
Abstract
Dopamine is the endogenous neurotransmitter produced by nigral neurons. Dopamine loss can trigger not only prominent secondary morphological changes, but also changes in the density and sensitivity of dopamine receptors; therefore, it is a sign of PD development. The reasons for dopamine loss are attributed to dopamine’s molecular instability due to it is a member of catecholamine family, whose catechol structure contributes to high oxidative stress through enzymatic and non-enzymatic oxidation. Oxidative stress in the brain easily leads to the lipid peroxidation reaction due to a high concentration of polyunsaturated fatty acids (PUFA), such as docosahexaenoic acid (DHA, C22:6/ω-3) and arachidonic acid (AA, C18:4/ω-6). Recent studies have shown that lipid hydroperoxides, the primary peroxidative products, could non-specifically react with primary amino groups to form N-acyl-type (amide-linkage) adducts. Therefore, based on the NH2-teminals in dopamine’s structure, the aims of this chapter are to describes the possibility that reactive LOOH species derived from DHA/AA lipid peroxidation may modify dopamine to form amide-linkage dopamine adducts, which might be related to etiology of Parkinson’s diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Backgrounds
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by a dramatic loss of dopaminergic neurons in the substantia nigra, and the subsequent deficiency of dopamine in the brain areas (Galvan and Wichmann 2008). Until now, very little is known about why and how the PD neurodegenerative process begins and progresses; however, an increasing body of evidence suggests that oxidative stress, mitochondrial dysfunction, and impairment of the ubiquitin-proteasome system may be involved in the pathogenesis of PD (Leroy et al. 1998; Schapira 2001; Balaban et al. 2005). Recent studies indicate that there are high levels of basal oxidative stress in the substantia nigra pars compacta (SNc) in the normal brain and this is increased in PD (Jenner 2003).
Oxidative stress in the brain easily leads to the lipid peroxidation reaction due to a high concentration of polyunsaturated fatty acids (PUFA), such as docosahexaenoic acid (DHA, C22:6/ω-3) and arachidonic acid (AA, C18:4/ω-6), which are present in the brain (Porter et al. 1995). The polyunsaturated fatty acids are located almost exclusively in the SN2-position of the phosphoglycerides found in the neural cell membranes. The beneficial physiological effects of DHA and AA have been frequently reported (Simopoulos 1999; Hadders-Algra 2008); however, the fatty acids are highly unsaturated, thus making them particularly susceptible to peroxidation. During the lipid peroxidation reaction, lipid hydroperoxides are generated as primary products. Subsequent decomposition leads to the formation of reactive mediates including aldehydes, which can covalently modify biomolecules. We have recently found that lipid hydroperoxides, the primary peroxidative products, can universally react with primary amino groups to form N-acyl-type (amide-linkage) adducts (Kato et al. 1997, 1999; Kato and Osawa 1998; Kawai et al. 2003, 2004, 2006). In our previous studies, the formation of linoleic acid-derived lysine modification adducts, N-(hexanoyl) lysine and N-(azelayl) lysine, and DHA-derived adducts, N-(succinyl) lysine and N-(propanoyl) lysine, have been identified in vitro or in vivo by LC-MS/MS or immunochemical analysis. In addition, the formation of N-(hexanoyl) lysine also was detected, as well as N-(glutaryl) lysine, during the reaction of oxidized arachidonic acid (AA) with the lysine residue. The N-acyl-type adducts are specific to the peroxidation of polyunsaturated fatty acids, therefore, their formations are the useful markers for the lipid peroxidation, protein modification and related dysfunction that occur in these fatty acids enriched tissues.
