
Abstract
The liver is a truly remarkable organ when one considers the diversity of its many functions. This organ has a central role in nutrient (carbohydrate, protein and lipid) and vitamin metabolism and serves as an intermediary between dietary sources of energy and the extrahepatic tissues, which utilize such energy. Another major function is the detoxification and elimination of xenobiotic toxins and drugs, as well as endogenous metabolites, such as bilirubin. The liver is responsible for the synthesis of biologically important proteins (i.e., albumin, transferrin, clotting factors, complement factors, apolipoproteins, etc.). The liver synthesizes and secretes the components of bile, including bile acids, which are important for the intestinal solubilization and absorption of the products of lipid digestion. Finally, the liver has an important role in immune function, due primarily to its resident macrophage, the Kupffer cell.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The liver is a truly remarkable organ when one considers the diversity of its many functions. This organ has a central role in nutrient (carbohydrate, protein and lipid) and vitamin metabolism and serves as an intermediary between dietary sources of energy and the extrahepatic tissues, which utilize such energy. Another major function is the detoxification and elimination of xenobiotic toxins and drugs, as well as endogenous metabolites, such as bilirubin. The liver is responsible for the synthesis of biologically important proteins (i.e., albumin, transferrin, clotting factors, complement factors, apolipoproteins , etc.). The liver synthesizes and secretes the components of bile, including bile acids, which are important for the intestinal solubilization and absorption of the products of lipid digestion. Finally, the liver has an important role in immune function, due primarily to its resident macrophage, the Kupffer cell.
2 Liver Anatomy and Blood Supply
In order to efficiently carry out its myriad functions, the liver has unique requirements with regard to anatomic location, blood supply, and structural organization. The liver is the largest solid organ in the body, accounting for 3–5 % of body mass. The liver is subdivided into two main lobes (right and left) and two accessory lobes (quadrate and caudate) (Fig. 10.1). These lobes are supplied with blood by the right and left branches of the portal vein and hepatic artery, and bile is drained by the right and left hepatic bile ducts. Of more significance from a functional standpoint is the segmental anatomy, whereby the liver is divided into eight segments, each with distinct blood supplies and biliary drainage without collateral circulation between segments (Fig. 10.2). The liver receives approximately 28 % of the body’s total blood flow and consumes 20 % of the total oxygen used by the body. The liver is situated as a “sentinel” for the interception and processing of nutrients, toxins and bile acids from the intestine because of its unique dual blood supply. The liver receives all of the venous blood draining from the splanchnic vascular bed by way of the portal vein (Fig. 10.3). Seventy-five percent of the incoming blood volume to the liver is supplied from the portal vein. Normally, the portal system is a low-pressure (5–10 mmHg) system designed to optimally deliver the venous blood for flow through the hepatic sinusoids to allow the most efficient exposure to the hepatocyte plasma membrane. The liver receives oxygenated arterial blood from the hepatic artery. A portion of the blood from the terminal branches of the hepatic arterial system flows through plexuses surrounding the interlobular bile ducts before entering the sinusoids. Individuals who have received liver transplants may survive with normal liver function after complete thrombosis of the hepatic artery because oxygen delivered by the portal venous blood is adequate. However, the dependence of the bile ducts on hepatic arterial blood is underscored by the fact that these individuals often experience degeneration and loss of bile ducts after arterial thrombosis.

Anterior (left) and posteroinferior (right) views of the liver (Reproduced with permission from Elsevier [14])

Segmental anatomy of the liver (Reproduced with permission from Elsevier [14])

(a) Prehepatic portal venous system. 1 portal vein, 2 splenic vein, 3 superior mesenteric vein, 4 inferior mesenteric vein, 5 coronary vein. (b) Normal portal venous system shown by splenoportography. The contrast material immediately opacifies the splenic and portal veins (arrows). The intrahepatic branches of the portal system are also well visualized (Reproduced with permission from Silverman and Roy [19])
2.1 Microanatomy of the Liver
The liver comprises several cells types in addition to hepatocytes . The hepatocytes make up approximately 80 % of the volume of the liver and approximately 60 % of the total hepatic cell population. Mesenchymal cells , including Kupffer cells, stellate (fat-storing or Ito) cells, and sinusoidal endothelial cells, comprise 30–35 % of the total population, and bile ductular epithelial cells contribute 3–5 %. Liver cells are arranged in a polarized fashion on a basement membrane in the liver lobule forming cords of cells (Fig. 10.4). As shown in Fig. 10.5, hepatocytes are complex cells containing the necessary subcellular machinery to function in synthesis, processing and secretion, as well as degradation and oxidation of a variety of substrates. This is reflected by the presence of highly developed endoplasmic reticulum, mitochondria, peroxisomes, lysosomes, Golgi complex and cytoskeleton. Hepatocytes have a specialized cell membrane domain called the canaliculus (Fig. 10.5), which functions in the secretion of bile constituents from the hepatocyte. Newly formed bile coalesces in bile ductules (<20 μm diameter), which empty into progressively larger biliary channels to finally empty into the common bile duct.

Liver lobule. The central vein (CV) lies in the center of the figure, surrounded by anastomosing cords of block-like hepatocytes . Around the periphery are six portal areas (PA) consisting of branches of the portal vein, the hepatic artery, and the bile duct (Reproduced with permission from Jones and Spring-Mills [12])

Hepatocyte cellular (a) ultrastructure and (b) organization. BC bile canaliculus, G Golgi apparatus, L lysosome, M mitochondrion, MV microvilli, N nucleus, NE nuclear envelope, P peroxisome, PM plasma membrane, RER rough endoplasmic reticulum. (a: Reproduced with permission from Hanover [9]; b: Reproduced with permission [10])
Other non-hepatocyte (non-parenchymal) cells in the liver include stellate (fat-storing or Ito) cells, Kupffer cells, pit cells and sinusoidal endothelial cells. Stellate cells are located in the perisinusoidal space, usually in intimate contact with endothelial cells via cytoplasmic processes and are concentrated in periportal areas. As will subsequently be discussed, these cells function in the storage of vitamin A. Stellate cells also produce extracellular matrix components, as well as matrix degradative enzymes, and appear to play a role in the remodeling and maintenance of the extracellular matrix. However, when stimulated by cytokines and other factors, these cells are also responsible for hepatic fibrosis leading to cirrhosis . Stellate cells also are capable of Ca2+-dependent contractility and may function in regulation of sinusoidal blood flow. Kupffer cells are the liver’s resident macrophages and are located within the sinusoidal lumen. These cells phagocytize dead cells, as well as debris and microorganisms brought in by the portal blood from the intestine. When activated by substances such as bacterial endotoxin, Kupffer cells secrete cytokines, such as tumor necrosis factor-α, interleukin-1, and interleukin-6, which can modulate several hepatocyte functions, including synthesis of acute phase reactants (fibrinogen , α-1-antitrypsin, and serum amyloid A),albumin, and apolipoproteins , as well as lipid metabolism. These soluble factors also affect the metabolism of sinusoidal endothelial cells. Pit cells are intrasinusoidal lymphocytes with natural killer (NK) cell activity and play a role in surveillance for viral infection and malignancy. Sinusoidal endothelial cells line the hepatic sinusoids and are fenestrated to allow macromolecular permeation (Fig. 10.6). These cells can respond to humoral factors to change the size of these fenestrations to regulate permeability. These endothelial cells also are very metabolically active and have numerous other functions, including active transport , participation in coagulation, fibrinolysis, and immune responses. Sinusoidal endothelial cells secrete nitric oxide (NO) (vasodilation) and endothelin (vasoconstriction) and function in tandem with the stellate cell to regulate sinusoidal vascular tone and pressure.

