
Abstract
The discovery of retinoic acid receptors arose from research into how vitamins are essential for life. Early studies indicated that Vitamin A was metabolized into an active factor, retinoic acid (RA), which regulates RNA and protein expression in cells. Each step forward in our understanding of retinoic acid in human health was accomplished by the development and application of new technologies. Development cDNA cloning techniques and discovery of nuclear receptors for steroid hormones provided the basis for identification of two classes of retinoic acid receptors, RARs and RXRs, each of which has three isoforms, α, β and ɣ. DNA manipulation and crystallographic studies revealed that the receptors contain discrete functional domains responsible for binding to DNA, ligands and cofactors. Ligand binding was shown to induce conformational changes in the receptors that cause release of corepressors and recruitment of coactivators to create functional complexes that are bound to consensus promoter DNA sequences called retinoic acid response elements (RAREs) and that cause opening of chromatin and transcription of adjacent genes. Homologous recombination technology allowed the development of mice lacking expression of retinoic acid receptors, individually or in various combinations, which demonstrated that the receptors exhibit vital, but redundant, functions in fetal development and in vision, reproduction, and other functions required for maintenance of adult life. More recent advancements in sequencing and proteomic technologies reveal the complexity of retinoic acid receptor involvement in cellular function through regulation of gene expression and kinase activity. Future directions will require systems biology approaches to decipher how these integrated networks affect human stem cells, health, and disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Retinoic Acid
- Glucocorticoid Receptor
- Nuclear Receptor
- Nuclear Hormone Receptor
- Estrogen Response Element
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction: In Quest of a Mechanism of Action for Vitamin A
The idea that essential factors other than proteins, fat, starch, sugar, or minerals are present in food was a novel concept before the late 1880s. Ultimately this notion was verified by a series of human dietary supplementation studies and controlled experiments in animal models conducted between 1880 and 1920, which demonstrated that removal of these factors from the diet caused debilitating illnesses and death. The first discoveries in the field were made by Christiaan Eijkman and Frederick Gowland Hopkins who found that rice polishings contain substances preventing beriberi. These investigators received the Nobel prize for their work in 1929 [17]. In 1912, Casimer Funk identified the active fraction, which was named water-soluble factor b, later described as thiamine [16]. Since this substance, which was vital for life belonged to a class of organic compounds called amines, he named it “vitamine” (vital amines) [98].
During this time, two groups, one led by Elmer McCollum and the other by Thomas Osborne and Lafayette Mendel, independently provided evidence for another essential substance that was named fat-soluble factor a [113]. By 1920, several other low abundance dietary factors were also being described, and Jack Drummond argued to the American Chemical Society that since there was no evidence for the presence of an amine in all of these “vitamines”, it would be easiest for classification purposes to drop the -e on Funk’s general reference to vital factors and refer to them as Vitamin A, Vitamin B, Vitamin C, etc. [29].
The structure of vitamin A, also known as retinol, was first reported by Paul Karrer and his collaborators in 1931 [55, 111], confirmed by the group of Heilbron the following year [44], and crystallized in 1937 [47]. The molecule is composed of 20 carbon atoms arranged as a beta-ionone ring with a conjugated isoprene tail that terminates with an alcohol functional group (Fig. 1.1).

Retinol and its main metabolites
Ongoing studies revealed that vitamin A serves as a precursor for active derivatives that impart two very different physiologic effects: (1) an aldehyde derivative (11-cis retinal), which is the active chromophore of vision [2, 7, 48, 128], and (2): an acid derivative (all-trans retinoic acid), which has the ability to reverse developmental defects in vitamin A deficient (VAD) animals [4, 5, 123] (Fig. 1.1). Further experiments by Arens and van Dorp suggested that retinoic acid (RA) could not be converted into vitamin A in vivo and thus, they concluded that RA was itself a hormone involved in cell growth and in development. Since that time, several other active vitamin A metabolites have been identified, and active compounds have been synthesized. In 1976, all of them were grouped as retinoids [117].
Clues as to how retinol and retinoic acid work inside cells came from studies performed with other fat soluble (lipophilic) hormones such as estrogens and glucocorticoids. In the 1960s, advancements in methods to synthesize radiolabelled hormones with sufficient specific activity for in vivo use and in techniques to count tritium in animal tissues [51] allowed the identification of binding proteins for these hormones [52], and also suggested that there was a link between their physiological action and transcription in the nucleus. This was the first indication that lipophilic hormones regulate gene transcription through nuclear receptors functioning as transcription factors.
More support for this concept came from Pierre Chambon’s finding that administration of estradiol to immature chickens elicited an increase in liver aggregate polymerase that preceded an induction of protein synthesis [19, 130]. Subsequent studies found that estradiol induces translocation of an estrogen binding protein from the cytoplasm to the nucleus [53] and that steroid hormones induce transcription of specific subsets of genes [82, 96]. The generality of this phenomena in the animal kingdom was shown by studies in flies demonstrating that insect ecdysteroids induce alterations in chromatin dynamics, observed as puffing of chromosomes [6]. The final pieces of the puzzle came together in the 1980s with the discovery that glucocorticoid receptor proteins could be proteolytically cleaved into independently-functional, ligand-binding (LBD) and DNA-binding (DBD) domains [132], and that the DBD could bind to specific DNA sequence elements conferring glucocorticoid regulation of adjacent genes [20, 109].
At the same time, experimental evidence was accumulating that vitamin A and its derivatives also influence RNA and protein synthesis [24, 54, 134] and can bind intracellular proteins [8, 90]. In 1978, the group of Frank Chytil purified and characterized cytosolic proteins that bound retinol (CRBPI and CRBPII) or retinoic acid (CRABPI and CRABPII) [23, 91, 92]. However, subsequent studies performed by the same group revealed that CRBPs and CRABPs are present essentially in the cytosol and that these proteins merely serve as vehicles or shuttles transferring the ligand into the nucleus to specific binding sites on the chromatin [70, 71, 120, 121]. Also at that time, there appeared to be clear evidence that other retinoid receptors were present in the nucleus that could modulate the transcription of specific genes [43].
History: Cloning of the Nuclear Retinoic Acid Receptors
In the 1970s, progress in genetic technologies increased rapidly with promising possibilities for gene mapping. New procedures for DNA hybridization [27] and gene transfer [131] made possible gene localization to specific regions of chromosomes and gene cloning into DNA plasmids. A huge advance in molecular genetic studies was also provided by the purification and characterization of a microbial reverse transcriptase enzyme that could be engineered to generate DNA complementary (cDNA) to RNA in animals [116, 122]. This ability to generate DNA sequences representing processed RNA circumvented the problems of non-coding introns and exon splicing, and allowed mRNA isolated from cells to be copied into cDNA sequences that could then be individually cloned into plasmids [112] or bacteriophages to create libraries of all the DNA translation products expressed in a particular cell type.
