors inhibit AP1 activation by regulating extracellular signal-regulated kinase and CBP recruitment to an AP1-responsive promoter. Mol Cell Biol 22:4522–4534
Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE et al (2007) Identification and analysis of functional elements in 1 % of the human genome by the ENCODE pilot project. Nature 447:799–816
Blanco JC, Minucci S, Lu J, Yang XJ, Walker KK, Chen H, Evans RM, Nakatani Y, Ozato K (1998) The histone acetylase PCAF is a nuclear receptor coactivator. Genes Dev 12:1638–1651
Brown K, Chen Y, Underhill TM, Mymryk JS, Torchia J (2003) The coactivator p/CIP/SRC-3 facilitates retinoic acid receptor signaling via recruitment of GCN5. J Biol Chem 278:39402–39412
Chakravarti D, LaMorte VJ, Nelson MC, Nakajima T, Schulman IG, Juguilon H, Montminy M, Evans RM (1996) Role of CBP/P300 in nuclear receptor signalling. Nature 383:99–103
Chambon P (1996) A decade of molecular biology of retinoic acid receptors. Faseb J 10:940–954
Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR (1999) Regulation of transcription by a protein methyltransferase. Science 284:2174–2177
Chen JD, Evans RM (1995) A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 377:454–457
Chen JY, Penco S, Ostrowski J, Balaguer P, Pons M, Starrett JE, Reczek P, Chambon P, Gronemeyer H (1995) RAR-specific agonist/antagonists which dissociate transactivation and AP1 transrepression inhibit anchorage-independent cell proliferation. EMBO J 14:1187–1197
Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, Helt G et al (2005) Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308:1149–1154
Chepelev I, Wei G, Wangsa D, Tang Q, Zhao K (2012) Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res 22:490–503
Cho YS, Kim EJ, Park UH, Sin HS, Um SJ (2006) Additional sex comb-like 1 (ASXL1), in cooperation with SRC-1, acts as a ligand-dependent coactivator for retinoic acid receptor. J Biol Chem 281:17588–17598
Chu C, Qu K, Zhong FL, Artandi SE, Chang HY (2011) Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell 44:667–678
Colley SM, Leedman PJ (2011) Steroid receptor RNA activator—A nuclear receptor coregulator with multiple partners: insights and challenges. Biochimie 93:1966–1972
Cooper C, Vincett D, Yan Y, Hamedani MK, Myal Y, Leygue E (2011) Steroid receptor RNA activator bi-faceted genetic system: heads or tails? Biochimie 93:1973–1980
Danko CG, Hah N, Luo X, Martins AL, Core L, Lis JT, Siepel A, Kraus WL (2013) Signaling pathways differentially affect RNA polymerase II initiation, pausing, and elongation rate in cells. Mol Cell 50:212–222
De Carvalho DD, Binato R, Pereira WO, Leroy JM, Colassanti MD, Proto-Siqueira R, Bueno-Da-Silva AE, Zago MA, Zanichelli MA, Abdelhay E et al (2011) BCR-ABL-mediated upregulation of PRAME is responsible for knocking down TRAIL in CML patients. Oncogene 30:223–233
de The H, Vivanco-Ruiz MM, Tiollais P, Stunnenberg H, Dejean A (1990) Identification of a retinoic acid responsive element in the retinoic acid receptor beta gene. Nature 343:177–180
Dedieu S, Lefebvre P (2006) Retinoids interfere with the AP1 signalling pathway in human breast cancer cells. Cell Signal 18:889–898
Delacroix L, Moutier E, Altobelli G, Legras S, Poch O, Choukrallah MA, Bertin I, Jost B, Davidson I (2010) Cell-specific interaction of retinoic acid receptors with target genes in mouse embryonic fibroblasts and embryonic stem cells. Mol Cell Biol 30:231–244
Dilworth FJ, Fromental-Ramain C, Yamamoto K, Chambon P (2000) ATP-driven chromatin remodeling activity and histone acetyltransferases act sequentially during transactivation by RAR/RXR In vitro. Mol Cell 6:1049–1058
DiSepio D, Sutter M, Johnson AT, Chandraratna RA, Nagpal S (1999) Identification of the AP1-antagonism domain of retinoic acid receptors. Mol Cell Biol Res Commun: MCBRC 1:7–13
Durand B, Saunders M, Leroy P, Leid M, Chambon P (1992) All-trans and 9-cis retinoic acid induction of CRABPII transcription is mediated by RAR-RXR heterodimers bound to DR1 and DR2 repeated motifs. Cell 71:73–85
Easwaran V, Pishvaian M, Salimuddin, Byers S (1999) Cross-regulation of beta-catenin-LEF/TCF and retinoid signaling pathways. Curr Biol 9:1415–1418