Scanning electron photomicrograph of a sinusoid illustrating sieve plates comprising clusters of endothelial fenestrae. Areas of endothelium lacking fenestrae (asterisks) overlay processes of stellate cells. E endothelial cell, SD space of Disse containing microvilli extending from hepatic parenchymal cells (H) (Reproduced with permission from McCuskey [13])
The liver lobule is a histologic and functional unit of the liver (Fig. 10.4). A typical lobule is hexagonal in cross section with interlobular portal triads (each containing branches of the portal vein, hepatic artery, and bile duct) at the angles of the hexagon. In the center of the lobule is the central vein or terminal hepatic venule. Hepatocytes are arranged as single cell plates radiating from the central vein to the portal triads in the periphery of the lobule. These plates are located between the endothelial-lined sinusoids, which serve as vascular channels to carry blood from the vessels in the portal triad at the periphery of the lobule to the central vein in the center of the lobule. As discussed previously, the endothelial cells lining the sinusoids are fenestrated and highly permeable to macromolecules. This allows optimal exposure of hepatocyte plasma membranes to the mixed arterial-portal venous blood, as well as allowing the unimpeded movement of larger particles, such as lipoproteins, to and from the hepatocytes .
Another conceptual unit of liver microanatomy defined by Rappaport is the hepatic acinus (Fig. 10.7). In this structural model the portal triad is at the center of the acinus, and the terminal hepatic venules are at the periphery. Cells are grouped into zones based upon their distance from the blood vessels in the center of the acinus (hepatic artery and portal vein branches). The closest, zone 1 cells, are the first to receive oxygenated, nutrient-rich blood and to undergo regeneration, and are the last to develop hypoxic injury. The opposite is true of zone 3 cells. Zone 2 cells are intermediate.

Hepatic acinus . This three-dimensional structure is the microvascular unit of the liver parenchyma. The acinar axis is formed by the terminal hepatic venules (TPV), the hepatic arteriole (HA), and the bile ductule (BD). The perfusion of this unit is unidirectional from the acinar axis to the acinar periphery, where two or more terminal hepatic venules (THV) empty the acinus. The arbitrary division of the acinus into functional zones is represented by 1, 2 and 3 (Reproduced with permission from Gumucio and Chianale [8])

An anastomosing network of hepatocyte canaliculi in the hepatic lobule. The terminal portion of a rat biliary tree shows a bile ductule, a canal of Hering (arrow), and the fine anastomosing network of bile canaliculi (Reproduced with permission from Jones [11])
There is an anastomosing network of hepatocyte canaliculi in the hepatic lobule, which coalesces to drain bile into the interlobular bile duct in the portal triad through the canal of Hering (Fig. 10.8). Bile flows into larger intrahepatic ducts and finally into the right and left hepatic ducts, which exit the liver , merge in the porta hepatis, and form the common hepatic duct. This duct is joined by the cystic duct from the gallbladder to form the common bile duct, which empties into the duodenum.
An important structural-functional concept in the liver is that of hepatocyte heterogeneity and metabolic zonation (Fig. 10.9). For example, zone 1 hepatocytes are most active in glucose synthesis and release, oxidative energy metabolism, amino acid utilization, and bile acid and bilirubin excretion. Zone 3 hepatocytes are most active in glucose uptake and utilization, biotransformation, and ammonia detoxification . Although lobular blood flow patterns may be responsible for at least part of this zonation, in experiments in which flow patterns in rat liver were reversed, some, but not all, of these zonal metabolic patterns were reversed. For example, gluconeogenesis shifted from zone 1 to zone 3, and glycolysis and ketogenesis shifted from zone 3 to zone 1. However, glucuronidation and monooxygenation remained localized to zone 3.