In 1985, these technologies were used by the group of Pierre Chambon to clone the human estrogen [129] and glucocorticoid receptors [41] (Fig. 1.2). Phage libraries were infected into bacteria growing on a petri dish at densities that allowed individual phage to be separately identified by the clear areas they produced in the bacterial lawn, called plaques. The cDNA in the plaques was transferred to a nitrocellulose membrane, which was then screened with a radioactively labeled query probe (for example, a nucleotide sequence with homology to putative hormone receptors). When exposed to X-RAY film, plaques hybridizing with the radioactive probe produced a spot that allowed their identification and isolation. Then, the isolated cDNA’s could be used to produce large quantities of protein and could be genetically manipulated to mutate or excise coding regions for specific parts of the proteins, which allowed precise identification of functional areas.

Gene cloning using phage cDNA libraries
Cloning of RARs
The first retinoic acid receptor, RARα, was independently identified in 1987 by the laboratories of Pierre Chambon and Ronald Evans. Chambon’s group cloned RARα by using a consensus oligonucleotide probe corresponding to a highly conserved sequence in the DBD of several members of the nuclear receptor family (human and chicken estrogen receptor, human and rat glucocorticoid receptor, human progesterone receptor and viral oncogene erbA) [97]. This probe was radiolabeled and hybridized with a phage cDNA library created from the human breast cancer cell lines MCF-7 and T47D. Several phage plaques giving positive signals were obtained and rescreened with probes corresponding to the DBD of the human estrogen, progesterone and glucocorticoids receptors in order to eliminate clones of these receptors. The cDNA inserts of the remaining positive clones were subcloned into a plasmid vector and sequenced. One of the cDNA inserts contained an open reading frame encoding a 432 amino-acid protein with a predicted molecular mass of 47,682 Da. This sequence was referred to as hRAR.
Analysis of the cDNA-deduced hRAR protein sequence revealed that the regions corresponding to the DBD and the LBD possess a high degree of similarity with that of all other nuclear receptors. Several experiments were designed to define whether the cloned hRAR protein binds RA and whether, by analogy with the other members of the nuclear receptor family, it could act as a ligand-inducible transcription factor. A cDNA fragment encoding the LBD was subcloned into an expression vector and introduced in HeLa cells. Incubation of the extracts with radiolabelled ligands confirmed that the hRAR protein binds RA selectively and with high affinity. A chimeric receptor in which the DBD of hRAR is replaced by the DBD of the estrogen receptor was constructed and cotransfected into HeLa cells with a vit-tk-CAT reporter gene under the control of an estrogen response element. Addition of RA resulted in an increase in CAT reporter activity indicating that hRAR is a ligand-inducible trans-activator of transcription. Thus the first human nuclear retinoic acid receptor was cloned and is now named RARα.
A few month later, Evans’ group published that they had independently cloned the same receptor using a different probe corresponding to a novel sequence with striking similarity to the DBD of the steroid hormone receptors that they fortuitously identified in a human hepatocellular carcinoma [36]. Close on the heels of these groundbreaking publications, two independent groups led by Magnus Phfahl and Chambon used the same probe [9, 13, 25] and cloned a second human nuclear retinoic acid receptor, which depicted high homology with RARα. Both groups created chimeric proteins by swapping the DBD for that of the estrogen receptor and verified that RA could activate the chimeric protein to transactivate a reporter gene under the control of an estrogen responsive DNA sequence element. One group called the new receptor, RARε, based on its high expression in epithelial tissues [9], but the name proposed by the other group, RARβ, became the commonly known name for this second RAR [13]. Finally, efforts to clone the murine counterparts of the human RARα and RARβ receptors revealed the existence of a third RA receptor, RARγ [137]. DNA sequences of the mouse RARγ gene were then used to clone the human RARγ gene [58].
Subsequent research indicated that the three RARs are encoded by three distinct genes located in different chromosomes [50, 81]. Several isoforms of each RAR subtype were identified that differ in the N-terminal region as a result of alternative splicing or of the use of different promoters upstream of the gene. Two major isoforms were identified for RARα (RARα1 and RARα2) [67], four isoforms for RARβ (RARβ1, RARβ2, RARβ3, and RARβ4) [89, 138], and two isoforms for RARγ (RARγ1 and RARγ2) [37, 57]. Official classification of the nuclear receptors identified RARα as NR1B1, RARβ as NR1B2 and RARγ as NR1B3 [35].
Cloning of a Second Family of Nuclear Retinoid Receptors: The RXRs
In the late 1980s a novel strategy was used to isolate cDNA clones encoding DNA binding domains. The technique involves absorbing the proteins produced by a phage cDNA library onto nitrocellulose filters and probing the filters with radioactive, double-stranded DNA [114, 125]. In 1989, the Keiko Ozato group used this technique to isolate from mouse liver a cDNA clone encoding a protein capable of binding the conserved MHC class I regulatory element (CRE). Interestingly, this protein, H-2RIIBP (H-2 region II binding protein), had modular domains characteristic of the nuclear hormone receptors and could also bind estrogen response elements [42]. More research needed to be done however, to determine whether the H-2RIIBP protein was a nuclear hormone receptor. Then Mangelsdorf et al. performed a low stringency screen of human liver and kidney cDNA libraries using a probe corresponding to the DBD of RARα and isolated a novel nuclear receptor, which was substantially different from RARα and was referred to as hRXRα [79]. A few month later, the Michael Rosenfeld group screened a cDNA phage library from a thyroid tumor using a RAR response element and isolated an additional nuclear receptor, which exhibited remarkable homology to RXRα and which differed by only 2 amino acids from the H-2RIIBP protein. This protein was finally named RXRβ [133]. Subsequently, three murine RXRs (RXRα, RXRβ and RXRγ) encoded by different genes were isolated [78]. Several studies attempted to clone the human RXRγ gene, but without results. Nevertheless, a human genomic locus encoding RXRγ has been mapped in chromosome 1 using florescence in situ hybridization (FISH) [3]. According to the official classification of nuclear receptors, RXRs are now identified as NR2B1 (RXRα), NR2B2 (RXRβ), and NR2B3 (RXRγ) [35].
The interesting feature of the RXR proteins is that they can activate transcription in response to RA, but are unable to bind all-trans RA due to the fact that they do not share significant homology with the LBD of RARs. Instead, 9-cis retinoic acid was identified as a high affinity ligand for RXRs [45, 68]. However, today it is clear that 9-cis RA cannot be detected in most tissues, and the existence of a physiological RXR ligand is still being investigated [34]. Nevertheless, several synthetic compounds that bind RXRs and not RARs, rexinoids, have been designed and have provided useful tools [94].
Establishment of the Basis of RARs and RXRs Mechanism of Action (1990–1995)
Once they were cloned, the sequences of the different RAR and RXR cDNAs were analyzed, aligned and compared to that of the other nuclear receptors [61]. This analysis revealed that all nuclear hormone receptors including RARs and RXRs exhibit a modular structure composed of 6 conserved regions designated A–F (Fig. 1.3a) with different degrees of conservation [65]. Region C, which corresponds to the DBD is the most conserved region with 94–97 % identity between RARs and with 60 % identity between RARs and RXRs. Region E encompasses the LBD and is also well conserved between RARs (84–90 % identity), but differs considerably between RARs and RXRs (27 % identity). Finally the A and F regions differ markedly among the 3 RARs, and the F region is lacking in RXRs.

