
Abstract
The role of genetic diversity is crucial for future improvements to meet societal demand for food security under a climate change scenario. From this perspective, it is thus crucial to understand the structure and evolution of crop species and their wild relatives. The common bean (Phaseolus vulgaris L.) is the world’s most important food legume for direct use, and the demand for this crop can be expected to increase based on the current trends in population growth and bean consumption. The wild P. vulgaris has a Mesoamerican origin, and since its expansion, it has become distributed from northern Mexico to north-western Argentina, which has led to the formation of two major gene pools in these geographical regions. Domestication took place after the formation of these gene pools, and their structure is still clearly evident in both the wild and the domesticated forms. This evolutionary scenario renders P. vulgaris almost unique among crops, and therefore particularly useful to investigate crop domestication, as this process can be studied in the same species as a replicated experiment (i.e., in Mesoamerica and in the Andes). The present review offers an overview of the current knowledge on the evolutionary history of P. vulgaris L. including speciation, domestication, diversification, and crop expansion outside its centers of domestication in Mesoamerica and in the Andes. Within this context, we also present a description of the available genomic tools and the germplasm collections that are at present available for genetic studies on the common bean, while showing their potential for improvements to the productivity and quality of this crop.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Legumes represent an important component of agricultural food crops and they have a crucial role in both farming systems and the human diet, especially in developing countries. Globally, legumes complement cereal crops as a source of protein and minerals, with a harvested area of about one-tenth that collectively under cereals (Akibode and Maredia 2011).
The important role of food legumes in the farming systems is underlined by the demonstration that over the past 15 years overall legume production has increased at a greater rate than the growth rate of the world population (Akibode and Maredia 2011). Among pulse crops, Phaseolus is a large and diverse genus that comprises about 70 species from Central and North America (Freytag and Debouck 2002), five of which have been domesticated (P. vulgaris, P. dumosus, P. coccineus, P. acutifolius, P. lunatus), and with a few additional species that show signs of incipient domestication (Delgado-Salinas et al. 2006).
The common bean (Phaseolus vulgaris L.) is the world’s most important food legume for direct use, with a production of about 12 million metric tons per year. The leading countries in this production are Latin America and sub-Saharan Africa, where three-quarters of this crop is grown (http://faostat.fao.org/; Akibode and Maredia 2011).
Considering the current trends in population growth and bean consumption, the demand for P. vulgaris can be expected to increase (CIAT report 2001, http://webapp.ciat.cgiar.org/ciatinfocus/beans.htm; Akibode and Maredia 2011), and compelling questions about this species must be addressed in the future. Bean productivity, food quality, and resistance to biotic and abiotic factors, among others, would realistically be the aim for future investigations to meet the challenges posed by climate change and the fast increasing demand for food.
It has been shown that an acceleration of the rate of crop improvement can be achieved by taking advantage of high-throughput genomic technologies that are having significant effects on the management of gene banks and on the way germplasm collections are exploited (Tuberosa et al. 2011). Different tools and sources of genomic information on the bean genome are nowadays available to investigate the diversity present in this species, including molecular linkage maps, expressed sequence tags (EST) collections, bacterial artificial chromosome libraries, a physical map, and soon, a whole-genome sequence (McClean et al. 2008, 2013; Gepts et al. 2008; http://www.phytozome.net/commonbean.php). The advent of next-generation sequencing has revolutionized genomic and transcriptomic approaches to biology (Gupta et al. 2008; Mardis 2008). These new sequencing tools are also valuable for single nucleotide polymorphism (SNP) discovery and the detection of genetic markers in populations, which in turn, can be exploited in different studies (Davey et al. 2011).
Despite there still being limitations to contemporary common bean breeding, genomics-assisted techniques have been widely exploited in this species, and have enhanced the effectiveness of breeding programs and responses to selection (Beaver and Osorno 2009; Tuberosa et al. 2011). Marker-assisted selection is often routinely used for traits controlled by major loci, although marker-assisted selection for complex quantitative traits still remains a challenging task in breeding programs (Miklas et al. 2006; Tuberosa et al. 2011). Genome-wide association mapping is an approach that is being increasingly adopted to dissect out the genetic basis of target traits, and when it is applied to wild populations, it has substantial benefits for conservation genetics and ecology (Allendorf et al. 2010; Galeano et al. 2012). Concurrently, diversity analyses and evolution of the species can be understood by investigating domestication, local adaptation, genetic drift, and gene flow through novel genomic techniques (Davey et al. 2011; van Heerwaarden et al. 2011).
The lack of a whole genome sequence for the common bean has been a major limitation for such an important crop species. The forthcoming availability for the scientific community of the biotechnology tools that are available for other crops will enhance the competitiveness of this species. Realistically, the sequence will provide powerful tools to improve agronomic and nutritional traits, which is particularly important to maintain and improve the nutritional status of poor individuals. Future genomic studies will contribute to the gaining of insights into this important crop, such as comparative gene discovery in legumes, fine-mapping and candidate gene identification, and the identification of Phaseoleae domestication and adaptation genes.
2 Origin of the Common Bean
According to the geographical distribution of most of the species belonging to the Phaseolus genus, these are considered to be of Mesoamerican origin (Freytag and Debouck 1996, 2002; Delgado-Salinas et al. 1999, 2006). Delgado-Salinas et al. (2006) analyzed internal transcribed spacers (ITS) of the ribosomal DNA and the chloroplast trnK locus, and they showed that the Phaseolus crown clade is no older than ca. 4–6 My. The present-day form of Mexico was apparent by the Late Miocene (5 My ago), with a final major event of subduction volcanism that resulted in the modern Trans-Mexican Volcanic Belt. This strongly suggests that Phaseolus diversification took place during and after this major tectonic activity (Delgado-Salinas et al. 2006), and thus evolved well after the period when the land bridge connecting Mesoamerica and South America was formed, which was ca. 7 My ago (Coates et al. 2004). Delgado-Salinas et al. (2006) detected eight principal crown clades within Phaseolus, with the vulgaris group as the oldest, at ca. 4 My. This group includes four of the five domesticated species of the genus (P. vulgaris, P. dumosus, P. coccineus, P. acutifolius). The closest relatives to P. vulgaris are the Mesoamerican species P. dumosus and P. coccineus, and these three species together are partially intercrossable. The other domesticated species (P. lunatus, P. acutifolius) are more distantly related. On the basis of sequence data of the α-amylase inhibitor gene, P. vulgaris diverged from P. dumosus and P. coccineus ca. 2 My ago (Gepts et al. 1999).