Epping MT, Wang L, Edel MJ, Carlee L, Hernandez M, Bernards R (2005) The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell 122:835–847
Epping MT, Wang L, Plumb JA, Lieb M, Gronemeyer H, Brown R, Bernards R (2007) A functional genetic screen identifies retinoic acid signaling as a target of histone deacetylase inhibitors. Proc Natl Acad Sci USA 104:17777–17782
Fanjul A, Dawson MI, Hobbs PD, Jong L, Cameron JF, Harlev E, Graupner G, Lu XP, Pfahl M (1994) A new class of retinoids with selective inhibition of AP-1 inhibits proliferation. Nature 372:107–111
Fernandes I, Bastien Y, Wai T, Nygard K, Lin R, Cormier O, Lee HS, Eng F, Bertos NR, Pelletier N et al (2003) Ligand-dependent nuclear receptor corepressor LCoR functions by histone deacetylase-dependent and -independent mechanisms. Mol Cell 11:139–150
Franco PJ, Li G, Wei LN (2003) Interaction of nuclear receptor zinc finger DNA binding domains with histone deacetylase. Mol Cell Endocrinol 206:1–12
Fujiki R, Chikanishi T, Hashiba W, Ito H, Takada I, Roeder RG, Kitagawa H, Kato S (2009) GlcNAcylation of a histone methyltransferase in retinoic-acid-induced granulopoiesis. Nature 459:455–459
Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK (2007) Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell 25:57–70
Gianni M, Bauer A, Garattini E, Chambon P, Rochette-Egly C (2002) Phosphorylation by p38MAPK and recruitment of SUG-1 are required for RA-induced RAR gamma degradation and transactivation. EMBO J 21:3760–3769
Glass CK, Rosenfeld MG (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 14:121–141
Gurevich I, Aneskievich BJ (2009) Liganded RARalpha and RARgamma interact with but are repressed by TNIP1. Biochem Biophys Res Commun 389:409–414
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP et al (2009) Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458:223–227
Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L et al (2011) lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477:295–300
Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, Kraus WL (2011) A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 145:622–634
Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW et al (2009) Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459:108–112
Heintzman ND, Ren B (2009) Finding distal regulatory elements in the human genome. Curr Opin Genet Dev 19:541–549
Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA et al (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39:311–318
Hermanson O, Glass CK, Rosenfeld MG (2002) Nuclear receptor coregulators: multiple modes of modification. Trends Endocrinol Metab 13:55–60
Hong SH, David G, Wong CW, Dejean A, Privalsky ML (1997) SMRT corepressor interacts with PLZF and with the PML-retinoic acid receptor alpha (RARalpha) and PLZF-RARalpha oncoproteins associated with acute promyelocytic leukemia. Proc Natl Acad Sci USA 94:9028–9033
Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK et al (1995) Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 377:397–404
Hou Z, Peng H, White DE, Negorev DG, Maul GG, Feng Y, Longmore GD, Waxman S, Zelent A, Rauscher FJ 3rd (2010) LIM protein Ajuba functions as a nuclear receptor corepressor and negatively regulates retinoic acid signaling. Proc Natl Acad Sci USA 107:2938–2943
Hu Q, Tanasa B, Trabucchi M, Li W, Zhang J, Ohgi KA, Rose DW, Glass CK, Rosenfeld MG (2012) DICER- and AGO3-dependent generation of retinoic acid-induced DR2 Alu RNAs regulates human stem cell proliferation. Nat Struct Mol Biol
Hu X, Chen Y, Farooqui M, Thomas MC, Chiang CM, Wei LN (2004) Suppressive effect of receptor-interacting protein 140 on coregulator binding to retinoic acid receptor complexes, histone-modifying enzyme activity, and gene activation. J Biol Chem 279:319–325
Hua S, Kittler R, White KP (2009) Genomic antagonism between retinoic acid and estrogen signaling in breast cancer. Cell 137:1259–1271
Huq MD, Ha SG, Wei LN (2008) Modulation of retinoic acid receptor alpha activity by lysine methylation in the DNA binding domain. J Proteome Res 7:4538–4545
Huq MD, Tsai NP, Khan SA, Wei LN (2007) Lysine trimethylation of retinoic acid receptor-alpha: a novel means to regulate receptor function. Mol Cell Proteomics: MCP 6:677–688