Structure of RARs and of their DNA binding sites. a Schematic representation of the modular organization of RARs and RXRs with the functional domains. The conservation between RARα and RXRα is shown. b High resolution structure of the RXRα DNA binding domain NMR (mmdbId: 8588). c Description of the classical retinoic acid response elements (RAREs). d Structural changes induced upon RA binding. The crystal structures of the unliganded RXRα and liganded RARγ LBDs are shown with the binding domain for corepressors. Helices are represented as ribbons and labeled from H1 to H12. Adapted from Protein Data Bank 1lbd and 2lbd
During the same time, the promoter regions of endogenous genes that are controlled by RA were characterized, leading to the identification of specific cis-acting DNA elements that bind RARs (Fig. 1.3c). The first natural RA response element (RARE) identified was that in the RARβ2 promoter [26, 46, 119]. It consists of a direct repetition of 2 motifs (G/AGTTCA) separated by 5 base pairs and was called a DR5. During ensuing years, other RAREs that have different spacings between the direct repeats (DR1 and DR2) were discovered in the promoters of several other RA-responsive genes [31, 115].
RARs from crude cell extracts were able to bind RAREs, however several groups observed that high affinity binding required interactions with other nuclear factors [38, 66]. Chambon’s group purified the nuclear accessory factor that enhanced the binding of RARs to RAREs in vitro and discovered that this protein was RXRβ [66]. This group also showed that RXRs form heterodimers with RARs and that interaction regions overlap with the LBDs and the DBDs of both partners [66]. Concomitantly, Rosenfeld’s group also identified RXRβ as the factor that stimulates the binding of RARs to RAREs [133].
In 1994–1995, knowledge of the mechanism(s) of action of RAR/RXR heterodimers and most nuclear receptors increased tremendously due to the discovery of co-activators that bind specific sequences of the LBD in response to RA [30, 62, 126, 127] and characterization of the three-dimensional structures of the DBD [64] (Fig. 1.3b) and the LBD [12, 100] (Fig. 1.3d). Most notably, the description of the crystallographic structures of the LBD in the absence and presence of RA revealed that the on switch for gene transcription by liganded RARs relies on structural rearrangements that create binding surfaces for co-regulators [18, 87] (Fig. 1.3d).
During this same time, peptides corresponding to amino acid sequences specific for each RAR and RXR subtypes were synthesized, allowing generation of antibodies recognizing not only the RAR and RXR proteins produced in vitro (recombinant proteins), but also endogenous RARs and RXRs [32, 105]. In addition to their utility in localizing endogenous receptor expression in tissues, the antibodies had an additional interesting benefit of revealing that endogenous RARs and RXRs migrated to different positions on polyacrylamide gels when compared with their recombinant counterparts. This later finding suggested the native proteins had a higher relative molecular weight, possibly due to post translational modifications, such as phosphorylation, in vivo [33, 104, 106].
Genetic Evidence that RARs Transduce the Retinoid Signals in Vivo
Until the 1990s, the physiological functions of vitamin A and retinoids in vivo were mainly inferred from studies on vitamin A-deficient (VAD) animals. These studies demonstrated that vitamin A is required during pre- and post-natal development as well as in adult life. The VAD syndrome is associated with several congenital malformations during embryonic development and with defects in growth, vision, reproduction and maintenance of several tissues after birth. The cloning of RARs and RXRs raised the question of whether these nuclear receptors mediate the physiological effects of vitamin A and retinoids in vivo.
The development of the homologous recombination technology for targeted disruption of specific genes allowed the generation of mice lacking CRABPs, RARs or RXRs. Single or double knock out of CRABP I and II did not generate overt phenotypic abnormalities corroborating that these retinoic acid binding proteins are not required for the effects of RA during development [40, 60]. In contrast, knock out of RARα or RARγ exhibited congenital malformations and displayed some of the defects of the postnatal VAD syndrome with reduced postnatal viability [72, 74, 75]. Double mutants were also generated that recapitulated all of the fetal VAD syndrome malformations and that exhibited a dramatically reduced viability [73, 83]. All these results confirmed that the effects of vitamin A in development are indeed mediated by RARs. Subsequently, RXR knock out mice were also engineered, but the interpretation of the phenotypes was more complicated since RXR heterodimerize not only with RARs, but also with multiple other types of nuclear hormone receptors [56, 118]. Nevertheless, compound mutants in both RXR and RAR greatly exacerbated the VAD phenotypes [56] suggesting that the RAR/RXR heterodimers are the functional units that mediate vitamin A signaling in vivo.
Development of the Field: A Huge Explosion Has Occurred in the Field of RARs During the Last Two Decades
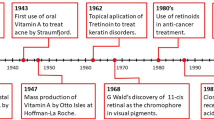
Between 1987 and 1995, RARs and RXRs were cloned and the basis for their mechanism of action was established (Fig. 1.4). During the next twenty years, up to now, knowledge in the field of RARs and RXRs has increased tremendously due to the development of novel genetic, biophysical and high throughput molecular technologies. Integration of these approaches has provided an in-depth view of the mechanism of action of RARs and of all nuclear receptors. These findings have been the subject of several recent comprehensive reviews, some of which are recapitulated in this book volume. In addition, several databases have been developed and are now publically available including [NURSA (http://www.nursa.org), Transcriptomine (http://www.nursa.org/transcriptomine) and IUPHAR (http://www.iuphar-db.org)].

Briefly, the novel findings discovered during the two last decades can be summarized as follows:
A large number of coactivator and corepressors have been identified and found to be components of multisubunit coregulator complexes exhibiting an ever-expanding diversity of enzymatic and epigenetic activities, exemplified by ATP-dependent nucleosome remodeling complexes, histone acetyl/deacetyl transferases (HATs and HDACs), histone methyl transferases (HMTs) and histone demethylases [39]. The development of the chromatin immunoprecipitation (ChIP) technique revealed that transcription of RAR-target genes is a dynamic process and that RA binding switches RARs from an inactive state to an active state by promoting the exchange of corepressor complexes for coactivators [95, 102]. The consummate effects of these coregulators are the modification, remodeling and decompaction of chromatin to pave the way for the recruitment of the transcription machinery [28]. Today new proteins and complexes are still continuously being discovered increasing our knowledge of the complexity of RAR-mediated transcription. Moreover, the combination of different biophysical methods [X-ray crystallography, small angle X-ray (SAXs), fluorescence resonance energy transfer (FRET), nuclear magnetic resonance (NMR)] and the use of several synthetic agonists or antagonists for RARs and RXRs is providing a view of the dynamic structure of RAR-RXR heterodimers associated with coactivators on different response elements [101].