Dopamine is the endogenous neurotransmitter produced in nigral neurons. Dopaminergic neuronal loss can trigger not only prominent secondary morphological changes, such as density reduction of the dendritic spines, but also changes in the density and sensitivity of dopamine receptors (Galvan and Wichmann 2008); therefore, it is a sign of PD development. The reasons for dopamine loss are attributed to molecular instability of dopamine. Some possible causes of dopamine are characteristics of dopaminergic neurons (Bove et al. 2005), such as dopamine degradation by monoamine oxidase A (MAO-A) (Gotz et al. 1994) or auto-oxidation (Hald and Lotharius 2005) and the reaction with amino acid cysteine (LaVoie and Hastings 1999). Dopamine is a member of catecholamine family. The catechol structure of dopamine contributes to vulnerability to oxidative stress. Additionally, the NH2-teminals in dopamine’s structure may represent another reactive spot, however, little experimental evidence have been proven. Based on our previously described reaction between lipid hydroperoxides and NH2 residues, the possibility that reactive LOOH species derived from lipid peroxidation may modify dopamine to form amide-linkage dopamine adducts was investigated.
2 Chemical Formation of DHA- and AA-Derived Dopamine Adducts
Lipid hydroperoxides, the primary products of lipid peroxidation, could non-specifically react with primary amino groups to form N-acyl-type (amide-linkage) adducts, and also within the chemical structure of dopamine, an amino residue is present. The DHA and AA-derived four amide-linkage dopamine adducts, succinyl dopamine (SUD) and propanoyl dopamine (PRD), hexanoyl dopamine (HED) and glutaroyl dopamine (GLD), were chemically synthesized respectively. The chemical structures of the authentic adducts were identified by NMR. The formation of these dopamine adducts was further confirmed by HPLC-MS/MS analysis. Collision-induced dissociation (CID) of the authentic adducts, SUD (m/z 254), PRD (m/z 210), HED (m/z 252) and GLD (m/z 268), produced the same daughter ions at m/z 91, and 137. SUD, PRD and HED also produced daughter ions at m/z 154, whereas GLD did not. The ion at m/z 137 was detected with the highest peak intensity in the fragments, and this ion was also identified to be derived from the dopamine spectra (Fig. 4.1).

Proposed chemical formation scheme and HPLC-MS/MS analysis of DHA- and AA-derived dopamine adducts. (a) Proposed reaction scheme of DHA- and AA-derived dopamine adduct formation. (b) The [MH]+ ion m/z 254, 210, 252, and 268 of SUD, PRD, HED, and GLD, respectively, were subjected to CID, and the daughter ions were scanned (left, upper). The proposed structures of individual ions are shown (right, upper). The chemical structure composition of the dopamine adducts is proposed by fragmental analysis (lower)
3 In Vitro and In Vivo Detection of Dopamine Adducts
In our previous studies, to determinate the in vitro formation of the dopamine adducts, the reaction of dopamine with DHA- or AA-hydroperoxides were carried out. The results showed that the four adducts including SUD, PRD, HED and GLD were successfully detected by HPLC-MS/MS among the reaction mixtures.
It has been reported that polyunsaturated fatty acids such as DHA and AA are significantly enriched in the brain (Tapiero et al. 2002), and that there are high levels of basal oxidative stress in the normal brain, which increases with aging (Lin and Flint 2006). To investigate whether the DHA- and AA- derived dopamine adducts can be formed in vivo, the brains of 7-week- and 27-week-old male F344/NSIc rats were removed and the homogenates were used. The detection of the dopamine adducts in the homogenates was carried out by HPLC-MS/MS. The whole adducts were detected in the 7- and 27-week rat brains in both the positive ion mode and negative ion mode of LC-MS/MS. The level of HED and PRD, which are derived from the CH3-teminous of AA and DHA, were more preferentially formed than that of SUD and PRD; however, no significant difference of adduct level was found between the 7- week and 27-week rats.