Among the five domesticated Phaseolus species, P. vulgaris is the most important economically, as it is the main grain legume for direct human consumption. It is a rich source of protein, vitamins, minerals and fiber, especially in less-developed countries (http://www.fao.org/index_en.htm, 2010; Brougthon et al. 2003). P. vulgaris is a true autogamous diploid species, with 22 chromosomes and a haploid genome size that is estimated to be between 587 Mbp and 637 Mbp (Arumuganathan and Earle 1991; Bennett and Leitch 1995, 2010).
Wild P. vulgaris is widely distributed from northern Mexico to north-western Argentina (Toro et al. 1990), and it is characterized by two major eco-geographical gene pools: those of Mesoamerica and the Andes. These two gene pools show parallel wild and domesticated geographical structures, as shown by several studies based on different datasets, including plant morphology (Singh et al. 1991b), seed proteins (Gepts et al. 1986; Gepts and Bliss 1985), allozymes (Koenig and Gepts 1989), restriction fragment length polymorphism (R Becerra-Velásquez and Gepts 1994), random amplified polymorphic DNA (RAPD, Freyre et al. 1996), amplified fragment length polymorphism (AFLP; Papa and Gepts 2003; Rossi et al. 2009), and simple sequence repeats (microsatellites, SSRs; Kwak and Gepts 2009).
In the 1980’s, a wild P. vulgaris population was discovered in northern Peru and Ecuador (Debouck et al. 1993). Kami et al. (1995) analyzed a portion of the gene that codes for the seed protein phaseolin, and they identified a new phaseolin type (type I) for this population from northern Peru–Ecuador that was not present in the other gene pools, thus indicating that this population is a new distinct wild gene pool. The type I phaseolin gene does not carry the tandem direct repeats that are present in Mesoamerican and Andean accessions. With the consideration that duplications that generate tandem direct repeats are more likely than deletions that specifically eliminate a member of a tandem direct repeat, Kami et al. (1995) suggested that type I phaseolin is ancestral to the other phaseolin sequences of P. vulgaris. This arises because duplications can occur in many locations along a sequence, whereas deletions can occur only at the site of the tandem direct repeats. Thus, the most credited hypothesis on the origin of the common bean was that from the core area of the western slopes of the Andes in northern Peru and Ecuador, from where the wild bean was dispersed northwards (Colombia, Central America and Mexico) and southwards (southern Peru, Bolivia and Argentina), which resulted in the Mesoamerican and Andean gene pools, respectively (Kami et al. 1995). However, recently, this hypothesis has been called into question by different studies (Rossi et al. 2009; Nanni et al. 2011; Bitocchi et al. 2012, 2013; Desiderio et al. 2013). In particular, Bitocchi et al. (2012) clearly indicated a Mesoamerican origin of the common bean by investigating the nucleotide diversity at five different gene fragments on a wide sample of wild P. vulgaris that is representative of its geographical distribution.
The first evidence towards this statement was the occurrence of a bottleneck prior to domestication for the Andean gene pool. This is supported by the higher genetic diversity detected for the Mesoamerican gene pool, as compared to the Andean gene pool, which resulted in a 90 % loss of diversity for the Andean gene pool (Bitocchi et al. 2012). This trend had already been reported in earlier (Freyre et al. 1996; Koenig and Gepts 1989) and more recent studies (Kwak and Gepts 2009; Rossi et al. 2009; Nanni et al. 2011; Bitocchi et al. 2013; Desiderio et al. 2013). However, the genetic diversity reduction using sequence data was about two-fold, 13-fold and three-fold higher than those in a comparable sample of P. vulgaris genotypes using AFLP data (45 %; Rossi et al. 2009), SSR data (7 %; Kwak and Gepts 2009) and chloroplast (cp)SSR data (26 %; Desiderio et al. 2013), respectively. This is clear evidence of the crucial role of marker mutation rates for describing the diversity of plant populations (Thuillet et al. 2005). In particular, the loss of diversity detected with cpSSRs is intermediate between the SSRs and AFLPs, as is their mutation rate (Provan et al. 1999; Marshall et al. 2002). Indeed, as showed in several studies (Glémin and Bataillon 2009, Rossi et al. 2009; Nanni et al. 2011; Bitocchi et al. 2012, 2013; Desiderio et al. 2013), in populations that have experienced a bottleneck, the differences in loss of diversity estimates using different markers are related to their different mutation rates: in markers characterized by high mutation rates, such as SSRs, the recovery of the diversity lost after a bottleneck is faster than for markers with lower mutation rates, such as sequence data.
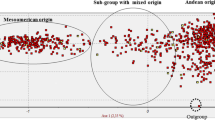
The second novel outcome of the analysis carried out using sequence data was the population structure identified in Mesoamerica. Indeed, before the study of Bitocchi et al. (2012), even if it was known that the wild Mesoamerican gene pool was characterized by a high population structure (Papa and Gepts 2003), a clear distinction into groups had never been found, and thus Mesoamerica was usually considered as a single gene pool. The main reason for this was probably related to the nature of the markers used; indeed, previous studies did not clearly detect any population subdivisions in Mesoamerica due to hybridization and recombination between the different groups, which reduced the discriminatory power of the multilocus molecular markers used (Kwak and Gepts 2009; Rossi et al. 2009). As sequence data are less prone to these factors, Bitocchi et al. (2012) showed that the Mesoamerican accessions can indeed be split into four distinct genetic groups: B1, B2, B3 and B4 (Fig. 20.1). The B1 group was represented by accessions distributed across all of the geographical area, from the north of Mexico down to Colombia. The other three groups were composed of only Mexican accessions. The B2 group was spread from central to southern Mexico, while the B3 and B4 groups were present in a wide area of central Mexico (Fig. 20.1). Investigations into the relationships between these different groups have shown that, remarkably, there is no clear distinction between the Mesoamerican and Andean wild gene pools, while different relationships of the Mesoamerican groups with the north Peru–Ecuador and Andean gene pools were found (Bitocchi et al. 2012). In particular, the Andean wild accessions were more related to the Mesoamerican B3 accessions, and the northern Peru–Ecuador accessions to the Mesoamerican B4 accessions (Fig. 20.1). The Bitocchi et al. (2012) study shows clear evidence of a Mesoamerican origin of the common bean, which was most likely located in Mexico, which is consistent with the known distribution of most of the close relatives of P. vulgaris. Thus, both of the gene pools from South America originated through different migration events from the Mesoamerica populations of central Mexico. These results are strongly supported by those obtained at chloroplastic DNA level on a partially overlapping sample of wild accessions (Desiderio et al. 2013). Bitocchi et al. (2012) suggested that the wild common bean from northern Peru and Ecuador is a relict population that only represents a fraction of the genetic diversity of the ancestral population. Considering that the results of Kami et al. (1995) that indicated that phaseolin type I (PhI) is an ancestral phaseolin are relatively robust, the absence of this phaseolin type in Mesoamerica would be due to its extinction in this gene pool, or alternatively, it might still be present, but just not included in the samples analyzed in the literature.