Subsequent to the sequencing of the human genome, the development of genome-wide profiling technologies such as RNA-seq (high throughput qPCR sequencing) and ChIP-seq (chromatin immunoprecipitation coupled with deep sequencing) has allowed the identification of novel RA response elements with different spacings [88] and novel RA-target genes [59, 77]. The genome-wide integrative analysis of RAR/RXR binding and transcriptional regulation has also provided a dynamic view of RA signaling [84]. Recently, RARs have been found to target the expression of microRNAs, introducing yet another level of complexity in the regulation of RAR-regulated gene transcription [93, 135, 136]. These recent approaches and findings are identifying novel mechanisms of action for RARs and are opening up promising new avenues for research and development.
During the last two decades, the concept arose that posttranslational modifications such as phosphorylation and ubiquitination are crucial for RARs activity [2]. Early studies using phosphopeptide analysis performed with radioactivity and large amounts of recombinant receptors resulted in the identification of a number of phosphorylation sites on RARs and RXRs [1, 63, 103, 107]. Subsequently, the emergence of new methods for enrichment of phosphopeptide samples and development of phosphospecific antibodies provided the ability to analyze phosphorylation of endogenous RARs in response to their cognate ligand or signaling pathways [14]. Now it is clear that RARs, as well as several other proteins, are rapidly phosphorylated in response to RA, subsequent to RA-induced activation of kinase cascades via a pool of RARs that are present in membrane lipid rafts [80, 99]. A concept that is gaining support is that phosphorylations induce subtle changes in the conformation of the receptors that modulate the association/dissociation of new coregulators [21, 108]. Another developing concept is that phosphorylation is a signal for the degradation of RARs by the ubiquitin proteasome system, a process that signals the end of transcription [11].
In situ analysis of endogenous RAR protein expression profiles became possible with the generation of purified highly-specific antibodies [15, 124]. Moreover, the development of novel conditional gene targeting strategies based on the use of the Cre recombinase allowed the generation of somatic mutations in individual genes in a specific cell type and at a given time in the life of a transgenic mouse [85, 86]. This novel strategy has yielded remarkable advances in understanding the roles played by RARs and RXRs because it circumvents the limitations of previous transgenic approaches that led to early lethality and that were compromised by redundancies in receptor isoform expression in tissues under investigation [49, 69].
Future Directions
Today, 25 years after the cloning of RARs (Fig. 1.4), knowledge continues to evolve in the field of retinoid biology. The structures of RAR/RXR heterodimers bound to DNA have been solved, but they are still lacking the N-terminal domain which exhibits a quasi absence of defined secondary structures but confers considerable flexibility to RARs. The integration of data from several sources and from high-resolution biophysical approaches should provide the structure of the RAR/RXR heterodimers as full-length proteins bound to DNA with their coregulators intact.
A driving goal for future studies will be the discovery of the rules for cell fate specification integrated into a systems biology view of RAR/RXR actions and RA signaling. Application of computational models and programs to reconstruct differentiation-related gene networks obtained from different cell types should allow the prediction of RA-regulated gene network intricacies and the identification of key factors that direct cells towards a particular differentiation phenotype. Stem cells, which are pluripotent cells capable of generating all the differentiated cell types present in the body and which are responsive to RA, are currently, a most promising tool for such cell fate studies. Extrapolation of data generated from stem cell differentiation models should have applicability to a deeper understanding RAR-dysfunctional diseases that interfere with normal cell homeostasis and redirect normal cells to a more primitive, mitosis-driven state.
Recent findings are stretching the boundaries of our understanding of vitamin A action. These newer studies are indicating that the effects of vitamin A retinol and RA are not mediated only by RAR/RXR heterodimers and transcriptional processes, and this is opening up new avenues in the field. As an example, it has been found that RA can activate other nuclear receptors such as the peroxisome proliferator-activated receptor β/δ [110], providing a rationale for the long-noted, but poorly understood function of vitamin A in regulating energy balance. Moreover, recent findings are hinting that RA, as well as vitamin A itself, can have extranuclear, non-transcriptional effects and can activate kinase-signaling pathways [2, 10]. Consequently, one can speculate that, in addition to affecting the transcriptome, RA and retinol could also affect the phospho-proteome. Next generation, dual linear ion trap mass spectrometers coupled with Orbitrap technology should allow the identification of new panels of proteins that are phosphorylated in response to retinol or RA. The future objectives should be to integrate the RA-induced variations in the phospho-proteome with the transcriptome. Such an integrative study should pave the way to breakthroughs in disease-related research. The recent observation that RARs are present in the cytosol of specific cell types [22, 76] continues to open new areas in the mechanisms of action of vitamin A and RA.
Great progress has been made in deciphering how specific molecules and signaling pathways interact to mediate vitamin A/RA action. But much is left to be done to fully understand the complexities of their action at the cellular and sub-cellular levels and of their regulation in time and space throughout the life of an organism.
Abbreviations
- ChIP:
-
Chromatin immunoprecipitation
- ChIP-seq:
-
Chromatin immunoprecipitation coupled with deep sequencing
- cDNA:
-
Complementary DNA
- CRABP:
-
Cellular retinoic acid binding protein