Metabolic zonation of the liver and direction of liver perfusion. The predominant zonal location of some metabolic pathways depends on the direction of liver perfusion. This has been demonstrated for gluconeogenesis, glycolysis, and ketogenesis . Monooxygenation and glucuronidation were not affected by the direction of perfusion. TPV terminal portal venule, THV terminal hepatic venule (Reproduced with permission from Gumucio and Chianale [8])
3 Evaluation of Liver Function
Clinicians routinely rely on the performance of a battery of “liver function tests” to assess the degree of liver injury and functional impairment in various disease states. However, many of these tests really do not measure function at all, but rather indirectly reflect degree of hepatocellular or bile ductular injury. It is also important to be aware of all of the non-hepatic factors, as well as inherent limitations of the tests themselves, which may affect the reliability of these tests. Table 10.1 lists many of these tests and their uses. One test, the serum lactate dehydrogenase (LDH) level, has such poor specificity for liver disease that it is not included.
3.1 Measures of Cellular Injury
One of the most common groups of tests is the serum aminotransaminases, alanine aminotransferase (ALT; serum glutamic pyruvic transaminase or SGPT) and aspartate aminotransferase (AST; serum glutamic oxaloacetate transaminase or SGOT). These enzymes catalyze the reversible transfer of the alpha amino group of the amino acids alanine and aspartic acid, respectively, to the α-keto group of α-ketoglutaric acid. These enzymes are released into the circulation whenever there is hepatocyte plasma membrane damage or necrosis. Whereas ALT is localized exclusively in the cytosol, 20 % of AST is in the cytosol and 80 % is found in the mitochondria. It is also important to note that AST is present in a variety of tissues other than liver , including heart, skeletal muscle, kidney, pancreas , red blood cells and brain. However, ALT is relatively specific for liver. Although serum levels of ALT and AST are elevated to some degree in almost all liver diseases, the highest levels are seen with acute hepatocellular injury, such as viral hepatitis, toxin-induced hepatic necrosis, and hypoxic liver injury. The decline in serum levels after such an insult, particularly viral hepatitis, may not necessarily reflect recovery, but may indicate massive necrosis with loss of viable hepatocyte mass available to release transaminases. The same is true in the “burned out” cirrhotic patient with chronic liver disease who also has little remaining viable liver cell mass. In the majority of other liver diseases, including cholestatic and chronic liver diseases and hepatic malignancy, serum transaminase levels are only slightly to moderately elevated. A ratio of serum AST to ALT of more than 2 is suggestive of alcoholic liver disease, if the ALT level is normal or only minimally elevated and the AST level is only modestly elevated. This observation may be due to relatively selective mitochondrial injury in alcoholic liver disease. If the AST is elevated significantly out of proportion to a minimally elevated ALT, one should look for extrahepatic reasons for the elevation, such as hemolysis, myocardial infarction or myopathy. Concomitant elevation of LDH may suggest hemolysis, and elevation of creatine phosphokinase (CPK) and aldolase would suggest muscle disease. Both ALT and AST levels may be low in pyridoxine deficiency. Serum AST levels are depressed in patients on long-term hemodialysis.
3.2 Measures of Cholestasis
Another group of tests is considered useful in the evaluation of cholestasis (bile excretory failure). Alkaline phosphatase is actually a broad family of enzymes found in a variety of tissues, including liver , bone, intestine, placenta, kidney and leukocytes, which hydrolyze organic phosphate esters. In the liver, the enzyme is associated with both sinusoidal and canalicular membranes, as well as the cytosol. Normally, liver and bone isozymes are found in the circulation. Normal serum levels are highly dependent on age, with high levels in childhood and puberty (due to active bone growth), a plateau in middle age, and another increase in old age. The various isozymes in serum can be biochemically fractionated to determine their source. The most striking elevations of alkaline phosphatase are seen in cholestatic liver disease , particularly extrahepatic biliary obstruction , and hepatic carcinoma. With regard to the mechanism of elevation in cholestasis, certain bile acids may induce synthesis of alkaline phosphatase at the translational level. Obviously, many types of bone disease, including biliary rickets seen in patients with chronic cholestasis who are vitamin D deficient, may elevate the bone component of the serum alkaline phosphatase level. The placental isozyme may account for serum elevation during pregnancy. Serum alkaline phosphatase may be spuriously low in the face of zinc deficiency.
Another enzyme used to evaluate cholestasis, γ-glutamyl transpeptidase (GGTP), is present throughout the liver and biliary tract , as well as other organs, including small intestine , kidney, testes, pancreas , spleen, heart and brain. This enzyme catalyzes the transfer of γ-glutamyl groups from glutathione and other peptides to other amino acids. The serum level of GGTP is a sensitive, though not specific, test for detecting biliary tract disease and generally correlates well with alkaline phosphatase levels. GGTP is inducible by drugs such as ethanol, phenytoin, phenobarbital, and warfarin . In addition, levels may be elevated in a number of conditions, including diabetes, chronic ethanol consumption, myocardial infarction, renal failure, pancreatic disease and chronic obstructive pulmonary disease. Another enzyme, 5′-nucleotidase, is found in liver, intestine, brain, heart, and pancreas. In liver it is primarily associated with canalicular and sinusoidal membranes, and elevation of the serum level is more specific for liver disease than either alkaline phosphatase or GGTP. However, this assay is often not available on a timely basis in many hospitals.
Although hepatic metabolism of bilirubin and bile acids will be discussed subsequently in detail, they are discussed here in the context of their diagnostic usefulness. Serum conjugated bilirubin levels are elevated in a number of disease states, including hepatitis, drug-induced liver injury, and obstructive and hepatocellular cholestasis. Although conjugated bilirubin elevation in cholestasis is to be expected in the face of biliary excretory failure, its elevation in hepatocellular injury is due to the fact that, except in cases such as massive hepatocellular necrosis, hepatic bilirubin conjugating activity is relatively preserved in the face of hepatocellular injury and impaired excretion with release of the conjugated bilirubin into the plasma compartment. Unlike unconjugated bilirubin, conjugated bilirubin is readily filtered by the kidney and excreted in the urine. Urobilinogen is formed by the action of intestinal bacteria on conjugated bilirubin. Up to 20 % of urobilinogen is absorbed and undergoes an enterohepatic circulation, and a small fraction is excreted in the urine. With liver dysfunction, a smaller fraction of absorbed urobilinogen is cleared by the liver and an increased amount is excreted in the urine. In the case of biliary obstruction , with decreased entry of bilirubin into the intestine, urinary urobilinogen levels fall.
Measurement of bile acids in serum can be a useful marker for cholestasis, which is more specific and sensitive than the bilirubin level. In fact, elevation of serum bile acid levels is implicit in the diagnosis of cholestasis. Measurements may include total bile acids or specific ones such as cholylglycine. Unfortunately, bile acid measurement does not reliably differentiate hepatocellular and obstructive cholestasis. Fasting and post-prandial bile acid levels may be used to evaluate the integrity of the enterohepatic circulation, because a slight elevation is expected after a meal despite efficient first-pass clearance by the liver under normal conditions. The post-prandial rise is even more pronounced in the face of mild liver disease, with sub-optimal first-pass clearance. However, it has not been confirmed that post-prandial measurements are more sensitive than those obtained with fasting for the diagnosis of liver disease. Analysis of bile acid metabolites in urine is used to diagnose inherited defects of bile acid metabolism, which may cause severe liver disease in the neonatal period.
3.3 Measures of Liver Synthetic Function
Measurement of serum proteins produced by the liver can provide an index of liver synthetic function. The liver synthesizes six coagulation factors, including factors I (fibrinogen ), II (prothrombin ), V, VII, IX, and X. Although these factors may be measured individually, the prothrombin time determination can indirectly assess these factors. This assay evaluates the extrinsic coagulation pathway and is prolonged when factors I, II, V, VII, and X are deficient, singly or in combination. In addition to liver synthetic failure, other causes for prolongation of the prothrombin time include congenital coagulation factor deficiency, consumptive coagulopathy with disseminated intravascular coagulation (DIC), drugs such as warfarin , and vitamin K deficiency (factors II, VII, IX, and X). In fact, one should always check for correction of the prothrombin time by parenterally administered vitamin K. Correction is rapid, usually within 24 h. In the case of liver disease, there is generally a large reserve for coagulation factor synthesis, such that prolongation occurs only when a large portion of the liver’s synthetic functional reserve is impaired, usually in the setting of acute hepatocellular necrosis or chronic liver disease with cirrhosis .
Albumin is the most abundant plasma protein synthesized by the liver , and the serum level can serve as an index of liver synthetic function. The half-life of serum albumin is 20 days, which limits its usefulness as a synthetic marker for acute liver disease. In order to use the serum albumin level as an indicator of liver synthetic function, one must be aware of the factors regulating serum levels, including synthesis, distribution, and catabolism. Factors regulating albumin synthesis include nutrition, plasma oncotic pressure, and liver dysfunction. Nutrition is an extremely important regulator, particularly the availability of amino acids, especially tryptophan . Abnormal loss of serum albumin through protein-losing enteropathy or nephrotic syndrome should always be considered. In fact, some degree of protein-losing enteropathy may accompany portal hypertension. In the cirrhotic patient with edema and ascites , the volume of distribution for albumin may be expanded, leading to reduced serum concentrations.
3.4 Quantitative Liver Function Tests
Several so-called “quantitative” liver function tests have been developed over the years that generally rely on the plasma clearance of various substances by the liver. Bromosulfophthalein (BSP) is an organic ion that was extensively used in the past. However, it is presently not available in the United States for intravenous injection. Indocyanine green is an inert dye that is exclusively eliminated by the liver, unchanged, into bile. Administered intravenously in low doses, it is a good indicator of liver blood flow. In higher doses, clearance is more dependent on processing of the dye by the liver. This test is particularly sensitive for detecting cirrhosis . The galactose elimination test is reflective of functional liver cell mass. It is particularly useful in following individual patients with progressive liver disease and declining hepatic function. Another quantitative test is the aminopyrine breath test . After hepatic uptake, this compound is demethylated by cytochrome P450. The labeled methyl group is eventually excreted in expired air as CO2. Measurement of the labeled CO2 provides an index of hepatic excretion. Non-radioactive 13C has replaced the formerly used radioactive tracer. This test has utility for the detection of cirrhosis, as well as a prognostic indicator for the survival of patients with cirrhosis. Another quantitative test is the lidocaine clearance test . This test is performed by injecting a standardized dose of lidocaine intravenously and measuring the appearance of its main metabolite, monoethylglycinexylidide (MEGX) in serum. MEGX is formed in the hepatocyte by cytochrome P450-dependent demethylation. Clinical studies indicate that the MEGX test may be useful in evaluating liver dysfunction, particularly in the area of liver transplantation, as an indicator for the need for transplantation, evaluation of graft function, and determination of outcome after transplantation. Currently, all of these tests are expensive, difficult to perform and not readily available in most hospitals for routine use.
3.5 Imaging Studies
Several imaging studies are used for the evaluation of liver disease. Plain films of the liver rarely provide useful information, except in the occasional case with an intrahepatic calcification or calcified gallstones. Ultrasonography can provide detailed anatomic data, as well as evaluate blood flow using a Doppler probe. Ultrasonography can detect gallstones, bile duct dilatation, abscesses and masses, such as tumors, down to 1–2 cm in size, and diagnose portal vein and hepatic vein (Budd-Chiari syndrome ) thrombosis/obstruction. Ultrasonography is less reliable for evaluation of diffuse parenchymal lesions, such as cirrhosis and fatty liver. Contrast-enhanced ultrasound (CEUS) uses an intravenous microbubble contrast agent. This imaging modality is most useful in evaluating focal lesions, including hemangiomas and focal nodular hyperplasia. However, CEUS currently has limited availability.
Greater resolution is possible with computed tomography (CT) scanning, which can be performed with and without intravenous and intestinal contrast administration. CT scanning is particularly useful in detection of abscesses and masses such as tumors. Angiography has largely been supplanted by newer imaging methods, but still may be useful for studying the hepatic vessels and blood supply, particularly in identifying the blood supply to tumors and evaluating the patient with portal hypertension.
Magnetic resonance imaging (MRI) is gaining widespread use for liver imaging. It is especially useful for detecting focal hepatic lesions, particularly in distinguishing hemangiomas from other lesions, and detecting excessive hepatic iron deposition in hemochromatosis . Imaging by MRI is also the most accurate method for assessing hepatic steatosis, as occurs in non-alcoholic fatty liver disease (NAFLD) associated with obesity and insulin resistance. Intraluminal injection of contrast into the biliary tree, either percutaneously (percutaneous transhepatic cholangiography) or endoscopically directly through the ampulla of Vater (endoscopic retrograde cholangiopancreatography or ERCP) is useful for evaluating the source of biliary tract obstruction. Biliary imaging by MRI (magnetic resonance cholangiopancreatography or MRCP) is useful for the diagnosis of biliary tract disease, such as primary sclerosing cholangitis (PSC), and is especially useful in children, because it carries less risk than ERCP and avoids radiation exposure. An MRCP image of the bile duct lesions in PSC is shown in Fig. 10.10. Intravenous cholangiography is rarely used because of its relatively poor imaging. Radionuclide imaging of the liver is useful in the detection of filling defects and diffuse hepatocellular disease. Hepatobiliary scans utilizing radionuclides, such as 99mTc-DISIDA, taken up by the liver and excreted into bile, are used to evaluate biliary excretory function. When used in conjunction with ultrasonography, these scans can often reliably differentiate intrahepatic from extrahepatic biliary obstruction .