Representation of the geographical distribution of the clusters identified by the Bayesian clustering analysis performed by Bitocchi et al. (2012). See legend for correspondence between colors and genetic clusters
3 Domestication of P. vulgaris
Domestication is a complex process that modifies a wild plant and makes it into a crop. In P. vulgaris, this involved several morphological and physiological changes, such as differences in growth habit (indeterminate vs determinate), seed dormancy (present vs not present), photoperiod sensitivity (short-day vs insensitivity), shape, color and size of the plant and its harvested parts, and the dissemination mechanisms (shattering vs non-opening pods). All of these structural and functional modifications shared among most crop species (the domestication syndrome) make them genetically different from their wild types, and confer better adaptation to different agro-ecosystems (Gepts and Papa 2002). The process of common-bean domestication has been studied in detail, and the major domestication traits have been mapped (Koinange et al. 1996). Koinange et al. (1996) performed a quantitative trait locus (QTL) analysis using a recombinant inbred population derived from a cross of wild × cultivated, and they found that the QTLs for the traits measured tended to cluster into several regions on the bean linkage map. Some of the candidate genes associated with the domestication process have been characterized (Anthony et al. 1990; Kwak et al. 2006, 2008; Repinski et al. 2012).
One of the consequences of domestication that is common to most crop species is the reduction of genetic diversity due to a founder effect (Glémin and Bataillon, 2009). In analyzing Mesoamerican and Andean wild and domesticated populations using AFLP markers, Rossi et al. (2009) observed a strong reduction in the genetic diversity due to domestication (wild vs domesticated samples) only in the Mesoamerica population (ΔH = 0.32). Markers that differ substantially in their mutation rates can show very different patterns of molecular diversity, and indeed, Kwak and Gepts (2009) used SSR markers to show a lower reduction in Mesoamerica (ca. 10 %).
The data from Nanni et al. (2011) from the analysis of a genomic sequence in the wild and domesticated common bean that is similar to SHATTERPROOF 1 (PvSHP1), the gene involved in the control of fruit shattering in Arabidopsis thaliana, offered the first estimates of the effects of domestication on nucleotide variation in this species, based on a relatively large and representative sample of genotypes. The loss of diversity in the domesticated accessions in the Andes was 54 %; in Mesoamerica, this loss of diversity ranged from 65–69 % when compared with only the wild accessions from Mexico, and with all of the Mesoamerican wild populations, respectively. These results have been confirmed more recently by the analysis of five gene fragments in 214 accessions (102 wild and 112 domesticated) of P. vulgaris (Bitocchi et al. 2013). Indeed, it was shown that the domestication of the common bean in Mesoamerica induced a severe reduction (72 %) in genetic diversity, which was consistently reproduced for all of the five genes studied (range, 44–98 %). Additionally, the pattern was also confirmed in the Andes data (loss of diversity, 27 %). However, the reduction in genetic diversity was three-fold greater in Mesoamerica compared with the Andes. As proposed by Bitocchi et al. (2013), this difference can be explained as the result of the bottleneck that occurred before domestication in the Andes (Rossi et al. 2009; Bitocchi et al. 2012), which strongly impoverished the Andean wild populations, leading to the minor effects of the subsequent domestication bottleneck (i.e., sequential bottleneck). These findings show the importance of considering the evolutionary history of a crop species as a major factor that influences its current level and structure of genetic diversity.
Papa et al. (2005) showed that genes for domestication are located in regions of high divergence between wild and domesticated P. vulgaris. Also, the regions linked to the domestication loci have probably been less exploited by farmers and breeders, and these are the ones where the highest diversity of the wild relatives is located. Several studies have clearly indicated that the use of wild relatives can have a tremendous impact on crop improvement (Tanskley and McCouch 1997; McCouch 2004); therefore, to better exploit the genetic diversity that is present in the wild relatives of a crop, knowledge of the locations of the genes involved in the domestication syndrome and the proportion of the genome affected by domestication appears to be crucial. This knowledge of the domestication loci is indeed useful in two main ways: for identification of markers that are tightly linked to undesirable genes (e.g., shattering); and for the possibility to identify the surrounding chromosomal regions that would be most likely to harbor the highest and historically less exploited diversity of the wild germplasm. Using the approach of a genome scan for the signature of domestication, Papa et al. (2007) estimated that a large fraction of the genome of the common bean appears to be under the effects of selection during domestication (about 16 %). Molecular analysis was carried out using 2,506 AFLP markers on 14 bulks of individuals (seven bulks of wild, and seven bulks of domesticated). For the allelic frequencies of the wild and domesticated populations based on the bulk analysis, and for each marker, in both datasets, the departure from the neutral expectation was evaluated using a method based on F ST , to identify loci that were putatively under selection. Moreover, AFLP markers analyzed on single genotypes were mapped on a P. vulgaris consensus map, and most of those that were putatively under the effects of selection due to the domestication loci were localized close to genes and QTLs that are linked to the domestication process.