- CRBP:
-
Cellular retinol binding protein
- DNA:
-
Deoxyribonucleic acid
- DBD:
-
DNA binding domain
- LBD:
-
Ligand binding domain
- NMR:
-
Nuclear magnetic resonance
- RA:
-
Retinoic acid
- RAR:
-
Retinoic acid receptor
- RARE:
-
Retinoic acid response element
- RNA:
-
Ribonucleic acid
- RNA-seq:
-
High throughput RNA sequencing
- RXR:
-
Retinoic X receptor
- VAD:
-
Vitamin A deficiency
References
Adam-Stitah S, Penna L, Chambon P, Rochette-Egly C (1999) Hyperphosphorylation of the retinoid X receptor alpha by activated c-Jun NH2-terminal kinases. J Biol Chem 274:18932–18941
Al Tanoury Z, Piskunov A, Rochette-Egly C (2013) Vitamin A and retinoid signaling: genomic and nongenomic effects: thematic review Series: Fat-Soluble Vitamins: Vitamin A. J Lipid Res 54:1761–1775
Almasan A, Mangelsdorf DJ, Ong ES, Wahl GM, Evans RM (1994) Chromosomal localization of the human retinoid X receptors. Genomics 20:397–403
Arens JF, Van Dorp DA (1946) Activity of vitamin A-acid in the rat. Nature 158:622
Arens JF, Van Dorp DA (1946) Synthesis of some compounds possessing vitamin A activity. Nature 157:190
Ashburner M, Chihara C, Meltzer P, Richards G (1974) Temporal control of puffing activity in polytene chromosomes. Cold Spring Harb Symp Quant Biol 38:655–662
Ball S, Goodwin TW, Morton RA (1948) Studies on vitamin A; the preparation of retinene1, vitamin A aldehyde. Biochem J 42:516–523
Bashor MM, Toft DO, Chytil F (1973) In vitro binding of retinol to rat-tissue components. Proc Natl Acad Sci USA 70:3483–3487
Benbrook D, Lernhardt E, Pfahl M (1988) A new retinoic acid receptor identified from a hepatocellular carcinoma. Nature 333:669–672
Berry DC, Noy N (2012) Signaling by vitamin A and retinol-binding protein in regulation of insulin responses and lipid homeostasis. Biochim Biophys Acta 1821:168–176
Bour G, Lalevee S, Rochette-Egly C (2007) Protein kinases and the proteasome join in the combinatorial control of transcription by nuclear retinoic acid receptors. Trends Cell Biol 17:302–309
Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D (1995) Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-alpha. Nature 375:377–382
Brand N, Petkovich M, Krust A, Chambon P, de The H, Marchio A, Tiollais P, Dejean A (1988) Identification of a second human retinoic acid receptor. Nature 332:850–853
Bruck N, Vitoux D, Ferry C, Duong V, Bauer A, de The H, Rochette-Egly C (2009) A coordinated phosphorylation cascade initiated by p38MAPK/MSK1 directs RARalpha to target promoters. EMBO J 28:34–47
Buchanan FQ, Rochette-Egly C, Asson-Batres MA (2011) Detection of variable levels of RARalpha and RARgamma proteins in pluripotent and differentiating mouse embryonal carcinoma and mouse embryonic stem cells. Cell Tissue Res 346:43–51
Carpenter KJ (2012) The discovery of thiamin. Ann Nutr Metab 61:219–223
Carpenter KJ, Sutherland B (1995) Eijkman’s contribution to the discovery of vitamins. J Nutr 125:155–163
Chambon P (1996) A decade of molecular biology of retinoic acid receptors. Faseb J 10:940–954
Chambon P (2004) How I became one of the fathers of a superfamily. Nat Med 10:1027–1031
Chandler VL, Maler BA, Yamamoto KR (1983) DNA sequences bound specifically by glucocorticoid receptor in vitro render a heterologous promoter hormone responsive in vivo. Cell 33:489–499
Chebaro Y, Amal I, Rochel N, Rochette-Egly C, Stote R, Dejaegere A (2013) Phosphorylation of the Retinoic Acid Receptor alpha induces a mechanical allosteric regulation and changes in internal dynamics. Plos Comp Biol 9:e1003012
Chen N, Napoli JL (2008) All-trans-retinoic acid stimulates translation and induces spine formation in hippocampal neurons through a membrane-associated RARalpha. Faseb J 22:236–245
Chytil F, Ong DE (1978) Cellular vitamin A binding proteins. Vitam Horm 36:1–32
De Luca L, Little EP, Wolf G (1969) Vitamin A and protein synthesis by rat intestinal mucosa. J Biol Chem 244:701–708
de The H, Marchio A, Tiollais P, Dejean A (1987) A novel steroid thyroid hormone receptor-related gene inappropriately expressed in human hepatocellular carcinoma. Nature 330:667–670
de The H, Vivanco-Ruiz MM, Tiollais P, Stunnenberg H, Dejean A (1990) Identification of a retinoic acid responsive element in the retinoic acid receptor beta gene. Nature 343:177–180
Deisseroth A, Nienhuis A, Turner P, Velez R, Anderson WF, Ruddle F, Lawrence J, Creagan R, Kucherlapati R (1977) Localization of the human alpha-globin structural gene to chromosome 16 in somatic cell hybrids by molecular hybridization assay. Cell 12:205–218
Dilworth FJ, Chambon P (2001) Nuclear receptors coordinate the activities of chromatin remodeling complexes and coactivators to facilitate initiation of transcription. Oncogene 20:3047–3054
Drummond JC (1920) The nomenclature of the so-called accessory food factors (Vitamins). Biochem J 14:660
Durand B, Saunders M, Gaudon C, Roy B, Losson R, Chambon P (1994) Activation function 2 (AF-2) of retinoic acid receptor and 9-cis retinoic acid receptor: presence of a conserved autonomous constitutive activating domain and influence of the nature of the response element on AF-2 activity. EMBO J 13:5370–5382
Durand B, Saunders M, Leroy P, Leid M, Chambon P (1992) All-trans and 9-cis retinoic acid induction of CRABPII transcription is mediated by RAR-RXR heterodimers bound to DR1 and DR2 repeated motifs. Cell 71:73–85
Gaub MP, Lutz Y, Ruberte E, Petkovich M, Brand N, Chambon P (1989) Antibodies specific to the retinoic acid human nuclear receptors alpha and beta. Proc Natl Acad Sci USA 86:3089–3093
Gaub MP, Rochette-Egly C, Lutz Y, Ali S, Matthes H, Scheuer I, Chambon P (1992) Immunodetection of multiple species of retinoic acid receptor alpha: evidence for phosphorylation. Exp Cell Res 201:335–346
Germain P, Chambon P, Eichele G, Evans RM, Lazar MA, Leid M, De Lera AR, Lotan R, Mangelsdorf DJ, Gronemeyer H (2006) International union of pharmacology. LXIII. Retinoid X receptors. Pharmacol Rev 58:760–772
Germain P, Staels B, Dacquet C, Spedding M, Laudet V (2006) Overview of nomenclature of nuclear receptors. Pharmacol Rev 58:685–704
Giguere V, Ong ES, Segui P, Evans RM (1987) Identification of a receptor for the morphogen retinoic acid. Nature 330:624–629
Giguere V, Shago M, Zirngibl R, Tate P, Rossant J, Varmuza S (1990) Identification of a novel isoform of the retinoic acid receptor gamma expressed in the mouse embryo. Mol Cell Biol 10:2335–2340