Magnetic resonance cholangiopancreatography from a patient with primary sclerosing cholangitis showing stenosis at the estuary of the left hepatic duct in the common hepatic duct (arrow). Stenoses are also present along the branches of the left hepatic duct (arrowheads) with mild prestenotic dilations (Reproduced with permission from Alexopoulou et al. [1])
3.6 Elastography
A promising technology for the non-invasive assessment of liver fibrosis is transient elastography. This technique can be performed with either ultrasonography or magnetic resonance and is based on the observation that the propagation of a waveform through a homogeneous tissue, such as liver, is proportional to its stiffness, which in the case of liver is most often due to fibrosis or cirrhosis . Increased stiffness causes the wave to propagate more quickly, which can be measured. Invalid measurements may result from improper positioning of the transducer over rib or lung or in obese patients. Elastography is a rapidly evolving technology, and its routine clinical utility remains to be proven.
3.7 Liver Biopsy
Examination of liver histology obtained by liver biopsy usually provides useful information for both diagnosis and prognosis. In addition to providing sections for light microscopy, tissue may be sent for special studies such as electron microscopy for the diagnosis of mitochondrial or peroxisomal disorders, immunostaining for antigens such as hepatitis B or α-1-antitrypsin, measurement of iron or copper content for diagnosis of hemochromatosis or Wilson disease, respectively, and specific enzyme assays for the diagnosis of metabolic disorders such as glycogen storage disease. Percutaneous needle biopsy is generally safe and provides adequate tissue. However, other approaches include the transjugular route when coagulopathy is present, laparoscopic biopsy to allow direct visualization of the liver, and open surgical wedge biopsy .
3.8 Other Testing
Additional disease-specific tests include detection of viral infections, such as hepatitis A, B or C and other hepatotropic viruses by polymerase chain reaction (PCR) and serology. Detection of specific autoantibodies is useful in the diagnosis of autoimmune liver diseases, including autoimmune hepatitis and primary sclerosing cholangitis . Genetic testing is widely available for inherited diseases such as alpha-1-antitrypsin deficiency, hemochromatosis , and tyrosinemia. Tumor markers, such as alpha-fetoprotein (AFP) and carbohydrate antigen 19-9 (CA19-9), are helpful in the diagnosis of hepatocellular carcinoma and cholangiocarcinoma, but are not specific for liver cancer.
A new class of biomarkers for liver disease undergoing intense scrutiny is microribonucleic acid (miRNA). MicroRNAs are small (19–25 nucleotide) non-coding single-stranded RNA molecules that regulate protein-coding genes and are involved in modulation of many cell functions, both normal and aberrant. They also function in complex gene regulatory networks. Their clinical utility resides in their usefulness as biomarkers for disease and as potential therapeutic targets. They may be measured in serum where they are protected from degradation by RNases. Specific patterns of changes in serum levels of miRNA species are observed in a variety of liver diseases, including viral hepatitis, malignancy, biliary atresia (a serious liver disease in infants) and in liver graft rejection after transplantation.
4 Hepatic Blood Flow, Cirrhosis, and Portal Hypertension
Under normal conditions, an important feature of hepatic blood flow is a very low outflow resistance from the sinusoids. The sinusoidal pressure is generally constant despite changes in blood flow. Maintenance of a low sinusoidal pressure prevents an imbalance of Starling forces, which would result in excessive movement of fluid, solutes and proteins through the highly permeable sinusoidal endothelial lining into the space of Disse. Another feature of the hepatic circulation is the regulation of hepatic arterial blood flow in response to changes in portal venous blood flow and sinusoidal pressure. This results in a reflex increase in hepatic arterial blood flow in the face of a decrease in portal venous flow or sinusoidal pressure, and a decrease in hepatic arterial flow in the face of an increase in sinusoidal pressure. Hepatic arterial vascular resistance is responsive to intrinsic, hormonal, and neural factors, all of which may play a regulatory role.
The hepatic circulation is also very sensitive to changes in systemic pCO 2 levels, much more so than to changes in pO2 levels. In animal models, higher systemic pCO2 levels result in increased hepatic arterial and portal venous flow due to decreased arterial resistance. Portal blood acidosis increases portal resistance and decreases arterial resistance in the liver , and portal alkalosis has the opposite effects. During digestion there is an increased oxygen extraction by the intestine with an accompanying increase in portal venous pCO2 and decrease in pH. These changes would be expected to increase hepatic arterial blood flow during a period of enhanced hepatic metabolic activity. Increased portal flow also occurs after a meal secondary to the accompanying splanchnic hyperemic response.
The normal portal venous pressure is 5–10 mmHg. Because the direct measurement of portal venous pressure in vivo is not generally practical, a wedged hepatic venous pressure is often used as an indirect measurement. Portal hypertension is defined as a wedged hepatic venous pressure that is more than 5 mmHg greater than the pressure in the inferior vena cava. Portal hypertension is also present when the splenic pulp pressure is greater than 15 mmHg, or the pressure in the portal vein directly measured in surgery is greater than 30 cm H2O. Doppler ultrasonography allows non-invasive measurement of portal venous flow. Portal hypertension results from either an increase in portal blood flow or any process that increases the resistance to portal blood flow at the level of the portal vein, liver , hepatic veins, or heart. Therefore, portal hypertension may result from increased portal blood flow (arteriovenous fistula ), pre-hepatic obstruction (portal vein thrombosis or compression by tumor), increased vascular resistance at the hepatic level (cirrhosis or congenital hepatic fibrosis), or post-hepatic venous obstruction/congestion (Budd-Chiari syndrome or right heart failure). Different types of portal hypertension may also be classified as pre-sinusoidal, sinusoidal, or post-sinusoidal, based upon information obtained from measurement of wedged hepatic venous pressure. However, technical limitations of this measurement and the fact that the pattern may change over time with the same disease process limit the usefulness of this system of classification. Table 10.2 lists causes of portal hypertension. The following discussion will generally be limited to portal hypertension resulting from hepatic cirrhosis.
A number of insults to the liver , including hepatitis (viral or autoimmune), toxins (alcohol, drugs, and environmental toxins), metabolic disease (Wilson disease, α-1-antitrypsin deficiency), and extrahepatic biliary obstruction (stones, tumor) may result in hepatic fibrosis and marked alteration of the normal hepatic microanatomy. Figure 10.11 shows a photomicrograph of a liver biopsy from a patient with advanced cirrhosis . Normal lobular architecture is abolished. Present are regenerative nodules surrounded by fibrous bands. Although vessels are identified in the fibrous bands, no central veins are present in the nodules. These changes result in inefficient perfusion of hepatocytes with intrahepatic shunting of blood and increased resistance to blood flow through the liver, contributing to the development of portal hypertension.