Dopamine is a natural neurotransmitter in the brain, and its deficiency is a sign of Parkinson’s disease (Ang 2006). Although the mechanism of neurodegeneration is not fully understood, some considerations include dopaminergic neuron abnormalities, dopamine degradation by monoamine oxidase A (MAO-A) or auto-oxidation and modification (Gotz et al. 1994; Hald and Lotharius 2005; LaVoie and Hastings 1999). The in vitro and in vivo detections of DHA- and AA-derived dopamine adducts established may indicate an additional clue to the causes of dopamine deficiency in PD. Although the level of the dopamine adducts was not obviously increased in the 27-week-old rat brain compared to the 7-week-old rat brain, 27 weeks represents only middle age for a rat and the level of basal oxidative stress is increased with age (Navarro and Boveris 2007; Forster et al. 1996; Boveris and Navarro 2008); therefore, further study should confirm these adduct formations in the brain using aging model rats such as 1 year age and more and also PD model animals.
4 Identification of HED as a Potent Inducer of Neuronal Apoptosis
In recent years, several dopamine oxidants and dopamine-modified adducts have been reported, such as neuromelanin (Wakamatsu et al. 2003), aminochrome (Graumann et al. 2002), 6-OHDA (Saner and Thoenen 1971) and 5-S-CDA (LaVoie and Hastings 1999), among them 6-OHDA has been generally known as a potent neurotoxin (Pezzella et al. 1997; Izumi et al. 2005; Maharaj et al. 2005). To test that some of these DHA- and AA-derived dopamine adducts could cause neuronal cell death. The effect of these dopamine adducts on the cell viability in SH-SY5Y cells was studied. The results showed that among the tested dopamine adducts, HED and PRD induced about 80 and 30 % of the cell death, respectively. On the other hand, SUD and GLD had almost no influence on the cell viability, suggesting the death of SH-SY5Y cells was induced only by the CH3-terminus-derived adducts, and not by the COOH-terminus-derived adducts. Of interest, two HED analogs, nonanoyl dopamine (NOD) and lauroyl dopamine (LAD), which were synthesized and characterized by more carbons than HED in the methyl terminus, also showed a significant toxicity to SH-SY5Y cells, suggesting that the number of carbon in the CH3-terminus-derived dopamine adducts might be associated with the adduct-induced cell death.
HED was a potent inducer of SH-SY5Y cell death compared to SUD, PRD and GLD. Because apoptosis is suggested to be involved in neurodegeneration, we then characterized whether HED-induced cell death in SH-SY5Y cells is apoptosis or not. The exposure to HED induces to a dose-dependent decrease in the viable cells. Moreover, the fragmented nuclei were found in cells exhibiting the typical morphological features of apoptosis. In addition, the gel electrophoresis of DNA from the SH-SY5Y cells exposed to HED also displayed nucleosomal DNA fragmentation. HED treatment also led to the time- and dose- dependent cleavage of PARP resulting in the accumulation of the 85-kDa fragment and decreasing in the 116-kDa protein, as well as in the accumulation of the active caspase-3, both which are hallmarks of apoptosis. Moreover, the pretreatment with the caspase-3 inhibitor significantly prevented SH-SY5Y cells from HED-induced DNA fragmentation, providing further evidence that HED induced a caspase-3-mediated apoptotic cell death.
Dopamine-derived metabolites have been reported to inflict damage on neuronal cells (Asanuma et al. 2003). For example, 6-hydroxydopamine (6-OHDA), a hydroxylated analogue of dopamine, has been demonstrated to induce apoptosis in several neuronal cell lines (Hanrott et al. 2006; Chalovich et al. 2006; Jia et al. 2008; Lee et al. 2008). In addition, dopamine autoxidation generating dopamine quinone can react with protein sulfhydryl groups leading to structural modifications of proteins and reduced levels of glutathione (GSH) (Berman and Hastings 1999). HED, an AA-derived dopamine adduct, caused significant cell death in SH-SY5Y cells. Furthermore, the events including DNA fragmentation, chromatin condensation, PARP cleavage and accumulation of active caspase-3 suggest that HED-induced cell death was apoptosis.