Kashyap V, Gudas LJ (2010) Epigenetic regulatory mechanisms distinguish retinoic acid-mediated transcriptional responses in stem cells and fibroblasts. J Biol Chem 285:14534–14548
Khetchoumian K, Teletin M, Tisserand J, Mark M, Herquel B, Ignat M, Zucman-Rossi J, Cammas F, Lerouge T, Thibault C et al (2007) Loss of Trim24 (Tif1alpha) gene function confers oncogenic activity to retinoic acid receptor alpha. Nat Genet 39:1500–1506
Kim JH, Lee JM, Nam HJ, Choi HJ, Yang JW, Lee JS, Kim MH, Kim SI, Chung CH, Kim KI et al (2007) SUMOylation of pontin chromatin-remodeling complex reveals a signal integration code in prostate cancer cells. Proc Natl Acad Sci USA 104:20793–20798
Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S et al (2010) Widespread transcription at neuronal activity-regulated enhancers. Nature 465:182–187
Lafyatis R, Kim SJ, Angel P, Roberts AB, Sporn MB, Karin M, Wilder RL (1990) Interleukin-1 stimulates and all-trans-retinoic acid inhibits collagenase gene expression through its 5′ activator protein-1-binding site. Mol Endocrinol 4:973–980
Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, Shiekhattar R (2013) Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 494:497–501
Lam MT, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, Benner C, Kaikkonen MU, Kim AS, Kosaka M et al (2013) Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 498:511–515
Langlais D, Couture C, Balsalobre A, Drouin J (2012) The Stat3/GR interaction code: predictive value of direct/indirect DNA recruitment for transcription outcome. Mol Cell 47:38–49
Laperriere D, Wang TT, White JH, Mader S (2007) Widespread Alu repeat-driven expansion of consensus DR2 retinoic acid response elements during primate evolution. BMC Genom 8:23
Le May N, Iltis I, Ame JC, Zhovmer A, Biard D, Egly JM, Schreiber V, Coin F (2012) Poly (ADP-ribose) glycohydrolase regulates retinoic acid receptor-mediated gene expression. Mol Cell 48:785–798
Le May N, Fradin D, Iltis I, Bougneres P, Egly JM (2012) XPG and XPF endonucleases trigger chromatin looping and DNA demethylation for accurate expression of activated genes. Mol Cell 47:622–632
Le May N, Mota-Fernandes D, Velez-Cruz R, Iltis I, Biard D, Egly JM (2010) NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol Cell 38:54–66
Lee HK, Park UH, Kim EJ, Um SJ (2007) MED25 is distinct from TRAP220/MED1 in cooperating with CBP for retinoid receptor activation. EMBO J 26:3545–3557
Lee S, Lee DK, Dou Y, Lee J, Lee B, Kwak E, Kong YY, Lee SK, Roeder RG, Lee JW (2006) Coactivator as a target gene specificity determinant for histone H3 lysine 4 methyltransferases. Proc Natl Acad Sci USA 103:15392–15397
Lee SW, Cho YS, Na JM, Park UH, Kang M, Kim EJ, Um SJ (2010) ASXL1 represses retinoic acid receptor-mediated transcription through associating with HP1 and LSD1. J Biol Chem 285:18–29
Leygue E (2007) Steroid receptor RNA activator (SRA1): unusual bifaceted gene products with suspected relevance to breast cancer. Nucl Receptor Signal 5:e006
Li HJ, Haque ZK, Chen A, Mendelsohn M (2007) RIF-1, a novel nuclear receptor corepressor that associates with the nuclear matrix. J Cell Biochem 102:1021–1035
Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X et al (2013) Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498:516–520
Lin F, Xiao D, Kolluri SK, Zhang X (2000) Unique anti-activator protein-1 activity of retinoic acid receptor beta. Cancer Res 60:3271–3280
Lindahl T, Wood RD (1999) Quality control by DNA repair. Science 286:1897–1905
Liu W, Ma Q, Wong K, Li W, Ohgi K, Zhang J, Aggarwal AK, Rosenfeld MG (2013) Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell 155:1581–1595
Loudig O, Babichuk C, White J, Abu-Abed S, Mueller C, Petkovich M (2000) Cytochrome P450RAI(CYP26) promoter: a distinct composite retinoic acid response element underlies the complex regulation of retinoic acid metabolism. Mol Endocrinol 14:1483–1497
Mahony S, Mazzoni EO, McCuine S, Young RA, Wichterle H, Gifford DK (2011) Ligand-dependent dynamics of retinoic acid receptor binding during early neurogenesis. Genome Biol 12:R2
Mangelsdorf DJ, Evans RM (1995) The RXR heterodimers and orphan receptors. Cell 83:841–850
Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P et al (1995) The nuclear receptor superfamily: the second decade. Cell 83:835–839
Mangelsdorf DJ, Umesono K, Kliewer SA, Borgmeyer U, Ong ES, Evans RM (1991) A direct repeat in the cellular retinol-binding protein type II gene confers differential regulation by RXR and RAR. Cell 66:555–561
Mengus G, May M, Carre L, Chambon P, Davidson I (1997) Human TAF(II)135 potentiates transcriptional activation by the AF-2s of the retinoic acid, vitamin D3, and thyroid hormone receptors in mammalian cells. Genes Dev 11:1381–1395
Moon M, Um SJ, Kim EJ (2012) CAC1 negatively regulates RARalpha activity through cooperation with HDAC. Biochem Biophys Res Commun 427:41–46
Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, Schreiber SL, Evans RM (1997) Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell 89:373–380
Nicholson RC, Mader S, Nagpal S, Leid M, Rochette-Egly C, Chambon P (1990) Negative regulation of the rat stromelysin gene promoter by retinoic acid is mediated by an AP1 binding site. EMBO J 9:4443–4454
Nouspikel T (2009) DNA repair in mammalian cells: nucleotide excision repair: variations on versatility. Cell Mol Life Sci: CMLS 66:994–1009
Onate SA, Tsai SY, Tsai MJ, O’Malley BW (1995) Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 270:1354–1357
Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK (2005) A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 437:759–763
Pascual G, Sullivan AL, Ogawa S, Gamliel A, Perissi V, Rosenfeld MG, Glass CK (2007) Anti-inflammatory and antidiabetic roles of PPARgamma. Novartis Found Symp 286:183–196; discussion 196–203
Passeron T, Valencia JC, Namiki T, Vieira WD, Passeron H, Miyamura Y, Hearing VJ (2009) Upregulation of SOX9 inhibits the growth of human and mouse melanomas and restores their sensitivity to retinoic acid. J Clin Invest 119:954–963
Perissi V, Aggarwal A, Glass CK, Rose DW, Rosenfeld MG (2004) A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell 116:511–526
Perissi V, Jepsen K, Glass CK, Rosenfeld MG (2010) Deconstructing repression: evolving models of co-repressor action. Nat Rev Genet 11:109–123
Perissi V, Rosenfeld MG (2005) Controlling nuclear receptors: the circular logic of cofactor cycles. Nat Rev Mol Cell Biol 6:542–554
Perissi V, Scafoglio C, Zhang J, Ohgi KA, Rose DW, Glass CK, Rosenfeld MG (2008) TBL1 and TBLR1 phosphorylation on regulated gene promoters overcomes dual CtBP and NCoR/SMRT transcriptional repression checkpoints. Mol Cell 29:755–766
Polak P, Domany E (2006) Alu elements contain many binding sites for transcription factors and may play a role in regulation of developmental processes. BMC Genom 7:133
Qiu J, Shi G, Jia Y, Li J, Wu M, Dong S, Wong J (2010) The X-linked mental retardation gene PHF8 is a histone demethylase involved in neuronal differentiation. Cell Res 20:908–918
Resche-Rigon M, Gronemeyer H (1998) Therapeutic potential of selective modulators of nuclear receptor action. Curr Opin Chem Biol 2:501–507
Rochette-Egly C, Plassat JL, Taneja R, Chambon P (2000) The AF-1 and AF-2 activating domains of retinoic acid receptor-alpha (RARalpha) and their phosphorylation are differentially involved in parietal endodermal differentiation of F9 cells and retinoid-induced expression of target genes. Mol Endocrinol 14:1398–1410
Rosenfeld MG, Lunyak VV, Glass CK (2006) Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev 20:1405–1428
Ross-Innes CS, Stark R, Holmes KA, Schmidt D, Spyrou C, Russell R, Massie CE, Vowler SL, Eldridge M, Carroll JS (2010) Cooperative interaction between retinoic acid receptor-alpha and estrogen receptor in breast cancer. Genes Dev 24:171–182
Schule R, Rangarajan P, Yang N, Kliewer S, Ransone LJ, Bolado J, Verma IM, Evans RM (1991) Retinoic acid is a negative regulator of AP-1-responsive genes. Proc Natl Acad Sci USA 88:6092–6096
Schule R, Umesono K, Mangelsdorf DJ, Bolado J, Pike JW, Evans RM (1990) Jun-Fos and receptors for vitamins A and D recognize a common response element in the human osteocalcin gene. Cell 61:497–504
Shao W, Halachmi S, Brown M (2002) ERAP140, a conserved tissue-specific nuclear receptor coactivator. Mol Cell Biol 22:3358–3372