Glass CK, Devary OV, Rosenfeld MG (1990) Multiple cell type-specific proteins differentially regulate target sequence recognition by the alpha retinoic acid receptor. Cell 63:729–738
Glass CK, Rosenfeld MG (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 14:121–141
Gorry P, Lufkin T, Dierich A, Rochette-Egly C, Decimo D, Dolle P, Mark M, Durand B, Chambon P (1994) The cellular retinoic acid binding protein I is dispensable. Proc Natl Acad Sci USA 91:9032–9036
Govindan MV, Devic M, Green S, Gronemeyer H, Chambon P (1985) Cloning of the human glucocorticoid receptor cDNA. Nucleic Acids Res 13:8293–8304
Hamada K, Gleason SL, Levi BZ, Hirschfeld S, Appella E, Ozato K (1989) H-2RIIBP, a member of the nuclear hormone receptor superfamily that binds to both the regulatory element of major histocompatibility class I genes and the estrogen response element. Proc Natl Acad Sci USA 86:8289–8293
Haq R, Chytil F (1988) Retinoic acid rapidly induces lung cellular retinol-binding protein mRNA levels in retinol deficient rats. Biochem Biophys Res Commun 156:712–716
Heilbron IM, Morton RA, Webster ET (1932) The structure of vitamin A. Biochem J 26:1194–1196
Heyman RA, Mangelsdorf DJ, Dyck JA, Stein RB, Eichele G, Evans RM, Thaller C (1992) 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell 68:397–406
Hoffmann B, Lehmann JM, Zhang XK, Hermann T, Husmann M, Graupner G, Pfahl M (1990) A retinoic acid receptor-specific element controls the retinoic acid receptor-beta promoter. Mol Endocrinol 4:1727–1736
Holmes HN, Corbet RE (1937) A crystalline Vitamin a concentrate. Science 85:103
Hubbard R, Wald G (1951) The mechanism of rhodopsin synthesis. Proc Natl Acad Sci USA 37:69–79
Imai T, Jiang M, Kastner P, Chambon P, Metzger D (2001) Selective ablation of retinoid X receptor alpha in hepatocytes impairs their lifespan and regenerative capacity. Proc Natl Acad Sci USA 98:4581–4586
Ishikawa T, Umesono K, Mangelsdorf DJ, Aburatani H, Stanger BZ, Shibasaki Y, Imawari M, Evans RM, Takaku F (1990) A functional retinoic acid receptor encoded by the gene on human chromosome 12. Mol Endocrinol 4:837–844
Jacobson HI, Gupta GN, Fernandez C, Hennix S, Jensen EV (1960) Determination of tritium in biological material. Arch Biochem Biophys 86:89–93
Jensen EV (1962) On the mechanism of estrogen action. Perspect Biol Med 6:47–59
Jensen EV, Suzuki T, Kawashima T, Stumpf WE, Jungblut PW, DeSombre ER (1968) A two-step mechanism for the interaction of estradiol with rat uterus. Proc Natl Acad Sci USA 59:632–638
Johnson BC, Kennedy M, Chiba N (1969) Vitamin A and nuclear RNA synthesis. Am J Clin Nutr 22:1048–1058
Karrer P, Morf R, Schöpp K (1931) Zur Kenntnis des Vitamins-A aus Fischtranen. Helv Chim Acta 14:1036–1040
Kastner P, Grondona JM, Mark M, Gansmuller A, LeMeur M, Decimo D, Vonesch JL, Dolle P, Chambon P (1994) Genetic analysis of RXR alpha developmental function: convergence of RXR and RAR signaling pathways in heart and eye morphogenesis. Cell 78:987–1003
Kastner P, Krust A, Mendelsohn C, Garnier JM, Zelent A, Leroy P, Staub A, Chambon P (1990) Murine isoforms of retinoic acid receptor gamma with specific patterns of expression. Proc Natl Acad Sci USA 87:2700–2704
Krust A, Kastner P, Petkovich M, Zelent A, Chambon P (1989) A third human retinoic acid receptor, hRAR-gamma. Proc Natl Acad Sci USA 86:5310–5314
Lalevee S, Anno YN, Chatagnon A, Samarut E, Poch O, Laudet V, Benoit G, Lecompte O, Rochette-Egly C (2011) Genome-wide in silico identification of new conserved and functional retinoic acid receptor response elements (direct repeats separated by 5 bp). J Biol Chem 286:33322–33334
Lampron C, Rochette-Egly C, Gorry P, Dolle P, Mark M, Lufkin T, LeMeur M, Chambon P (1995) Mice deficient in cellular retinoic acid binding protein II (CRABPII) or in both CRABPI and CRABPII are essentially normal. Development 121:539–548
Laudet V, Gronemeyer H (2001) Nuclear receptor factsbook. Acedemic, London
Le Douarin B, Zechel C, Garnier JM, Lutz Y, Tora L, Pierrat P, Heery D, Gronemeyer H, Chambon P, Losson R (1995) The N-terminal part of TIF1, a putative mediator of the ligand-dependent activation function (AF-2) of nuclear receptors, is fused to B-raf in the oncogenic protein T18. EMBO J 14:2020–2033
Lee HY, Suh YA, Robinson MJ, Clifford JL, Hong WK, Woodgett JR, Cobb MH, Mangelsdorf DJ, Kurie JM (2000) Stress pathway activation induces phosphorylation of retinoid X receptor. J Biol Chem 275:32193–32199
Lee MS, Kliewer SA, Provencal J, Wright PE, Evans RM (1993) Structure of the retinoid X receptor alpha DNA binding domain: a helix required for homodimeric DNA binding. Science 260:1117–1121
Leid M, Kastner P, Chambon P (1992) Multiplicity generates diversity in the retinoic acid signalling pathways. Trends Biochem Sci 17:427–433
Leid M, Kastner P, Lyons R, Nakshatri H, Saunders M, Zacharewski T, Chen JY, Staub A, Garnier JM, Mader S et al (1992) Purification, cloning, and RXR identity of the HeLa cell factor with which RAR or TR heterodimerizes to bind target sequences efficiently. Cell 68:377–395
Leroy P, Krust A, Zelent A, Mendelsohn C, Garnier JM, Kastner P, Dierich A, Chambon P (1991) Multiple isoforms of the mouse retinoic acid receptor alpha are generated by alternative splicing and differential induction by retinoic acid. EMBO J 10:59–69
Levin AA, Sturzenbecker LJ, Kazmer S, Bosakowski T, Huselton C, Allenby G, Speck J, Kratzeisen C, Rosenberger M, Lovey A et al (1992) 9-cis retinoic acid stereoisomer binds and activates the nuclear receptor RXR alpha. Nature 355:359–361
Li M, Indra AK, Warot X, Brocard J, Messaddeq N, Kato S, Metzger D, Chambon P (2000) Skin abnormalities generated by temporally controlled RXRalpha mutations in mouse epidermis. Nature 407:633–636
Liau G, Ong DE, Chytil F (1981) Interaction of the retinol/cellular retinol-binding protein complex with isolated nuclei and nuclear components. J Cell Biol 91:63–68
Liau G, Ong DE, Chytil F (1985) Partial characterization of nuclear binding sites for retinol delivered by cellular retinol binding protein. Arch Biochem Biophys 237:354–360
Lohnes D, Kastner P, Dierich A, Mark M, LeMeur M, Chambon P (1993) Function of retinoic acid receptor gamma in the mouse. Cell 73:643–658