5 Regulation of HED-Induced Apoptosis in SH-SY5Y Cells
What might be the signaling mechanism underlying the HED-induced apoptosis is our interests. It is well accepted that reactive oxygen species (ROS) generation is a key contributor to neuronal apoptosis induced by neurotoxin compounds (Chinopoulos and Adam-Vizi 2006). Hence, experiments were first carried out to assess the ROS generation induced by the HED treatment and the possibility that the HED-induced apoptosis is mediated via ROS generation in SH-SY5Y cells. HED led to increased ROS generation in the cells compared to the DMSO-treated cells, whereas the other three dopamine adducts, SUD, PRD and GLD, had a much less effect on the cells. Furthermore, a dose-dependent increase in the ROS generation was found by dichlorofluorescein (DCF) fluorescence staining. The pretreatment with NAC, a potent antioxidant, clearly inhibited the PARP cleavage, indicating that the ROS generation might be critically involved in the HED-induced apoptosis. It is widely accepted that mitochondrial dysfunction may play very important roles in neuronal cell death (Kluck et al. 1997). The cytochrome c release from mitochondria was found in HED-treated cells.
The precise mechanisms regulating apoptotic events in neuronal cells remain largely unclear; however, high levels of ROS generation and the increases in the mitochondrial permeability appear to be common occurrences in many forms of apoptotic neuronal cell death. The finding that HED induced a significant ROS generation and that NAC pretreatment clearly blocked the apoptosis suggests that ROS generation is an essential trigger for HED-induced apoptosis in the SH-SY5Y cells. The source of ROS generation has not been identified, however, the catechol ring is kept in the structure of HED like dopamine and 6-OHDA, therefore, the catechol oxidation might be one of the important causes for the ROS generation in the HED-treated SH-SY5Y cells. The regulation of neuronal apoptosis is generally characterized by the several signaling mediators such as p-53, Bcl-2 family proteins and cytochrome c release (Gorman et al. 2000). A significant release of cytochrome c from mitochondrial fraction in HED-treated SH-SY5Y cells was found, suggesting that the apoptosis may be critically mediated via a mitochondrial abnormality.
6 Monoamine Transporters Are Important in HED-Induced Apoptosis and ROS Generation
Monoamine transporters including the dopamine transporter (DAT), norepinephrine transporter (NET) and 5-HT transporter (5-HTT), which are of fundamental importance for proper signaling between neurons, have been reported to associate with experimental neurotoxins-induced toxicity (Kita et al. 2003). HED possesses a dopamine-based chemical structure, suggesting that the above-described HED cytotoxicity that occurred in the SH-SY5Y cells might be mediated by uptake of HED by monoamine transporters. The pretreatment with both GBR12909 and Imipramine, the inhibitors of DAT and NET/5-HHT, respectively, clearly inhibited the occurrence of the HED-induced PARP cleavage and active caspase-3 expression in the SH-SY5Y cells. Furthermore, the ROS generation by HED was also found to be suppressed in these two inhibitor-pretreated cells. The result that both monoamine transporter inhibitors showed markedly inhibitive effect on the HED-induced apoptosis and ROS generation suggested that HED might be primarily transported into the SH-SY5Y cells by the monoamine transporters, and inflicted damage on the cells.
To characterize whether the HED-induced cytotoxicity is specific to neuronal cells, our study investigated the effect of HED on apoptotic cell death and ROS generation in mouse embryonic fibroblast NIH-3T3 cells in comparison to that of the SH-SY5Y cells. A dose-dependent analysis revealed that HED led to no apoptotic cell death in the NIH-3T3 cells estimated by Hoechst 33258 and Propidium Iodide (PI) nuclear staining. A further quantitative analysis of the apoptotic cells by flow cytometry also indicated apoptosis in SH-SY5Y cells, whereas not in the NIH-3T3 cells. Moreover, no ROS generation was found in the HED-treated NIH-3T3 cells; on the other hand, the HED analogs, NOD and LAD, also induced only a slight ROS generation in the NIH-3T3 cells. Monoamine transporter is known to be absent in NIH-3T3 cells, which may indicate that the HED-induced cytotoxicity might be specific to neuronal cells.