The common bean was domesticated independently in Mesoamerica and in the Andes. Two independent domestication events in the Americas have been documented in several studies, and a large set of coherent data have been obtained using different approaches based on molecular markers and morphological characteristics (Gepts et al. 1986; Gepts and Bliss 1988; Koenig and Gepts 1989; Gepts and Debouk 1991; Singh et al. 1991a, b, c; Becerra Velasquez and Gepts 1994; Freyre et al. 1996; Tohme et al. 1996; Gepts 1998; Delgado-Salinas et al. 1999; Papa and Gepts 2003; Blair et al. 2006a, b; Diaz and Blair 2006; Angioi et al. 2009a; Kwak and Gepts 2009; Rossi et al. 2009; Nanni et al. 2011; Blair et al. 2012; Bitocchi et al. 2013). These two independent domestication events, one in Mesoamerica and one in the Andes, gave origin to two major domesticated gene pools (Papa et al. 2006; Acosta-Gallegos et al. 2007; Angioi et al. 2009a). Following domestication, the domesticated gene pools of the common bean appear to have been organized into four Mesoamerican (Durango, Jalisco, Mesoamerica, Guatemala) and three Andean (Nueva Granada, Peru, Chile) races (Singh et al. 1991c; Beebe et al. 2000, 2001). All of these races differ in ecological adaptation, geographical range, morpho-agronomic traits, allozyme alleles, and random amplified polymorphic DNA markers (Singh et al. 1991c; Beebe et al. 2000) and their origins are still controversial. It is not known if they are the results of multiple independent domestications within each region, or the result of a single domestication in each region followed by diversification under cultivation.
Indeed, a topic of discussion is whether multiple domestications have occurred within each gene pool and the role of gene flow and introgression. For the Mesoamerican gene pool, different studies have suggested both single (Gepts et al. 1986; Papa and Gepts 2003; Kwak and Gepts 2009; Kwak et al. 2009; Rossi et al. 2009) and multiple domestication events (Singh et al. 1991a, b, c; Beebe et al. 2000; Chacón et al. 2005). In the Andes, the situation is even less clear, because of the lack of geographic structure of the genetic diversity, which reduces the resolving power of the molecular studies. However, both single and multiple domestications have been suggested within the Andean gene pool (Beebe et al. 2001; Santalla et al. 2004; Chacón et al. 2005; Rossi et al. 2009).
Recently, and for the first time, Nanni et al. (2011) approached this question by analyzing nucleotide data, and these strongly support a single domestication event in Mesoamerica. However, the question could not be answered in the Andean gene pool because of the low level of diversity. Using multilocus sequence data to test multiple demographic models in domesticated P. vulgaris landraces, Mamidi et al. (2011) suggested that there was a single domestication event in each gene pool. This issue was also undertaken by Bitocchi et al. (2013), by analyzing nucleotide data from five gene fragments, and they clearly indicated a single domestication event for the Mesoamerican gene pool, and suggested a similar scenario for the Andean gene pool.
Other important common bean domestication matters that are still under debate are the identification of the presumed geographic center of domestication, and the domestication dating. Bitocchi et al. (2013) addressed this question and they suggested the Oaxaca valley in Mesoamerica (but see Kwak et al. 2009), and southern Bolivia and northern Argentina in South America, as the origins of common bean domestication.
These results, although encouraging, should be considered with caution, and further efforts are needed to investigate these aspects more deeply, mainly because of the low genetic diversity of the Andean gene pool and because other events might have had roles in shaping the common bean diversity in the areas investigated, such as gene flow between wild and domesticated common bean.
Finally, an important aspect is the occurrence of gene flow between wild and domesticated forms. Using AFLP markers, Papa and Gepts (2003) analyzed the genetic structure of wild and domesticated populations of P. vulgaris from Mexico (with different levels of sympatry). Their results highlighted that the wild and domesticated forms are not genetically isolated, as they show moderate and asymmetric gene flow (> 3-fold higher from domesticated to wild, than vice versa). In the presence of gene flow, the marked phenotypic differences between the two forms growing in sympatry are explained by the selection acting against the domesticated alleles in a wild context, and against the wild alleles in an agroecosystem.
Thus, the common bean scenario is characterized by two independent domestication events that gave origins to two clearly differentiated gene pools, and by the co-existence of the wild and domesticated populations, and because crosses between wild and domesticated forms are possible and give fertile and vital progeny, this has made P. vulgaris an almost unique and important model among crops for the study of genes and QTLs involved in the domestication process.
4 Diffusion and Evolution of P. vulgaris Out of the American Centers of Origin
The expansion and the pathways of distribution of the bean out of the American domestication centers were very complex. This also involved several introductions from the New World that were combined with exchanges between continents, and among several countries within continents. In the Old World, the breakdown of the spatial isolation between these two gene pools (Mesoamerican and Andean) increased the potential for their hybridization and introgression. The amplitude of agro-ecological conditions experienced by this crop also dramatically increased, giving new opportunities for both natural and human-mediated selection. Several continents and countries have been proposed as the secondary centers of diversification for P. vulgaris, including Europe (Santalla et al. 2002; Angioi et al. 2010, 2011; Gioia et al. in press), Brazil (Burle et al. 2010), central-eastern and southern Africa (Martin and Adams 1987a, b; Asfaw et al. 2009; Blair et al. 2010) and China (Zhang et al. 2008).
4.1 The Contribution of the Mesoamerican and Andean Gene Pools to Bean Germplasm Collections from Different Parts of the World
While we must acknowledge that the amount of available data is larger for Europe than for other secondary centers of diversification, it is also clear that the proportions of the Mesoamerican and Andean gene pools can vary considerably across different continents, as also among countries within continents (Fig. 20.2).