Normal (a) and cirrhotic (b) liver . Note the fibrous bands (arrows) and regenerative nodules (N), which lack central veins (hematoxylin-eosin, original magnification 100×; Courtesy of Dr. John Hart)
The interplay of hepatic sinusoidal endothelial cells and stellate cells is a major determinant of the vascular changes that occur in the liver in cirrhosis and associated portal hypertension. There are likely two components to the increase in resistance to flow through the hepatic sinusoids. One is structural and related to the deposition of collagen in the sinusoids, as well as distortion by cirrhotic nodules, resulting in a largely irreversible increase in resistance to portal flow and increased portal pressure. The stellate cell becomes activated in response to exposure to inflammation and chemokines and is responsible for collagen and matrix molecule deposition. The other component is dynamic and related to dysregulation of vascular tone. Stellate cells and stellate cell-derived myofibroblasts also are capable of Ca2+-dependent contractility around the sinusoids to regulate vascular tone. These cells respond to endothelial cell secretion of NO mediating vasodilation and endothelins and other molecules modulating vasoconstriction. In disease states with stellate cell proliferation and activation, this balance is tipped in favor of vasoconstriction, contributing to increased resistance to blood flow through the liver. Future therapeutic targets may be induction of NO production by endothelial nitric oxide synthase (eNOS) and inhibition of endothelins in the liver.
The increase in intrahepatic vascular resistance in portal hypertension is accompanied by increased splanchnic blood flow. In response to the rise in portal pressure due to increased intrahepatic resistance, porto-systemic collateral blood vessels, such as esophageal and gastric varices, develop in response to increased release of vascular endothelial growth factor (VEGF) and other factors, and shunting contributes to a fall in splanchnic resistance and development of a hyperdynamic circulation. Another mechanism for the drop in splanchnic resistance is an increase in NO production in this vascular bed, in contrast to the local decrease in NO in the hepatic sinusoidal bed. The resultant increase in flow into the portal circulation in the face of increased resistance in the liver results in further elevation of the portal pressure.

Collateral blood flow in portal hypertension (Reproduced with permission from Black [4])

Esophageal varices. (a) Fiberoptic esophagoscopy reveals esophageal varices in the distal esophagus of a patient with cirrhosis and portal hypertension. (b) Esophageal varix (V) after endoscopic ligation with a rubber band (arrow)
In the patient with cirrhosis , collateral circulatory pathways develop to allow portosystemic shunting of blood. These collaterals may include prominent dilated superficial abdominal veins, caput medusae (tortuous periumbilical veins), recanalization of the umbilical vein remnant, and rectal varices, as shown in Fig. 10.12. Of the greatest clinical significance is the formation of esophageal varices , thin-walled superficial veins in the distal esophagus , which can result in life-threatening hemorrhage (Fig. 10.13a). Varices and portal hypertensive gastropathy may develop in the stomach , which are also often a source of bleeding. The factors that predispose varices to bleed are not well known. Irritation from gastroesophageal reflux, a popular theory in the past, is probably not a common event. The initiation of hemorrhage is probably a complex event dependent on factors such as wall tension (T), transmural pressure [intravariceal pressure (P 1 ) – esophageal lumen pressure (P 2 )], vessel radius (r), and wall thickness (W) as related to each other by the following equation involving Laplace’s law :
Varices probably burst open and bleed when the wall tension can no longer contain the forces expanding the varices. Large varices with thin walls would be expected to have a greater tendency to bleed, compared to smaller varices with thinner walls at the same transmural pressure.
Bleeding esophageal varices may be treated by endoscopic injection of a sclerosing solution (sclerotherapy) or placement of bands around the varices (banding, Fig. 10.13b). Acute reduction in portal hypertension is possible by the intravenous infusion of vasopressin or octreotide (a somatostatin analogue), which results in splanchnic arteriolar constriction and temporary reduction in portal venous pressure. Chronic reduction of portal hypertension is possible with administration of β-adrenergic antagonists, particularly propranolol, although not all patients are responsive. This therapy appears to work predominantly by enhancing splanchnic arteriolar vasoconstriction by blocking β1-receptor-mediated vasodilatation. These therapeutic modalities have replaced balloon tamponade using a Sengstaken-Blakemore tube. Another approach to treat the underlying portal hypertension is surgical creation of a portosystemic shunt between the portal system and systemic venous system to reduce portal blood pressure. The technique of transjugular intrahepatic portosystemic shunting (TIPS) via interventional radiology is widely used as an emergency procedure to reduce portal pressure to avoid surgery and as a bridge to liver transplantation.
Portal hypertension also results in splenic congestion and splenomegaly , often causing functional hypersplenism . Hypersplenism results in sequestration of red blood cells, white blood cells, and platelets with resultant depression of counts of one to all of these cell types in peripheral blood. Occasionally, the magnitude of this reduction in circulating cell number is great enough to result in significant anemia (red blood cells), risk for infection (leukocytes), or risk of bleeding (platelets).
Chronic portosystemic shunting can also result in chronic intermittent encephalopathy resulting from the entry of ammonia and other toxins, which would normally be cleared by the liver , from the intestine into the systemic circulation via collateral circulation and subsequently into the central nervous system . As might be expected, surgical shunting to treat portal hypertension often leads to worsening of encephalopathy.
Another consequence of cirrhosis and portal hypertension is the development of fluid in the peritoneal cavity (ascites ) and peripheral edema, as well as impaired renal function. The major factors contributing to these complications of cirrhosis and portal hypertension are summarized in Fig. 10.14. A discussion of the pathophysiology of these conditions provides an excellent insight into the important role of the liver in the maintenance of normal circulatory homeostasis. The increased lymph formation due to increased hydrostatic pressure in the splanchnic vascular bed as a result of portal hypertension, as well as increased hepatic lymph formation from locally increased vascular resistance, exceeds the capacity of the thoracic duct to return the lymph to the systemic circulation. This event, along with venous pooling in the splanchnic bed, results in a redistribution of the blood volume and ascites formation. One of the major factors resulting in the development of ascites is avid renal sodium retention in the patient with cirrhosis. A concept important to understanding this phenomenon is diminished effective blood volume. Effective plasma volume refers to that fraction of the total circulating volume that is effective in stimulating volume receptors. Decreased effective plasma volume is the result of several factors. Hypoalbuminemia, which often is present in cirrhosis, results in decreased plasma oncotic pressure and an imbalance in Starling forces favoring an extravascular shift of fluid. Other factors include a decrease in systemic vascular resistance caused by peripheral arteriovenous shunts, as well as circulating vasodilators, such NO, carbon monoxide, and others. The net result of these processes is a decreased effective plasma volume, despite the fact that the actual plasma volume is increased. Lack of appropriate stimulation of volume receptors sets in motion several compensatory mechanisms to correct what is inappropriately perceived as a reduced vascular volume. These mechanisms include activation of the renin-angiotensin system with resultant hyperaldosteronism and renal sodium retention. Decreased aldosterone clearance by the cirrhotic liver also contributes to the hyperaldosteronism. In addition, increased renin-angiotensin and sympathetic nervous system activity leads to increased renal vascular resistance and a decreased glomerular filtration rate (GFR). Other humoral factors may also contribute to decreased renal blood flow and increased tubular reabsorption of sodium, including renal prostaglandins , the kallikrein -kinin system, circulating natriuretic factor , and atrial natriuretic factors .