Monoamine transporters are of fundamental importance for proper signaling between neurons. Plasma membrane transporters, the major subclass of intracellular transporters (Gethe et al. 2006), include the dopamine transporter (DAT), norepinephrine transporter (NET), and 5-HT transporter (5-HTT). In this study, pretreatment with inhibitors of DAT, NET and 5-HTT significantly suppressed ROS generation and apoptosis events induced by HED. In the case of 6-OHDA, similar to HED, a high affinity for several catecholaminergic plasma membrane transporters, such as DAT and NET, is also essential for its entrance into the neuronal cells to inflict damage. The dependence of monamine transporter is considered to be due to a structural similarity between the HED, dopamine and norepinephrine. The necessity of the monoamine transporter in HED-induced cytotoxicity was further demonstrated by the result that HED could not induce apoptotic cell death and ROS generation in the monoamine transporter-absent NIH-3T3 cells, which also indicates that HED may selectively induce cytotoxicity in different cell lines.
7 Conclusion and Note
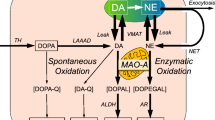
Four amide-linkage adducts of dopamine with DHA and AA were synthesized and the in vivo formation during the reaction of lipid hydroperoxides with dopamine were also revealed. HED, an AA-derived dopamine adduct, as a potent neurotoxin based on the significant induction of ROS generation and apoptosis in human neuroblastoma SH-SY5Y cells. The mechanism of HED-induced apoptosis has not been fully established in this study; however, it seems to be mediated by ROS generation, mitochondrial abnormalities, and monoamine transporter (Fig. 4.2).

Proposed mechanism of HED-induced apoptosis in SH-SY5Y cells
In fact, either DHA or AA is located almost exclusively in the SN2-position of phosphoglycerides found in the neural cell membranes (Ma et al. 2007; Beermann et al. 2005); however, free fatty acid levels are reported to increase with aging due to an increasing degradation by PLA2 (Rosenberger et al. 2004; Qu et al. 2003; Rapoport 1999), a phospholipase A2, which selectively acts on phosphoglycerides (Diez et al. 1994). DHA is the most enriched polyunsaturated fatty acid in the brain, and it has been implicated that DHA concentration is decreased in AD brain (Bazan et al. 2002); hence, the DHA-derived dopamine adducts formed in this study may be useful biomarkers for not only PD but also AD.
References
Ang SL (2006) Transcriptional control of midbrain dopaminergic neuron development. Development 133:3499–3506
Asanuma M, Miyazaki I, Ogawa N (2003) Dopamine- or L-DOPA-induced Neurotoxicity: the role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox Res 5:165–176
Balaban RS, Nemoto S, Finkel T (2005) Mitochondria, oxidants, and aging. Cell 120:483–495
Bazan NG, Palacios-Pelaez R, Lukiw WJ (2002) Hypoxia signaling to genes: significance in Alzheimer’s disease. Mol Neurobiol 26:283–298
Beermann C, Möbius M, Winterling N, Schmitt JJ, Boehm G (2005) sn-position determination of phospholipid-linked fatty acids derived from erythrocytes by liquid chromatography electrospray ionization ion-trap mass spectrometry. Lipids 40:211–218
Berman SB, Hastings TG (1999) Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson’s disease. J Neurochem 73:1127–1137
Bove J, Prou D, Perier C, Przedborski S (2005) Toxin-induced models of Parkinson’s disease. NeuroRx 2:484–494