Distribution of the Mesoamerican and Andean gene pools and their hybrids in Europe and other continents. Pie charts show the Mesoamerican (blue) and Andean (red) gene-pool frequencies (%). Hybrid percentages are indicated next to country names, within brackets. Europe (Angioi et al. 2010) (sample size, 307): Iberian peninsula (53), Italy (32), central-northern Europe (74), eastern Europe (69), south-eastern Europe (79). East Africa (111) (Gepts and Bliss, 1988). Ethiopia (99) and Kenya (89) (Asfaw et al. 2009). Central Africa (355) (Blair et al. 2010). Brazil (279) (Burle et al. 2010). China (299) (Zhang et al. 2008)
Pioneering studies carried out using the phaseolins showed that both the Mesoamerican and Andean gene pools are present in Europe, with a higher frequency for the Andean types (66–76 %; Gepts and Bliss 1988; Lioi 1989), as was subsequently confirmed (76 %) by Logozzo et al. (2007). Recently, to trace the distribution of the domesticated Mesoamerican and Andean gene pools in Europe, Angioi et al. (2010) used cpSSRs, nuclear markers (phaseolin and three indel-spanning markers of PvSHP1; Nanni et al. 2011), and morphological seed traits. This study was conducted on a large European collection, and it confirmed that the largest fraction of the European germplasm was of Andean origin (67 %) (Fig. 20.2). The Andean type has been shown to be the most frequent in three European macro-areas: the Iberian peninsula, Italy, and central-northern Europe. The prevalence of the Andean type has also often been confirmed on a local scale (e.g., Limongelli et al. 1996; Escribano et al. 1998; Piergiovanni et al. 2000a, b; Sicard et al. 2005; Angioi et al. 2009b). However, in the eastern part of Europe, the proportion of the Mesoamerican type tends to increase, with a maximum of 46 % in Greece (Fig. 20.2).
Overall, this suggests that there was high gene flow among the different regions of Europe and/or homogeneous selection (either anthropic or ‘natural’). Nonetheless, in some areas, founder effects and/or selection might also have acted. Recently, using methods for the identification of outlier loci for selection, Santalla et al. (2010) provided evidence that selective forces might have had significant roles (particularly for seed size, flowering time, growth habits, pest resistance).
Burle et al. (2010) assessed the genetic diversity and the structure of a sample of 279 geo-referenced common bean landraces from Brazil using nuclear SSR markers, Phaseolin, PvTFL1y, APAand SCAR markers. They showed that the Mesoamerican and Andean gene pools were both present in Brazil, although the Mesoamerican was four-fold more frequent than the Andean. This is surprising, given the closer proximity of Brazil to the Andes. To explain these data, Burle et al. (2010) formulated both selection and demographic hypotheses. Similarities in climate and soil between the two areas might explain the success and diffusion of the Mesoamerican bean germplasm in Brazil. Moreover, multiple introductions of Mesoamerican germplasm in pre- and post-conquest times (Gepts et al. 1988) might have had a considerable impact on the establishing of this pattern.
In Africa, the Mesoamerican and Andean gene pools are approximately equal in frequency (Fig. 20.2) (Martin and Adams 1987a; Gepts and Bliss 1988; Asfaw et al. 2009; Blair et al. 2010). However, there are striking differences between different countries. In Kenya (Asfaw et al. 2009), east Africa (Gepts and Bliss 1988) and southern Africa (Martin and Adams 1987a), the Andean type is the most frequent, while in Ethiopia (Asfaw et al. 2009) and central Africa (Blair et al. 2010), the Mesoamerican type predominates. Interestingly, the study of Asfaw et al. (2009) revealed that with some exceptions, the clustering of the accessions was based on the country of origin (Kenya or Ethiopia), with an overall F ST between countries of 0.06 (P < 0.001). In particular, the divergence is much greater for the Andean genotypes than for the Mesoamerican (F ST = 0.34 and 0.04, respectively; P < 0.001). This suggests that there are at least partially independent seed (and perhaps social) networks in Kenya and Ethiopia, with no strong trans-national bean-seed exchange. An additional reason for the divergence between the Kenyan and Ethiopian germplasm might have arisen through different farmer selection preferences, according to ecological adaptation, cooking value, and market orientation (Wortmann et al. 1998; Asfaw et al. 2009). The predominance of Mesoamerican types in central Africa has been attributed to several reasons: the recent increase in root rot, to which the Andean beans are less resistant (especially determinate types); the higher yield per plant that can often be obtained from Mesoamerica genotypes; and the input of germplasm from national programs (Blair et al. 2010).
China is a large producer of dry beans, and is the most important producer of snap beans in the World, through its intensive horticultural systems that are based on family farms. An analysis of a 229 landraces collection revealed higher prevalence of the Mesoamerican type in China (Zhang et al. 2008). At present, it is believed that there were only a limited number of introductions of the common bean into China (Zheng 1997; Zhang et al. 2008). Thus, one explanation for the prevalence of the Mesoamerican types might be that the few founding populations were biased towards a high frequency of the Mesoamerican type.
4.2 The Diversity of the ‘out-of-America’ Germplasm, and Their Divergence from the American Source Population
Under the bottleneck model, it is expected that dissemination from the center of origin will led to a reduction in genetic diversity. Considering the benchmark as the data obtained by Kwak and Gepts (2009) using nuclear SSR markers to characterize domesticated accessions from the centers of origin, it emerges that overall for the two gene pools the reduction in diversity has been strong for Brazil, intermediate for China, and low or nearly absent for Africa (Table 20.1). This appears counter intuitive, in that it would be expected that the reduction in diversity is in some way proportional to the distance from the center of origin; i.e., that the reduction in diversity in Brazil would be lower, for example, than in China and Africa. This discrepancy probably arises because the dissemination of Phaseolus over the last few centuries was tightly linked to the intense commercial activities and the routes that went all around the world, with the possibility that each continent (and country) has been both source and/or sink of bean germplasm several times, and in different historic periods.
A more comprehensive picture was obtained for Europe by Angioi et al. (2010), through direct comparisons of the levels of diversity between two collections using cpSSR markers: one American and one European. Angioi et al. (2010) concluded that the intensity of the cytoplasmic bottleneck that resulted from the introduction of the common bean into Europe was very low or absent (a loss of cpSSR diversity of ca. 2 %).
At the nuclear level, Papa et al. (2006) inferred a much higher loss of diversity consequent to the introduction of the common bean into Europe (ca. 30 %). However, Angioi (2006) studied the PvSHP1 nuclear markers and observed that the number of haplotypes and the genetic diversity were both higher in America than in Europe, supporting the hypothesis of a bottleneck at the nuclear level of greater intensity than for cpSSRs.
The lack of a cytoplasmic bottleneck in Europe is somewhat surprising, because cpSSR markers are very sensitive indicators of such phenomena, due to their uniparental inheritance, hypervariabilty and haploidy (Provan et al. 2001; Ebert and Peakall 2009; Angioi et al. 2009a). The most likely explanation for this is that the founding common bean populations that colonized Europe were highly variable in their cytoplasmic DNA or that different releases may have had different source populations.