Lohnes D, Mark M, Mendelsohn C, Dolle P, Dierich A, Gorry P, Gansmuller A, Chambon P (1994) Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development 120:2723–2748
Lufkin T, Lohnes D, Mark M, Dierich A, Gorry P, Gaub MP, LeMeur M, Chambon P (1993) High postnatal lethality and testis degeneration in retinoic acid receptor alpha mutant mice. Proc Natl Acad Sci USA 90:7225–7229
Luo J, Pasceri P, Conlon RA, Rossant J, Giguere V (1995) Mice lacking all isoforms of retinoic acid receptor beta develop normally and are susceptible to the teratogenic effects of retinoic acid. Mech Dev 53:61–71
Maghsoodi B, Poon MM, Nam CI, Aoto J, Ting P, Chen L (2008) Retinoic acid regulates RARalpha-mediated control of translation in dendritic RNA granules during homeostatic synaptic plasticity. Proc Natl Acad Sci USA 105:16015–16020
Mahony S, Mazzoni EO, McCuine S, Young RA, Wichterle H, Gifford DK (2011) Ligand-dependent dynamics of retinoic acid receptor binding during early neurogenesis. Genome Biol 12:R2
Mangelsdorf DJ, Borgmeyer U, Heyman RA, Zhou JY, Ong ES, Oro AE, Kakizuka A, Evans RM (1992) Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes Dev 6:329–344
Mangelsdorf DJ, Ong ES, Dyck JA, Evans RM (1990) Nuclear receptor that identifies a novel retinoic acid response pathway. Nature 345:224–229
Masia S, Alvarez S, de Lera AR, Barettino D (2007) Rapid, nongenomic actions of retinoic acid on phosphatidylinositol-3-kinase signaling pathway mediated by the retinoic acid receptor. Mol Endocrinol 21:2391–2402
Mattei MG, Riviere M, Krust A, Ingvarsson S, Vennstrom B, Islam MQ, Levan G, Kautner P, Zelent A, Chambon P et al (1991) Chromosomal assignment of retinoic acid receptor (RAR) genes in the human, mouse, and rat genomes. Genomics 10:1061–1069
McKnight GS, Palmiter RD (1979) Transcriptional regulation of the ovalbumin and conalbumin genes by steroid hormones in chick oviduct. J Biol Chem 254:9050–9058
Mendelsohn C, Lohnes D, Decimo D, Lufkin T, LeMeur M, Chambon P, Mark M (1994) Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 120:2749–2771
Mendoza-Parra MA, Walia M, Sankar M, Gronemeyer H (2011) Dissecting the retinoid-induced differentiation of F9 embryonal stem cells by integrative genomics. Mol Syst Biol 7:538
Metzger D, Chambon P (2001) Site- and time-specific gene targeting in the mouse. Methods 24:71–80
Metzger D, Clifford J, Chiba H, Chambon P (1995) Conditional site-specific recombination in mammalian cells using a ligand-dependent chimeric Cre recombinase. Proc Natl Acad Sci USA 92:6991–6995
Moras D, Gronemeyer H (1998) The nuclear receptor ligand-binding domain: structure and function. Curr Opin Cell Biol 10:384–391
Moutier E, Ye T, Choukrallah MA, Urban S, Osz J, Chatagnon A, Delacroix L, Langer D, Rochel N, Moras D, Benoit G, Davidson I (2012) Retinoic acid receptors recognise the mouse genome through binding elements with diverse spacing and topology. J Biol Chem 287:26328–26341
Nagpal S, Zelent A, Chambon P (1992) RAR-beta 4, a retinoic acid receptor isoform is generated from RAR-beta 2 by alternative splicing and usage of a CUG initiator codon. Proc Natl Acad Sci USA 89:2718–2722
Ong DE, Chytil F (1974) Multiple retinol binding proteins in rabbit lung. Biochem Biophys Res Commun 59:221–229
Ong DE, Chytil F (1978) Cellular retinoic acid-binding protein from rat testis. Purification and characterization. J Biol Chem 253:4551–4554
Ong DE, Chytil F (1978) Cellular retinol-binding protein from rat liver. Purification and characterization. J Biol Chem 253:828–832
Pelosi A, Careccia S, Lulli V, Romania P, Marziali G, Testa U, Lavorgna S, Lo-Coco F, Petti MC, Calabretta B, Levrero M, Piaggio G, Rizzo MG (2013) miRNA let-7c promotes granulocytic differentiation in acute myeloid leukemia. Oncogene 32:3648–3654
Perez E, Bourguet W, Gronemeyer H, de Lera AR (2012) Modulation of RXR function through ligand design. Biochim Biophys Acta 1821:57–69
Perissi V, Rosenfeld MG (2005) Controlling nuclear receptors: the circular logic of cofactor cycles. Nat Rev Mol Cell Biol 6:542–554
Peterkofsky B, Tomkins GM (1968) Evidence for the steroid-induced accumulation of tyrosine-aminotransferase messenger RNA in the absence of protein synthesis. Proc Natl Acad Sci USA 60:222–228
Petkovich M, Brand NJ, Krust A, Chambon P (1987) A human retinoic acid receptor which belongs to the family of nuclear receptors. Nature 330:444–450
Piro A, Tagarelli G, Lagonia P, Tagarelli A, Quattrone A (2010) Casimir funk: his discovery of the vitamins and their deficiency disorders. Ann Nutr Metab 57:85–88
Piskunov A, Rochette-Egly C (2012) A retinoic acid receptor RARalpha pool present in membrane lipid rafts forms complexes with G protein alphaQ to activate p38MAPK. Oncogene 31:3333–3345
Renaud JP, Rochel N, Ruff M, Vivat V, Chambon P, Gronemeyer H, Moras D (1995) Crystal structure of the RAR-gamma ligand-binding domain bound to all-trans retinoic acid. Nature 378:681–689
Rochel N, Ciesielski F, Godet J, Moman E, Roessle M, Peluso-Iltis C, Moulin M, Haertlein M, Callow P, Mely Y, Svergun DI, Moras D (2011) Common architecture of nuclear receptor heterodimers on DNA direct repeat elements with different spacings. Nat Struct Mol Biol 18:564–570
Rochette-Egly C (2005) Dynamic combinatorial networks in nuclear receptor-mediated transcription. J Biol Chem 280:32565–32568
Rochette-Egly C, Adam S, Rossignol M, Egly JM, Chambon P (1997) Stimulation of RAR alpha activation function AF-1 through binding to the general transcription factor TFIIH and phosphorylation by CDK7. Cell 90:97–107
Rochette-Egly C, Gaub MP, Lutz Y, Ali S, Scheuer I, Chambon P (1992) Retinoic acid receptor-beta: immunodetection and phosphorylation on tyrosine residues. Mol Endocrinol 6:2197–2209
Rochette-Egly C, Lutz Y, Pfister V, Heyberger S, Scheuer I, Chambon P, Gaub MP (1994) Detection of retinoid X receptors using specific monoclonal and polyclonal antibodies. Biochem Biophys Res Commun 204:525–536
Rochette-Egly C, Lutz Y, Saunders M, Scheuer I, Gaub MP, Chambon P (1991) Retinoic acid receptor gamma: specific immunodetection and phosphorylation. J Cell Biol 115:535–545
Rochette-Egly C, Oulad-Abdelghani M, Staub A, Pfister V, Scheuer I, Chambon P, Gaub MP (1995) Phosphorylation of the retinoic acid receptor-alpha by protein kinase A. Mol Endocrinol 9:860–871