Schematic representation of the major pathophysiological mechanisms leading to renal sodium retention and ascites in cirrhosis
All of these factors in the patient with cirrhosis and portal hypertension, particularly those affecting renal blood flow, can result in renal failure, which is termed the hepatorenal syndrome . This syndrome usually occurs in the setting of end-stage liver disease and may be precipitated by overzealous diuresis or paracentesis for ascites or variceal bleeding. It is characterized by the development of oliguria with decreased GFR in the face of preservation of tubular function, as reflected by a normal or increased fractional excretion of sodium and increased water reabsorption. The most striking pathological event in the kidney is marked vasoconstriction and diminution in arterial blood flow, particularly that supplying the cortex. Kidneys from individuals with hepatorenal syndrome dying from liver failure demonstrate a normal arterial tree on angiogram and have histologically normal blood vessels when removed from the patient. Furthermore, these kidneys function normally when transplanted into normal individuals with renal failure. This suggests that the renal abnormalities in the hepatorenal syndrome are functional and due to local and/or circulating factors.
Complications of ascites in the patient with cirrhosis include spontaneous bacterial peritonitis and impaired ventilation in extreme cases. Treatment of ascites in the cirrhotic patient should be dictated by knowledge of the pathophysiology. The cornerstone of treatment is sodium restriction. In fact, in many patients, this one maneuver, if complied with, may result in a striking diuresis. Since hyperaldosteronism is a prominent abnormality, treatment with the aldosterone antagonist, spironolactone, is often effective and can be combined with a loop diuretic if needed. However, one should be cautious, because aggressive diuresis may precipitate hepatorenal syndrome or encephalopathy. Complete elimination of ascitic fluid should never be the goal, but reducing it to make the patient more comfortable and to eliminate any respiratory compromise should be the objectives. Intravenous infusion of albumin in the patient with hypoalbuminemia may facilitate a transient diuresis. Refractory cases may require paracentesis , particularly in the face of respiratory compromise. The placement of shunts to drain ascitic fluid back into the systemic circulation has been used, but is fraught with complications, especially infection.
5 Hepatic Sex Hormone Metabolism in Cirrhosis
The role of the liver in sex hormone metabolism is underscored in the cirrhotic male patient, particularly in those with alcoholic cirrhosis . Hypogonadism is frequently present, with testicular atrophy and decreased libido. The testicular atrophy reflects a decrease in seminiferous tubule mass, most likely the result of chronic ethanol toxicity . Leydig cell dysfunction is also present and reflected by decreased production of testosterone. Cirrhosis results in an increase in plasma levels of sex hormone-binding globulin, and the combination of reduced total levels of testosterone and increased protein binding results in significantly reduced levels of free, biologically active hormone. Pre-menopausal women with alcoholic cirrhosis have reduced levels of estradiol and progesterone and accompanying loss of breast and pelvic fat and anovulation . Pathologic changes in the ovaries may be at least partially due to ethanol toxicity, as is postulated in the testes in males. At the hypothalamic-pituitary level, there is reduced basal gonadotropin secretion and a reduced luteinizing hormone (LH) response to LH-releasing factor. Males with cirrhosis frequently exhibit feminization manifested by gynecomastia, palmar erythema, cutaneous spider angiomata, and a female escutcheon. Although the mechanisms of this estrogenization are not completely known, hepatic clearance of estrogens is reduced and may contribute to elevated circulating levels. Another mechanism may involve the reabsorption of estrogens and androgens excreted in bile with accompanying inefficient first-pass clearance by the liver in the face of portosystemic shunting and hepatocellular dysfunction. This allows exposure of estrogen-sensitive peripheral tissues to these estrogens. There also appears to be increased peripheral conversion of reabsorbed androgens to estrogens. Finally, there may be increased sensitivity of peripheral tissues to estrogens in these individuals.
Clinical Correlations
Case Study 1
A 55-year-old white male is brought to the emergency room by family members with a history of passing three large, foul-smelling, black, tarry stools over the past 12 h. He has also been “acting strangely” over the past 2 days and has been verbally abusive and very agitated, more so than usual when intoxicated. Family members report that the patient has a history of “drinking heavily” for the past 25 years. He often will consume a pint of whiskey per day. There has also been an inversion of sleep patterns. Today, he has been very sleepy and difficult to arouse. They also report that they have noticed a large increase in the size of his abdomen over the past several weeks, and his pants no longer fit. There is no past history of blood transfusion, intravenous drug use, or hepatotoxic drug exposure.
Physical examination: Respirations 22, temperature 37.2 °C rectally, pulse 96, and blood pressure 122/81. When tilted up, the pulse rate goes up to 116, and the blood pressure drops to 106/66. The patient is sleepy, but arousable with vigorous stimulation. However, he is very combative and uncooperative when aroused. Pupils are equal and reactive to light. The chest is clear to auscultation. There is bilateral breast enlargement. Cardiac examination reveals a regular rate and rhythm and a grade II/VI systolic ejection murmur. The abdomen is distended and exhibits shifting dullness, indicating ascites . There are dilated superficial abdominal veins. The liver edge cannot be palpated but has a span of 12 cm by percussion. The spleen is palpable 8 cm below the left costal margin. There is reduced pubic hair in a female distribution. Rectal examination reveals black stool strongly positive for occult blood. Skin examination reveals several spider angiomata.
Laboratory studies: Hematocrit 28 %; hemoglobin 9.2 g/dL; white blood cell count 5,400 cells/mm3 (4,500–11,000); 136,000 platelets/mm3 (150,000–350,000); prothrombin time 18 s (control 12.4 s); albumin 2.8 g/dL (3.7–5.6); total bilirubin 2.8 mg/dL (0.1–1.0); direct bilirubin 2.4 mg/dL (0–0.2); ALT 200 IU/L (10–30), AST 280 IU/L (15–30); GGTP 180 IU/L (14–25); alkaline phosphatase 50 IU/L (30–94); blood ammonia 277 μmol/L (29–57); sodium 130 mEq/L (135–145), potassium 2.9 mEq/L (3.5–5.0), chloride 91 mEq/L (94–106); carbon dioxide 33 mEq/L (22–29); blood urea nitrogen (BUN) 77 mg/dL (5–25); and creatinine 0.2 mg/dL (0.6–1.2).
Questions:
-
1.
What is the most likely source of bleeding (melena) in this patient?
Answer: With the history of long-term heavy ethanol abuse, bleeding esophageal varices due to portal hypertension from alcoholic cirrhosis would be a likely source. The physical findings discussed below would also support this diagnosis. It should be remembered that ethanol itself is an irritant and can produce gastritis with bleeding. Also, there is a higher than usual incidence of peptic ulcer disease in individuals with cirrhosis.
-
2.
What is the cause of the altered mental state?
Answer: The history and present findings are certainly compatible with hepatic encephalopathy , particularly the inversion of the sleep/wake cycle, somnolence, and concomitant gastrointestinal bleeding, which often exacerbates hepatic encephalopathy because of the increased intestinal ammonia load. However, ethanol abuse alone can produce an organic brain syndrome . Electrolyte derangements can cause altered mentation and should always be checked. Wernicke-Korsakoff psychosis may develop in alcoholics in the setting of thiamine deficiency. Also, abuse of other psychoactive drugs should be considered, often on an unintentional basis because of confusion as to proper dosing of prescribed medication. Remember that the patient must be conscious and cooperative to allow elicitation of asterixis and that asterixis is not specific for hepatic encephalopathy.
-
3.
What findings support a diagnosis of cirrhosis ?