4.3 Introgression Between the Mesoamerican and Andean Gene Pools
Hybrids between the Mesoamerican and Andean gene pools are very important for plant breeding, which often finds the need to recombine Mesoamerican and Andean traits (Johnson and Gepts 1999, 2002). Indeed, hybridization can result in the production of novel genotypes and phenotypes (e.g., seed size, nutritional quality, resistance to pathogens; Angioi et al. 2010; Blair et al. 2010; Santalla et al. 2010) that do not occur in either of the parental taxa. Evolutionary novelty can result either from a combination of different traits from both of the parents, or from traits in a hybrid that transgresses the parental phenotypes (transgressive segregation) (Allendorf and Luikart 2007). Hybridization (with introgression) outside of America had more chances due to the breakdown of the geographical barriers and the isolation that existed between the gene pools in the centers of origin.
In general, distinguishing hybrids at the morphological level is not easy. The use of molecular genetic markers thus greatly simplifies the identification and description of hybrids.
A powerful method for the detection of hybridization events is the integration of cytoplasmic and nuclear analyses (Provan et al. 2001; Ebert and Peakall 2009; Angioi et al. 2009a). Using this approach Angioi et al. (2010) found that at least 33 % of the landraces in the collection were hybrids. Interestingly, in a previous study, and using a different marker system, Santalla et al. (2002) also estimated a high percentage of hybrids in their collection from the Iberian peninsula (25 %). In addition to the molecular results, the individuals identified as hybrids also showed evidence of hybridization from the analysis of seed traits. Indeed, seed size and coat traits tend to vary with the level of introgression between the two gene pools, with relatively good agreement. The complementation of cytoplasmic and nuclear analysis has also been applied with success at local scales in Italy, in the Marche region (12 % hybrids; Sicard et al. 2005) and in Sardinia (4 %; Angioi et al. 2009b).
Moreover, in adopting a maximum likelihood approach, Angioi et al. (2010) estimated that about 11 % of their ‘pure’ Mesoamerican and Andean individuals (derived from recombination from crosses between parents that belong to the two different gene pools) can be regarded as ‘hidden’ hybrids. Thus, 44 % of their collection appeared to be derived from at least one hybridization event, with a frequency of hybridization between gene pools ranging from 0.12 to 0.15 % per year.
Several other studies have analyzed hybridization among gene pools (Fig. 20.2) using molecular markers and different statistical approaches. In Brazil, Burle et al. (2010) estimated a hybridization percentage of 4.4 % based on phaseolin analysis, although this was reduced to 0.74 % based on Structure analysis of nuclear SSR markers (Pritchard et al. 2000). In Africa, the identification of hybrids was based on their intermediate positions between the two gene pools in neighbor-joining trees, and this varied from 1 to 10 % in different countries (Asfaw et al. 2009; Blair et al. 2010). In China, in considering the results of principal coordinate analysis and admixture values based on Structure analysis, Zhang et al. (2008) estimated 7 % as hybrids. As a conclusion, all of these studies based on clustering methods (distance-based, such as neighbor-joining, or model-based, such as Structure) have indicated a number of hybrids as between ca. 1 % and 10 %, which is much less than the estimates for Europe.
Such differences in hybrid frequency might be real: the co-occurrence of the two gene pools in the same continent or country does not necessarily imply that they had the potential for hybridization; i.e. the two gene pools might have had different levels of sympatry (so different chances of hybridization) in different places. However, it is also possible that the various molecular approaches have different statistical powers for the detection of hybrids. In particular, using approaches that involve clustering methods, parental type and F1 hybrids can be readily identified if many loci are examined (Allendorf and Luikart 2007). However, to distinguish between F2, backcrosses, or later-generation hybrids with model-based Bayesian methods can be challenging, even if many loci are examined and when divergence between parental populations is high (Vähä and Primmer 2006; Allendorf and Luikart 2007). On the contrary, contrasting cytoplasmic and nuclear markers might lead to the unraveling of not only recent, but also some ‘historic’ hybridization events between the two gene pools.
5 Genomic Tools and Germplasm Collections
Given its phylogenetic position in the Phaseoloids (Stefanovic et al. 2009), the common bean is considered a model organism for comparative legume genomics. It is closely related to other economically important members of the papilionid legumes, including cowpea (Vigna unguiculata), pigeon pea (Vigna radiata) and soybean (Glycine max).
The common bean and soybean diverged nearly 20 million years ago, around the time of the major duplication event in soybean (Lavin et al. 2005; Schlueter et al. 2004). Synteny analysis indicates that most segments of any single common bean linkage group are highly similar to two soybean chromosomes (Galeano et al. 2009). McClean et al. (2010) successfully tested the assumption that the common bean genome is a diploid version of the soybean, with a comparison of all of the mapped genes from bean (McConnell et al. 2010) against all of the scaffold sequences (20 pseudochromosomes) from the soybean genome. For these reasons, P. vulgaris has proven to be helpful as a model for understanding the larger soybean genome (about 1,100 Mbp), and a comparative genomics approach to gene discovery is practicable for these two evolutionarily related species.
Due to its importance as a grain legume for the human diet (FAO 2010; Broughton et al. 2003; Carvalho et al. 2012), the value of the common bean is best seen through its role as a societal crop, and its improvement is of constant concern (Singh 2001).
With the aim to create new varieties for farmers and consumers, the international consortium for Phaseolus genomics “Phaseomics” was founded, to develop bean genomics, transcriptomics and proteomics (Broughton et al. 2003, http://www.Phaseolus.net).
Two common bean whole genome sequences (Mesoamerican and Andean; each about 600 Mbp) will soon be released by a group of US (http://www.phytozome.net/commonbean.php) (McClean et al. 2013) and Ibero-American laboratories (http://mazorka.langebio.cinvestav.mx/ Phaseolus/). While waiting for this to become available, other methods are needed to develop and facilitate these genomic studies. Here, advanced high-throughput genotyping techniques will provide new insights for association mapping studies in the investigation of variants associated with important traits.