Boveris A, Navarro A (2008) Brain mitochondrial dysfunction in aging. IMUBM Life 60:308–314
Buhmann C, Arlt S, Kontush A, Moeller-Bertram T, Sperber S, Oechsner M, Stuerenburg HJ, Beisiegel U (2004) Plasma and CSF markers of oxidative stress are increased in Parkinson’s disease and influenced by antiparkinsonian medication. Neurobiol Dis 15:160–170
Chalovich EM, Zhu JH, Caltagarone J, Bowser R, Chu CT (2006) Functional repression of cAMP response element in 6-hydroxydopamine-treated neuronal cells. J Biol Chem 281:17870–17881
Chinopoulos C, Adam-Vizi V (2006) Calcium, mitochondria and oxidative stress in neuronal pathology. Novel aspects of an enduring theme. FEBS J 273:433–450
Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Agid Y, Lees A, Jenner P, Marsden CD (1989) Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J Neurochem 52:381–389
Dexter DT, Holley AE, Filtter WD, Slater TF, Wells FR, Daniel SE, Lees A, Jenner P, Marsden CD (1994) Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Mov Disord 9:92–97
Diez E, Chilton FH, Stroup G, Mayer RJ, Winkler JD, Fonteh AN (1994) Fatty acid and phospholipid selectivity of different phospholipase A2 enzymes studied by using a mammalian membrane as substrate. Biochem J 301:721–726
Fahn S, Cohen G (1992) The oxidant stress hypothesis in Parkinson’s disease: evidence supporting it. Ann Neurol 32:804–812
Forster MJ, Dubey A, Dawson KM, Stutts WA, Lal H, Sohal RS (1996) Age-related losses of cognitive function and motor skills in mice are associated with oxidative protein damage in the brain. Proc Natl Acad Sci USA 93:4765–4769
Galvan A, Wichmann T (2008) Pathophysiology of parkinsonism. Clin Neurophysiol 119:1459–1474
Gethe U, Andersen PH, Larsson OM, Schousboe A (2006) Neurotransmitter transporters: molecular function of important drug targets. Trends Pharmacol Sci 27:375–383
Gorman AM, Ceccatelli S, Orrenius S (2000) Role of mitochondria in neuronal apoptosis. Dev Neurosci 22:348–358
Gotz ME, Kunig G, Riederer P, Youdim MB (1994) Oxidative stress: free radical production in neural degeneration. Pharmacol Ther 63:37–122
Graumann R, Paris I, Martinez-Alvarado P, Rumanque P, Perez-Pastene C, Cardenas SP, Marin P, Diaz-Grez F, Caviedes R, Caviedes P, Segura-Aguilar J (2002) Oxidation of dopamine to aminochrome as a mechanism for neurodegeneration of dopaminergic systems in Parkinson’s disease. Possible neuroprotective role of DT-diaphorase. Pol J Pharmacol 54:573–579
Hadders-Algra M (2008) Prenatal long-chain polyunsaturated fatty acid status: the importance of a balanced intake of docosahexaenoic acid and arachidonic acid. J Perinat Med 36:101–109
Hald A, Lotharius J (2005) Oxidative stress and inflammation in Parkinson’s disease: is there a causal link? Exp Neurol 176:154–162
Hanrott K, Gudmunsen L, O'Neill MJ, Wonnacott S (2006) 6-hydroxydopamine-induced apoptosis is mediated via extracellular auto-oxidation and caspase 3-dependent activation of protein kinase Cdelta. J Biol Chem 281:5373–5382
Izumi Y, Sawada H, Sakka N, Yamamoto N, Kume T, Katsuki H, Shimohama S, Akaike A (2005) p-Quinone mediates 6-hydroxydopamine-induced dopaminergic neuronal death and ferrous iron accelerates the conversion of p-quinone into melanin extracellularly. J Neurosci Res 79:849–860
Jenner P (2003) Oxidative stress in Parkinson’s disease. Ann Neurol 53:S26–S38
Jia Z, Zhu H, Misra HP, Li Y (2008) Potent induction of total cellular GSH and NQO1 as well as mitochondrial GSH by 3H-1,2-dithiole-3-thione in SH-SY5Y neuroblastoma cells and primary human neurons: protection against neurocytotoxicity elicited by dopamine, 6-hydroxydopamine, 4-hydroxy-2-nonenal, or hydrogen peroxide. Brain Res 1197:159–169