Shao W, Rosenauer A, Mann K, Chang CP, Rachez C, Freedman LP, Miller WH Jr (2000) Ligand-inducible interaction of the DRIP/TRAP coactivator complex with retinoid receptors in retinoic acid-sensitive and—resistant acute promyelocytic leukemia cells. Blood 96:2233–2239
Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, Li M, Chambon P (2011) Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 145:224–241
Si J, Mueller L, Collins SJ (2007) CaMKII regulates retinoic acid receptor transcriptional activity and the differentiation of myeloid leukemia cells. J Clin Invest 117:1412–1421
Smith WC, Nakshatri H, Leroy P, Rees J, Chambon P (1991) A retinoic acid response element is present in the mouse cellular retinol binding protein I (mCRBPI) promoter. EMBO J 10:2223–2230
Suzukawa K, Colburn NH (2002) AP-1 transrepressing retinoic acid does not deplete coactivators or AP-1 monomers but may target specific Jun or Fos containing dimers. Oncogene 21:2181–2190
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY (2010) Long noncoding RNA as modular scaffold of histone modification complexes. Science 329:689–693
Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG (1997) The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature 387:677–684
Venteclef N, Jakobsson T, Ehrlund A, Damdimopoulos A, Mikkonen L, Ellis E, Nilsson LM, Parini P, Janne OA, Gustafsson JA et al (2010) GPS2-dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1 and LXRbeta in the hepatic acute phase response. Genes Dev 24:381–395
Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F et al (2009) ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457:854–858
Voegel JJ, Heine MJ, Zechel C, Chambon P, Gronemeyer H (1996) TIF2, a 160 kDa transcriptional mediator for the ligand-dependent activation function AF-2 of nuclear receptors. EMBO J 15:3667–3675
Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA et al (2011) Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 474:390–394
Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA et al (2011) A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472:120–124
Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K (2009) Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138:1019–1031
Yang-Yen HF, Zhang XK, Graupner G, Tzukerman M, Sakamoto B, Karin M, Pfahl M (1991) Antagonism between retinoic acid receptors and AP-1: implications for tumor promotion and inflammation. New Biol 3:1206–1219
Yang L, Lin C, Liu W, Zhang J, Ohgi KA, Grinstein JD, Dorrestein PC, Rosenfeld MG (2011) ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell 147:773–788
Yang Q, Sakurai T, Kakudo K (2002) Retinoid, retinoic acid receptor beta and breast cancer. Breast Cancer Res Treat 76:167–173
Zhang XK, Liu Y, Lee MO (1996) Retinoid receptors in human lung cancer and breast cancer. Mutat Res 350:267–277
Zhao X, Patton JR, Davis SL, Florence B, Ames SJ, Spanjaard RA (2004) Regulation of nuclear receptor activity by a pseudouridine synthase through posttranscriptional modification of steroid receptor RNA activator. Mol Cell 15:549–558
Zhao X, Patton JR, Ghosh SK, Fischel-Ghodsian N, Shen L, Spanjaard RA (2007) Pus3p- and Pus1p-dependent pseudouridylation of steroid receptor RNA activator controls a functional switch that regulates nuclear receptor signaling. Mol Endocrinol 21:686–699
Zhou XF, Shen XQ, Shemshedini L (1999) Ligand-activated retinoic acid receptor inhibits AP-1 transactivation by disrupting c-Jun/c-Fos dimerization. Mol Endocrinol 13:276–285
Acknowledgments
We thank members in the Rosenfeld laboratory for suggestions and comments on this manuscript, and the editing efforts of Rachel Pardee. M.G.R. is an investigator with HHMI.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Liu, Z., Hu, Q., Rosenfeld, M.G. (2014). Complexity of the RAR‐Mediated Transcriptional Regulatory Programs. In: Asson-Batres, M., Rochette-Egly, C. (eds) The Biochemistry of Retinoic Acid Receptors I: Structure, Activation, and Function at the Molecular Level. Subcellular Biochemistry, vol 70. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-9050-5_10
Download citation
DOI: https://doi.org/10.1007/978-94-017-9050-5_10
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-9049-9
Online ISBN: 978-94-017-9050-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)