DNA sequences are available for many crops; however, apart from the ongoing model genome projects for Medicago truncatula and Lotus japonicus (Young et al. 2005), and the recently completed soybean genome (Schmutz et al. 2010), there is comparatively little sequence data for other legumes, including the common bean. Nearly, all of the evidence regarding genetic diversity in the common bean is based on multilocus molecular markers (see Papa et al. 2006, and Acosta-Gallegos et al. 2007, for reviews). Only a few studies have investigated nucleotide diversity in this important crop species, and particularly for wild populations. These have included the sequence diversity of the phaseolin locus in wild accessions (Kami et al. 1995), and of three non-coding regions of the dihydroflavonol 4-reductase and chalcone isomerase genes in landraces and modern cultivars (McClean et al. 2004; McClean and Lee 2007). Studies by Nanni et al. (2011) and Bitocchi et al. (2012, 2013) investigated the nucleotide diversity for five different genes from a wide sample of wild and domesticated P. vulgaris that is representative of its geographical distribution. A larger amount of sequence data (over 500 genes) was obtained in a study by McConnell et al. (2010) for the two parents of one of the major mapping populations of P. vulgaris, ‘BAT93’ × ‘JaloEPP558’ (Freyre et al. 1998). This provided enrichment of the genetic map and allowed investigation of macrosynteny between the common bean and the model organisms of A. thaliana, M. truncatula and L. japonicus. McConnell et al. (2010) exploited over 2,686 P. vulgaris contiguous sequences that were generated by Ramirez et al. (2005), from which they obtained useful sequence data for both BAT93 and JaloEEP558 for 534 gene fragments. Of these 534 fragments, 395 were polymorphic between BAT and Jalo, and 300 were mapped and assigned to the 11 linkage groups of P. vulgaris. As an important consequence of this study, these markers have become useful for other Mesoamerican × Andean populations.
Linkage maps have been developed from crosses both between and within Mesoamerican and Andean gene pools (see Kelly et al. 2003, for a review). To date, a collection of over 25 linkage maps have been developed in the common bean. Molecular linkage maps are essential for many purposes, such as gene mapping, QTL analysis, linkage disequilibrium analysis, and synteny, and consequently, to find genes with particular agronomic and economic traits, for their application to plant breeding.
The availability of large sets of annotated sequences has arisen through the identification, sequencing and validation of gene expression, and these will help in the development of the accurate and complete structural annotation of the common bean genome, and in the identification of the genetic basis of agriculturally important traits. To date, there have been several relevant publications in the common bean regarding transcriptome sequencing and bioinformatics analyses (Table 20.2).
Ramirez et al. (2005) provided an initial platform for the functional genomics of the common bean. They identified almost 8,000 unique genes that were assembled from more than 20,000 ESTs sequenced from various cDNA libraries. These were derived from the Mesoamerican common bean genotype Negro Jamapa 81, and included nitrogen-fixing root nodules, phosphorus-deficient roots, developing pods, and leaves, and from the leaves of the Andean genotype G19833. They showed the utility of mining EST collections in the common bean for SNPs and provided new tools for genomic studies in this species. These sequences have enriched the collection of ESTs for this important crop, and have provided new understanding of bean metabolism, development, and adaptation to stress. The common bean EST sequences represent the foundation for genome-wide transcript studies, and they are a source of defined molecular markers for mapping bean linkage groups and anchoring physical maps.
Melotto et al. (2005) obtained over 5,000 sequences from three cDNA libraries from a common bean breeding line, from 19-day-old trifoliate leaves, 10-day-old shoots, and 13-day-old shoots inoculated with Colletotrichum lindemuthianum. They finally identified 3,126 unigenes, and of these only 314 showed similarity to sequences from the existing database.
Tian et al. (2007) constructed a suppression subtractive cDNA library to identify genes involved in response to phosphorous starvation. They characterized the differentially expressed genes into five functional groups, and by comparison with the GenBank non-redundant database, they were able to further classify 72 genes.
Over 6,000 new common bean ESTs were obtained by Thibivilliers et al. (2009), again using a subtractive cDNA library, which was constructed from a rust-resistant cultivar. As main result, they identified sequences that were up-regulated in response to susceptible and resistant host-pathogen interactions.
Blair et al. (2011) obtained a total of 4,219 unigenes from two cDNA libraries from the drought tolerant Mesoamerican genotype BAT477 and the acid-soil-tolerant Andean genotype G19833.
Several new genomics technologies have emerged in recent years, including next generation sequencing (Mardis 2008), high-throughput marker genotyping, and -omics technologies. These provide powerful tools for the understanding of genome variations in crop species at the DNA, RNA and protein levels, and particularly for nonmodel plant species (Vera et al. 2008). Next-generation sequencing (Mardis 2008) has revolutionized the “-omic era”, allowing the analysis of millions of reads in a very little time and at much reduced cost.
Kalavacharla et al. (2011) provided new genomic information by sequencing a large number of cDNA libraries from different plant tissues using the Roche 454-FLX pyrosequencing platform: leaves, flowers and roots from a common bean cultivar, and pods derived from the BAT93 breeding line, one of the parents of the core common bean mapping populations. They identified 59,295 common bean unigenes, 31,664 of which were newly discovered sequences. In this way they obtained a substantial transcriptome dataset for common bean and increased the number of P. vulgaris ESTs deposited in gene bank by 150 %, which is very useful for functional genomics research. They also detected a high number of microsatellites (SSRs): 1,516 and 4,517, in Roche 454-FLX system-derived and genomic sequences, respectively.
All of these efforts have provided significant resources for the discovery of new genes, for the development of molecular markers for future genetic linkage and QTL analyses, and for comparative studies with other legumes. They will also help in the discovery and understanding of the genes that underlie agriculturally important traits in the common bean.
Next-generation sequencing has significantly increased the speed at which SNPs can be discovered. These provide an ideal marker system for genetic research in many crops, which can be used as molecular markers for research. Furthermore, several high-throughput platforms have been developed that allow rapid and simultaneous genotyping of up to a million SNP markers (Yan et al. 2010).