Kato Y, Makino Y, Osawa T (1997) Formation of N ε-(hexanonyl) lysine in protein exposed to lipid hydroperoxide A plausible marker for lipid hydroperoxide-derived protein modification. J Lipid Res 38:1334–1346
Kato Y, Mori Y, Makino Y, Morimitsu Y, Hiroi S, Ishikawa T, Osawa T (1999) Formation of N ε-(hexanonyl) lysine in protein exposed to lipid hydroperoxide A plausible marker for lipid hydroperoxide-derived protein modification. J Biol Chem 274:20406–20414
Kato Y, Osawa T (1998) Detection of oxidized phospholipid-protein adducts using anti-15-hydroperoxyeicosatetraenoic acid-modified protein antibody: contribution of esterified fatty acid-protein adduct to oxidative modification of LDL. Arch Biochem Biophys 351:106–114
Kawai Y, Fujii H, Kato Y, Kodama M, Naito N, Uchida K, Osawa T (2004) Esterified lipid hydroperoxide-derived modification of protein: formation of a carboxyalkylamide-type lysine adduct in human atherosclerotic lesions. Biochem Biophys Res Commun 313:271–276
Kawai Y, Fujii H, Okada M, Tsuchie Y, Uchida K, Osawa T (2006) Formation of Nepsilon-(succinyl)lysine in vivo: a novel marker for docosahexaenoic acid-derived protein modification. J Lipid Res 47:1386–1398
Kawai Y, Kato Y, Fujii H, Mkino Y, Mori Y, Naito N, Osawa T (2003) Immunochemical detection of a novel lysine adduct using an antibody to linoleic acid hydroperoxide-modified protein. J Lipid Res 44:1124–1131
Kita T, Wagner GC, Nakashima T (2003) Current research on methamphetamine-induced neurotoxicity: animal models of monoamine disruption. J Pharmacol Sci 92:178–195
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD (1997) The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 275:1132–1136
LaVoie MJ, Hastings TG (1999) Peroxynitrite‐and nitrite‐induced oxidation of dopamine: implications for nitric oxide in dopaminergic cell loss. J Neurochem 73:2546–2554
Lee YM, Park SH, Shin DI, Hwang JY, Park B, Park YJ, Lee TH, Chae HZ, Jin BK, Oh TH, Oh YJ (2008) Oxidative modification of peroxiredoxin is associated with drug-induced apoptotic signaling in experimental models of Parkinson disease. J Biol Chem 283:9986–9998
Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH (1998) The ubiquitin pathway in Parkinson’s disease. Nature 395:451–452
Lin MT, Flint BM (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 433:787–795
Liu XB, Shibata T, Hisaka S, Osawa T (2008) DHA hydroperoxides as a potential inducer of neuronal cell death: a mitochondrial dysfunction-mediated pathway. J Clin Biochem Nutr 43:26–33
Long EK, Murphy TC, Leiphon LJ, Watt J, Morrow JD, Milne GL, Howard JR, Picklo MJ Sr (2008) Trans-4-hydroxy-2-hexenal is a neurotoxic product of docosahexaenoic (22:6; n-3) acid oxidation. J Neurochem 105:714–724
Ma QL, Teter B, Ubeda OJ, Morihara T, Dhoot D, Nyby MD, Tuck ML, Frautschy SA, Cole GM (2007) Omega-3 fatty acid docosahexaenoic acid increases SorLA/LR11, a sorting protein with reduced expression in sporadic Alzheimer's disease (AD): relevance to AD prevention. J Neurosci 27:14299–14307
Maharaj H, Mahara DS, Scheepers M, Mokokong R, Daya S (2005) l-DOPA administration enhances 6-hydroxydopamine generation. Brain Res 1063:180–186
Mariani E, Polidori MC, Cherubini A, Mecocci P (2005) Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. J Chromatogr B 827:65–75