Samarut E, Amal I, Markov G, Stote R, Dejaegere A, Laudet V, Rochette-Egly C (2011) Evolution of nuclear retinoic acid receptors alpha (RARa) phosphorylation sites. Serine gain provides fine-tuned regulation. Mol Biol Evol 28:2125–2137
Scheidereit C, Geisse S, Westphal HM, Beato M (1983) The glucocorticoid receptor binds to defined nucleotide sequences near the promoter of mouse mammary tumour virus. Nature 304:749–752
Schug TT, Berry DC, Shaw NS, Travis SN, Noy N (2007) Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 129:723–733
Semba RD (2012) On the ‘discovery’ of vitamin A. Ann Nutr Metab 61:192–198
Sim GK, Kafatos FC, Jones CW, Koehler MD, Efstratiadis A, Maniatis T (1979) Use of a cDNA library for studies on evolution and developmental expression of the chorion multigene families. Cell 18:1303–1316
Simoni RD, Hill RH, Vaughan M (2002) Nutritional biochemistry and the discovery of Vitamins: the work of Elmer Verner McCollum. J Biol Chem 277:e8
Singh H, LeBowitz JH, Baldwin AS Jr, Sharp PA (1988) Molecular cloning of an enhancer binding protein: isolation by screening of an expression library with a recognition site DNA. Cell 52:415–423
Smith WC, Nakshatri H, Leroy P, Rees J, Chambon P (1991) A retinoic acid response element is present in the mouse cellular retinol binding protein I (mCRBPI) promoter. EMBO J 10:2223–2230
Spiegelman S, Watson KF, Kacian DL (1971) Synthesis of DNA complements of natural RNAs: a general approach. Proc Natl Acad Sci USA 68:2843–2845
Sporn MB, Dunlop NM, Newton DL, Smith JM (1976) Prevention of chemical carcinogenesis by vitamin A and its synthetic analogs (retinoids). Fed Proc 35:1332–1338
Sucov HM, Dyson E, Gumeringer CL, Price J, Chien KR, Evans RM (1994) RXR alpha mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev 8:1007–1018
Sucov HM, Murakami KK, Evans RM (1990) Characterization of an autoregulated response element in the mouse retinoic acid receptor type beta gene. Proc Natl Acad Sci USA 87:5392–5396
Takase S, Ong DE, Chytil F (1979) Cellular retinol-binding protein allows specific interaction of retinol with the nucleus in vitro. Proc Natl Acad Sci USA 76:2204–2208
Takase S, Ong DE, Chytil F (1986) Transfer of retinoic acid from its complex with cellular retinoic acid-binding protein to the nucleus. Arch Biochem Biophys 247:328–334
Temin HM, Baltimore D (1972) RNA-directed DNA synthesis and RNA tumor viruses. Adv Virus Res 17:129–186
Van Dorp DA, Arens JF (1946) Biological activity of vitamin A acid. Nature 158:60
Vernet N, Dennefeld C, Rochette-Egly C, Oulad-Abdelghani M, Chambon P, Ghyselinck NB, Mark M (2006) Retinoic acid metabolism and signaling pathways in the adult and developing mouse testis. Endocrinology 147:96–110
Vinson CR, LaMarco KL, Johnson PF, Landschulz WH, McKnight SL (1988) In situ detection of sequence-specific DNA binding activity specified by a recombinant bacteriophage. Genes Dev 2:801–806
Voegel JJ, Heine MJ, Zechel C, Chambon P, Gronemeyer H (1996) TIF2, a 160 kDa transcriptional mediator for the ligand-dependent activation function AF-2 of nuclear receptors. EMBO J 15:3667–3675
vom Baur E, Zechel C, Heery D, Heine MJ, Garnier JM, Vivat V, Le Douarin B, Gronemeyer H, Chambon P, Losson R (1996) Differential ligand-dependent interactions between the AF-2 activating domain of nuclear receptors and the putative transcriptional intermediary factors mSUG1 and TIF1. EMBO J 15:110–124
Wald G (1948) The synthesis from vitamin A1 of retinene 1 and of a new 545 m-mu chromogen yielding light-sensitive products. J Gen Physiol 31:489–504
Walter P, Green S, Greene G, Krust A, Bornert JM, Jeltsch JM, Staub A, Jensen E, Scrace G, Waterfield M et al (1985) Cloning of the human estrogen receptor cDNA. Proc Natl Acad Sci USA 82:7889–7893
Weill JD, Busch S, Chambon P, Mandel P (1963) The effect of estradiol injections upon chicken liver nuclei ribonucleic acid polymerase. Biochem Biophys Res Commun 10:122–126
Willecke K, Ruddle FH (1975) Transfer of the human gene for hypoxanthine-guanine phosphoribosyltransferase via isolated human metaphase chromosomes into mouse L-cells. Proc Natl Acad Sci USA 72:1792–1796
Wrange O, Gustafsson JA (1978) Separation of the hormone- and DNA-binding sites of the hepatic glucocorticoid receptor by means of proteolysis. J Biol Chem 253:856–865
Yu VC, Delsert C, Andersen B, Holloway JM, Devary OV, Naar AM, Kim SY, Boutin JM, Glass CK, Rosenfeld MG (1991) RXR beta: a coregulator that enhances binding of retinoic acid, thyroid hormone, and vitamin D receptors to their cognate response elements. Cell 67:1251–1266
Zachman RD (1967) The stimulation of RNA synthesis in vivo and in vitro by retinol (vitamin A) in the intestine of vitamin A deficient rats. Life Sci 6:2207–2213
Zardo G, Ciolfi A, Vian L, Billi M, Racanicchi S, Grignani F, Nervi C (2012) Transcriptional targeting by microRNA-polycomb complexes: a novel route in cell fate determination. Cell Cycle 11:3543–3549
Zardo G, Ciolfi A, Vian L, Starnes LM, Billi M, Racanicchi S, Maresca C, Fazi F, Travaglini L, Noguera N, Mancini M, Nanni M, Cimino G, Lo-Coco F, Grignani F, Nervi C (2012) Polycombs and microRNA-223 regulate human granulopoiesis by transcriptional control of target gene expression. Blood 119:4034–4046
Zelent A, Krust A, Petkovich M, Kastner P, Chambon P (1989) Cloning of murine alpha and beta retinoic acid receptors and a novel receptor gamma predominantly expressed in skin. Nature 339:714–717
Zelent A, Mendelsohn C, Kastner P, Krust A, Garnier JM, Ruffenach F, Leroy P, Chambon P (1991) Differentially expressed isoforms of the mouse retinoic acid receptor beta generated by usage of two promoters and alternative splicing. EMBO J 10:71–81
Acknowledgments
We thank Gabriel R Batres for assistance with the design and preparation of Fig. 1.4.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Benbrook, D.M., Chambon, P., Rochette-Egly, C., Asson-Batres, M.A. (2014). History of Retinoic Acid Receptors. In: Asson-Batres, M., Rochette-Egly, C. (eds) The Biochemistry of Retinoic Acid Receptors I: Structure, Activation, and Function at the Molecular Level. Subcellular Biochemistry, vol 70. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-9050-5_1
Download citation
DOI: https://doi.org/10.1007/978-94-017-9050-5_1
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-9049-9
Online ISBN: 978-94-017-9050-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)