Answer: The suspicion of esophageal varices and the presence of dilated superficial abdominal veins as evidence of portosystemic collaterals and the presence of ascites and splenomegaly indicate portal hypertension. There are signs of hyperestrogenism and hypogonadism (gynecomastia, spider angiomata, and a female escutcheon). Palmar erythema may not be obvious in the face of blood loss and hypovolemia. In the setting of heavy ethanol abuse, alcoholic cirrhosis would be most likely. However, other considerations would include chronic active viral (HBV or HCV) hepatitis with cirrhosis and primary hemochromatosis . Much less likely would be etiologies such as Wilson disease or α-1-antitrypsin deficiency. There is no history of drug or toxin exposure, which might result in chronic liver disease and development of cirrhosis, although the medical history in such a patient may not be reliable. The serum GGTP elevation is most likely due to induction by chronic ethanol ingestion.
-
4.
How would you interpret the laboratory tests reflective of renal function in this patient?
Answer: The low serum sodium may reflect a relative dilutional hyponatremia in the face of total body sodium overload due to the renal sodium retention present in cirrhosis . Hypokalemia is often present in the cirrhotic patient due to respiratory and/or metabolic alkalosis driving potassium into cells and chronic renal loss caused by hyperaldosteronism. These individuals are almost always total body potassium-depleted. The presence of the hypokalemia may further exacerbate the alkalosis and have adverse effects on renal and muscle function. It is very difficult to interpret the BUN and creatinine as indices of renal function in this patient. The BUN may be elevated because of reduced vascular volume (pre-renal azotemia), increased urea production from blood in the gastrointestinal (GI) tract, or renal failure (acute tubular necrosis or hepatorenal syndrome ). The creatinine level may be high, reflecting renal failure, or factitiously low in the face of a markedly reduced muscle mass in the cirrhotic patient. Close monitoring of urine output during replenishment of blood volume and evaluation of renal tubular function using indices such as the fractional excretion of sodium and urine osmolality and examination of the urine sediment may be useful.
-
5.
How should one proceed with the treatment of this patient?
Answer: Because the patient has significant hypovolemia, as demonstrated by the positive “tilt” test, adequate venous access should be established (preferably a central line) and blood should be sent for type and crossmatch immediately. Volume expansion should be started immediately; 5 % albumin would be a good choice. Also, vitamin K should be administered parenterally, and fresh frozen plasma infusion would provide volume as well as correction of coagulopathy. Active variceal bleeding may be treated with a vasopressin or octreotide infusion followed by upper GI endoscopy with variceal banding. Encephalopathy may be treated by removing the source of GI bleeding (esophageal varices ) and administering lactulose or a non-absorbable antibiotic, such as rifaximin , by nasogastric tube.
Case Study 2
A 5-year-old female is brought to the emergency room with a history of vomiting blood twice and passing three large, tarry, foul-smelling stools in the last 6 h. She has remained alert and complained only of nausea. She has been healthy up to this time with no complaints of abdominal pain or jaundice. She was born prematurely at 32 weeks gestation and spent 2 weeks in the neonatal ICU with respiratory distress and sepsis. However, since then she has been healthy with no other significant illness.
Physical examination: Respirations 22, temperature 37.6 °C rectally, pulse 112, blood pressure 96/55, weight 18.2 kg (50th percentile), and height 108 cm (50th percentile). This is an anxious, pale female child who is alert and cooperative. There is no jaundice. Her physical examination is generally unremarkable without stigmata of chronic liver disease or cirrhosis except for the abdominal examination, which reveals an enlarged spleen palpable 4–5 cm below the left costal margin. There is no hepatomegaly or ascites . Rectal examination yields guaiac-positive stool. The neurologic examination is normal, and asterixis cannot be elicited.
Laboratory studies: Hematocrit 24 %; hemoglobin 8.1 g/dL; white blood cell count 3,600 cells/mm3 (5,000–15,500); 78, 000 platelets/mm3 (150,000–350,000); prothrombin time 13.0 (control 12.4 s); albumin 4.5 g/dL (3.7–5.6); total bilirubin 1.2 mg/dL (0.1–1.0); direct bilirubin 0.4 mg/dL (0–0.2); ALT 26 IU/L (10–30), AST 22 IU/L (15–30); GGTP 21 IU/L (14–25); alkaline phosphatase 296 IU/L (100–300); blood ammonia 77 μmol/L (29–57); sodium 139 mEq/L (135–145), potassium 3.9 mEq/L (3.5–5.0), chloride 101 mEq/L (94–106); carbon dioxide 26 mEq/L (22–29); blood urea nitrogen (BUN) 39 mg/dL (5–25); and creatinine 0.6 mg/dL (0.3–0.7).
Questions:
-
1.
What is the most likely source of this girl’s bleeding?
Answer: In a child with upper GI bleeding of this magnitude without significant liver dysfunction and with a past history of neonatal intensive care and splenomegaly , portal hypertension and bleeding esophageal varices secondary to cavernous transformation of the portal vein would be very likely. This form of post-hepatic portal hypertension often is a sequela of portal vein thrombosis from placement of an umbilical vein catheter and/or omphalitis in the neonatal period. The term “cavernous transformation” refers to the characteristic appearance of this lesion on angiogram demonstrating collaterals and recanalization in the region of the obstruction. This diagnosis can often be made by ultrasonography with Doppler flow analysis. Although these patients often have significant portal hypertension, they rarely have ascites , and they are rarely encephalopathic because of their normal liver function. This finding contrasts with hepatic (cirrhosis ) or pre-hepatic (Budd-Chiari syndrome ) portal hypertension, which is frequently complicated by ascites and liver dysfunction. Other causes of portal vein obstruction would include thrombosis due to a disorder in blood coagulation such as anti-phospholipid syndrome or protein C deficiency, extrinsic compression by tumor or other mass, or abdominal trauma. The majority of cases in otherwise healthy children are idiopathic. Indeed, in this patient esophageal varices were documented and sclerosed by endoscopy, and cavernous transformation of the portal vein was diagnosed by ultrasonography. Cavernous transformation of the portal vein demonstrated by venous phase superior mesenteric arteriography in a child is shown in Fig. 10.15.
Fig. 10.15 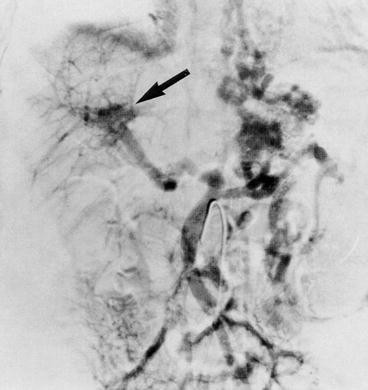
Venous phase of a superior mesenteric arteriogram showing cavernous transformation of the portal vein (arrow) (Reproduced with permission from Brady et al. [5])
-
2.
Why does this patient have elevated blood ammonia?
Answer: This patient has modest hyperammonemia because of the increased intestinal ammonia load from the variceal bleeding along with decreased hepatic first-pass portal blood clearance secondary to the portal vein obstruction and collateral circulation. The preservation of normal hepatic function prevents higher ammonia levels and clinical encephalopathy.
-
3.
What is the significance of the elevated BUN in this patient?
Answer: In the face of normal hepatic function and pre-existing normal renal function, the BUN is most likely elevated secondary to increased BUN production from GI bleeding and volume depletion leading to pre-renal azotemia. Although variceal bleeding and hypovolemia can lead to acute tubular necrosis, the normal creatinine level would not be consistent with renal failure. However, during hospitalization urine output and renal function should be monitored closely, particularly in the face of continued bleeding.
-
4.
Why are the white blood cell count and platelet count depressed?
Answer: The presence of splenomegaly in an individual with portal hypertension often leads to hypersplenism , with sequestration of red blood cells, leukocytes and platelets. In this patient it is difficult to determine how much of the present anemia was pre-existing from hypersplenism and how much is due to blood loss from variceal bleeding.
-
5.
How would you treat this patient?
Answer: The same measures to manage variceal bleeding discussed for the previous case would be used. However, treatment of coagulopathy and encephalopathy would not be necessary. These patients can often be managed by multiple sessions of endoscopic banding to obliterate their varices until they reach adolescence, when they will often have developed enough collateral circulation to prevent recurrence of varices. Occasionally, a surgical portosystemic shunt procedure is needed, but tends to be well tolerated without portosystemic encephalopathy because of the presence of normal liver function. Rarely is hypersplenism severe enough to be the sole indication for a surgical shunt procedure.