Maruyama W, Naoi M (2002) Cell death in Parkinson’s disease. J Neurol 249(Suppl 2):6–10
Montine KS, Quinn JF, Zhang J, Fessel JP, Roberts LJ 2nd, Morrow JD, Montine TJ (2004) Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem Phys Lipids 128:117–124
Navarro A, Boveris A (2007) The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol 292:C670–C686
Pezzella A, d’Ischia M, Napolitano A, Misuraca G, Prota G (1997) Iron-mediated generation of the neurotoxin 6-hydroxydopamine quinone by reaction of fatty acid hydroperoxides with dopamine: a possible contributory mechanism for neuronal degeneration in Parkinson’s disease. J Med Chem 40:2211–2216
Porter NA, Caldwell SE, Mills KA (1995) Mechanisms of free radical oxidation of unsaturated lipids. Lipids 30:277–290
Qu Y, Chang L, Klaff J, Seeman R, Balbo A, Rapoport SI (2003) Imaging of brain serotonergic neurotransmission involving phospholipase A2 activation and arachidonic acid release in unanesthetized rats. Brain Res Brain Res Protoc 12:16–25
Rapoport SI (1999) In vivo fatty acid incorporation into brain phospholipids in relation to signal transduction and membrane remodeling. Neurochem Res 24:1403–1415
Rosenberger TA, Villacreses NE, Hovda JT, Bosetti F, Weerasinghe G, Wine RN, Harry GJ, Rapoport SI (2004) Rat brain arachidonic acid metabolism is increased by a 6-day intracerebral ventricular infusion of bacterial lipopolysaccharide. J Neurochem 88:1168–1178
Saner A, Thoenen H (1970) Model experiments on the molecular mechanism of action of 6-hydroxydopamine. Mol Pharmacol 7:147–154
Schapira AH (2001) Causes of neuronal death in Parkinson’s disease. Adv Neurol 86:155–162
Shamoto-Nagai M, Maruyama W, Kato Y, Isobe K, Tanaka M, Naoi M, Osawa T (2003) An inhibitor of mitochondrial complex I, rotenone, inactivates proteasome by oxidative modification and induces aggregation of oxidized proteins in SH-SY5Y cells. J Neurosci Res 74:589–597
Shibata T, Iio K, Kawai Y, Shibata Y, Kawaguchi M, Toi S, Kobayashi M, Kobayashi M, Yamamoto K, Uchida K (2006) Identification of a lipid peroxidation product as a potential trigger of the p53 pathway. J Biol Chem 281:1196–1204
Siegel SJ, Bieschke J, Powers ET, Kelly JW (2007) The oxidative stress metabolite 4-hydroxynonenal promotes Alzheimer protofibril formation. Biochemistry 46:1503–1510
Simopoulos AP (1999) Essential fatty acids in health and chronic disease. Am J Clin Nutr 70(Suppl):560–569
Tapiero H, Ba GN, Couvreur P, Tew KD (2002) Polyunsaturated fatty acids (PUFA) and eicosanoids in human health and pathologies. Biomed Pharmacother 56:215–222
Wakamatsu K, Fujikawa K, Zucca FA, Zecca L, Ito S (2003) The structure of neuromelanin as studied by chemical degradative methods. J Neurochem 86:1015–1023
Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y (1996) Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci USA 93:2696–2701
Zecca L, Youdim MBH, Riederer P, Connor JR, Crichton RR (2004) Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci 5:863–873
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Liu, X., Yamada, N., Osawa, T. (2014). Amide-Type Adduct of Dopamine – Plausible Cause of Parkinson Diseases. In: Kato, Y. (eds) Lipid Hydroperoxide-Derived Modification of Biomolecules. Subcellular Biochemistry, vol 77. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-7920-4_4
Download citation
DOI: https://doi.org/10.1007/978-94-007-7920-4_4
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-7919-8
Online ISBN: 978-94-007-7920-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)