Case Study 3
A 19-year-old African-American male is referred to your clinic because of recent worsening of recurrent right upper quadrant pain that started 3 months ago. He has also noticed deepening yellowish discoloration of his sclerae. He relates that he has hemoglobin SS disease (sickle cell disease) that was diagnosed in childhood. He has had recurrent painful crises throughout his life, and his present symptoms were initially thought to represent a component of one of these crises. However, the pain is now persistent, and the jaundice is unrelenting. He cannot relate the pain to eating or a specific activity. His urine is now dark colored. He has had multiple hospitalizations for his sickle cell disease, but no other serious illnesses. He has two younger siblings with the same disease.
Physical examination: Respirations 20, temperature 37.3 °C rectally, pulse 72, and blood pressure 122/81. The patient is clearly uncomfortable, but in no acute distress. There is bilateral scleral icterus. The chest is clear to auscultation. Cardiac examination reveals a regular rate and rhythm and a grade I/VI systolic ejection murmur. The abdomen is soft and non-distended. There is mild right upper quadrant tenderness. The liver edge cannot be palpated but has a span of 12 cm by percussion. The spleen is not palpable. There is no abdominal mass or ascites . Further examination reveals no spider angiomata or other stigmata of chronic liver disease or cirrhosis .
Laboratory studies: Hematocrit 25 %; hemoglobin 8.3 g/dL; white blood cell count 10,200 cells/mm3 (4,500–11,000); 222,000 platelets/mm3 (150,000–350,000); reticulocyte count 6 %; prothrombin time 11.4 s (control 12.4 s); albumin 3.9 g/dL (3.7–5.6); total bilirubin 12.2 mg/dL (0.1–1.0); direct bilirubin 10.1 mg/dL (0–0.2); ALT 202 IU/L (10–30), AST 180 IU/L (15–30); GGTP 905 IU/L (14–25); alkaline phosphatase 1,140 IU/L (30–94).
Questions:
-
1.
To what hepatic complications are patients with sickle cell disease susceptible?
Answer: These individuals were prone to hepatitis C infection in the era before accurate testing was available for donated blood. They are also at increased risk for autoimmune liver disease. They may also develop liver disease secondary to sickled red blood cells lodging in hepatic sinusoids and producing ischemic changes in the hepatocytes . This may range across a spectrum from acute hepatic crisis with fever, right upper quadrant pain, tender hepatomegaly and jaundice with mild transaminase elevation and total bilirubin levels usually less than 13 mg/dL that is usually readily reversible, to a sequestration crisis with massive sequestration of red blood cells in the liver and severe anemia , to sickle cell intrahepatic cholestasis with severe liver impairment, often leading to liver failure. Multiple blood transfusions may lead to iron overload and liver injury. They are also at risk for thrombotic complications, such as hepatic vein thrombosis and Budd-Chiari syndrome . Finally, the increased production of bilirubin due to enhanced turnover of the abnormal cells containing hemoglobin SS, as reflected by the anemia and elevated reticulocyte count indicating increased red cell production by the bone marrow, frequently leads to formation of pigment gallstones. See the discussion of bilirubin metabolism in Chap. 12.
-
2.
What is the likely diagnosis in this patient?
Answer: The elevated alkaline phosphatase and GGTP suggest biliary obstruction . An ultrasound study is quickly obtained that shows multiple gallstones in the gallbladder, as well as a single stone in the common bile duct with significant dilatation of the duct proximal to the stone, indicating a high-grade obstruction.
-
3.
What is the recommended treatment?
Answer: After hydration and starting antibiotics to prevent bacterial cholangitis due to the obstruction, the patient undergoes an ERCP, and the endoscopist verifies the presence of a single stone in the common bile duct and is able to retrieve the stone with a basket. A few days later with further stabilization and improvement in the cholestasis, the patient undergoes a laparoscopic cholecystectomy to prevent any future stone formation and episodes of stones migrating from the gallbladder into the bile duct to cause obstruction.
Further Reading
Alexopoulou E, Xenophontos PE, Economopoulos N et al (2012) Investigative MR cholangiopancreatography for primary sclerosing cholangitis-type lesions in children with IBD. J Pediatr Gastroenterol Nutr 55:308–313
Arias IM (1994) The liver: biology and pathobiology, 3rd edn. Raven Press, New York, p 1344
Berzigotti A, Seijo S, Reverter E, Bosch J (2013) Assessing portal hypertension in liver diseases. Expert Rev Gastroenterol Hepatol 7(2):141–155
Black DD (1996) Liver physiology I: structure, functional assessment, and blood flow. In: Chang EB, Sitrin MD, Black DD (eds) Gastrointestinal, hepatobiliary, and nutritional physiology, 1st edn. Lippincott-Raven, New York
Brady L, Magilavy D, Black DD (1996) Portal vein thrombosis associated with antiphospholipid antibodies in a child. J Pediatr Gastroenterol Nutr 23:470–473
DeLeve LD (2009) The hepatic sinusoidal endothelial cell: morphology, function and pathobiology. In: Arias IM (ed) The liver: biology and pathobiology, 5th edn. Wiley-Blackwell, West Sussex, pp 373–388
Duarte-Rojo A, Altamirano JT, Feld JJ (2012) Noninvasive markers of fibrosis: key concepts for improving accuracy in daily clinical practice. Ann Hepatol 11(4):426–439
Gumucio JJ, Chianale J (1988) Liver cell heterogeneity and liver function. In: Arias IM, Jacoby WB, Popper H, Schachter D, Shafritz DA (eds) The liver: biology and pathobiology, 2nd edn. Raven Press, New York
Hanover JA (1988) Molecular signals controlling membrane traffic. In: Arias IM, Jacoby WB, Popper H, Schachter D, Shafritz DA (eds) The liver: biology and pathobiology, 2nd edn. Raven Press, New York
Hanover JA (1988) Introduction: organizational principles. In: Arias IM, Jacoby WB, Popper H, Schachter D, Shafritz DA (eds) The liver: biology and pathobiology, 2nd edn. Raven Press, New York
Jones AL (1990) Anatomy of the normal liver. In: Zakim D, Boyer TD (eds) Hepatology: a textbook of liver disease, 2nd edn. W. B. Saunders Co., Ltd, Philadelphia
Jones AL, Spring-Mills E (1977) In: Weiss L, Greep RO (eds) Histology, 4th edn. New York, McGraw-Hill Book Co
McCuskey RS (1994) Hepatic microvascular system. In: Arias IM, Boyer JL, Fausto N, Jakoby WB, Schachter DA, Shafritz DA (eds) The liver: biology and pathobiology, 3rd edn. Raven Press, New York
McCuskey R (2012) Anatomy of the liver. In: Boyer TD, Manns MP, Sanyal AJ (eds) Hepatology: a textbook of liver disease, 6th edn. Elsevier Saunders, Philadelphia
Moore CM, Van Thiel DH (2013) Cirrhotic ascites review: pathophysiology, diagnosis and management. World J Hepatol 5(5):251–263
Poynard T, Imbert-Bismut F (2012) Laboratory testing for liver disease. In: Boyer TD, Manns MP, Sanyal AJ (eds) Zakim and Boyer’s hepatology, 6th edn. Elsevier Saunders, Philadelphia, pp 201–215
Rockey DC, Friedman SL (2012) Hepatic fibrosis and cirrhosis. In: Boyer TD, Manns MP, Sanyal AJ (eds) Zakim and Boyer’s hepatology, 6th edn. Elsevier Saunders, Philadelphia, pp 64–85
Rojkind M, Reyes-Gordillo K (2009) Hepatic stellate cells. In: Arias IM (ed) The liver: biology and pathobiology, 5th edn. Wiley-Blackwell, West Sussex, pp 407–432
Silverman A, Roy CC (1983) Pediatric clinical gastroenterology, 3rd edn. C. V. Mosby Co., St. Louis
Wang XW, Heegaard NH, Orum H (2012) MicroRNAs in liver disease. Gastroenterology 142(7):1431–1443
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Black, D.D. (2014). Structure, Functional Assessment, and Blood Flow of the Liver. In: Leung, P. (eds) The Gastrointestinal System. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-8771-0_10
Download citation
DOI: https://doi.org/10.1007/978-94-017-8771-0_10
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-8770-3
Online ISBN: 978-94-017-8771-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)