However, as in other species for which the complete genome sequence is not yet available, in the common bean the use of next-generation sequencing for SNP discovery is much more difficult and costly. On this basis, Hyten et al. (2010) developed a method to improve the number of SNPs in common bean. This system was developed as a multi-tier reduced representation library, and it coupled sequences obtained from the Roche 454 platform (longer reads) with the Illumina genome analyzer (high-throughput) for SNP discovery, for which no whole genome sequence and normalized cDNA libraries are needed. They revealed 3,487 SNPs, 86 % of which were validated with Sanger sequencing.
The study of Cortes et al. (2011) was the first to explore SNP variations for diversity analysis in the common bean. Using KASPar technology (Cuppen 2007), they validated and accessed SNP diversity at 84 gene-based and 10 nongenic loci in a set of 70 genotypes, which included Andean and Mesoamerican accessions previously evaluated for SSRs (Blair et al. 2006b). They found that SNP markers are especially useful for inter–gene-pool comparisons, but not at the intra–gene-pool scale, where SSR markers are efficient (Sicard et al. 2005; Blair et al. 2006b, 2009; Angioi et al. 2009a; Kwak and Gepts 2009). Recently, Blair et al. (2013) developed an Illumina GoldenGate assay for common bean based on conserved legume gene sequences; they tested a total of 768 SNPs, 736 of which gave high quality reads and were scored in a wide sample of P. vulgaris accessions. Overall, they found the GoldenGate assay to be a useful genetic tool for rapid analysis of parental combinations, for germplasm studies, and for evaluation of association panels. The genes or genomic regions responsible for traits of interest can be identified either through conventional linkage mapping or through new genetic approaches, such as advanced-backcross QTL analysis, introgression libraries, multi-parent advanced generation intercross populations, and association genetics. These genes can be introgressed or pyramided to develop superior genotypes, using molecular breeding approaches, such as marker-assisted back crossing, marker-assisted recurrent selection, and genome-wide selection.
Genetic resources constitute a rich source of such ‘new’ genes. Important collections of common bean germplasm are maintained ex situ in the gene banks. The online portal Genesys (http://www.genesys-pgr.org) supplies information about the accessions of P. vulgaris stored in the gene banks. This portal indicates that there are over 83,000 accessions from 138 countries stored in 63 institutions around the World. The main accessions are landraces (ca. 61,000), improved cultivars (> 8,000), breeding materials (> 2,000), and wild forms (> 1,500). The largest and most diverse common bean collection in the world includes over 31,000 accessions, and it is at the Centro International de Agricultura Tropical (CIAT) in Colombia. These come from 104 countries, and in particular Mexico, Peru, Colombia and Guatemala, and also from Europe and Africa, and to a lesser extend from Asia. Another large common bean collection is at the United States Department of Agriculture (USDA- ARS) at Washington State University, where over 12,000 accessions from 94 countries are stored. The Leibniz Institute of Plant Genetics and Crop Plant Research (IPK) in Gatersleben (Germany) has the largest collection of Phaseolus genetic resources in Europe. About 8,000 accessions of P. vulgaris are registered, from 69 countries. An important second gene bank for the common bean in Europe is at ‘N.I. Vavilov’ Research Institute of Plant Industry (VIR, Russia), with 6,000 accessions from 90 countries.
6 Conclusions
The data reviewed above show that the improvements in P. vulgaris L. are of constant concern both as a societal crop and as a model species for comparative legume genomics. Different studies have been conducted to determine the diversity levels, the origin, the domestication processes and the evolution of this species.
Mesoamerica has been recently proposed to be the origin of P. vulgaris. Thus, the wild beans from South America originated through migration from the Mesoamerica populations. Several additional aspects of the evolution and domestication of the common bean have been widely highlighted using genomic tools, including the identification of single domestication events within both gene pools and the characterization of the spread of this crop out of America, with the parallel reduction of the genetic diversity and occurrence of hybridization between gene pools.
Diversity studies based on different molecular markers have highlighted that a large fraction of the bean genome appears to have been under selection during domestication. More data relative to the relationships between the wild and domesticated forms will likewise help in the mining of wild species for novel allelic variations and genes underlying primary agronomic traits.
At present, it appears clear that the forthcoming genome sequence of the common bean, together with the formerly available genomic tools and genetic resources, will become the paradigm to understand the structural and functional diversity of this crop. Alongside, marker-assisted selection programs and high-throughput selection of improved varieties will provide breeders with valuable instruments to achieve effective enhancement of this crop.
References
Acosta-Gallegos JA, Kelly JD, Gepts P (2007) Pre-breeding in common bean and use of genetic diversity from wild germplasm. Crop Sci 47:44–59
Akibode S, Maredia M (2011) Global and regional trends in production, trade and consumption of food legume crops. SPIA Report department of agricultural, food and resource economics, Michigan State University, East Lansing, MI
Allendorf FW, Luikart G (2007) Conservation and the genetics of populations. Blackwell, pp 642
Allendorf FW, Hohenlohe PA, Luikart G (2010) Genomics and the future of conservation genetics. Nat Rev Genet 11:697–709
Angioi SA (2006) Development and use of molecular tools to study the genetic diversity in Phaseolus vulgaris L. and Phaseolus coccineus L. PhD thesis, University of Turin, Italy
Angioi SA, Desiderio F, Rau D et al (2009a) Development and use of chloroplast microsatellites in Phaseolus spp. and other legumes. Plant Biol 11:598–612
Angioi SA, Rau D, Rodriguez M et al (2009b) Nuclear and chloroplast microsatellite diversity in Phaseolus vulgaris L. from Sardinia (Italy). Mol Breed 23:413–429
Angioi SA, Rau D, Attene G et al (2010) Beans in Europe: origin and structure of the European landraces of Phaseolus vulgaris L. Theor Appl Gen 121:829–843
Angioi SA, Rau D, Nanni L et al (2011) The genetic make–up of the European landraces of the common bean. Plant Genet Resour 9:197
Anthony JL, Vonder Haar RA, Hall TC (1990) Nucleotide sequence of an alpha–phaseolin gene from Phaseolus vulgaris. Nucleic Acids Res 18:3396
Arumuganthan K, Earle E (1991) Nuclear DNA content of some important plant species. Plant Mol Biol Rep 9:208–218
Asfaw A, Blair MW, Almekinders C (2009) Genetic diversity and population structure of common bean (Phaseolus vulgaris L.) landraces from the East African highlands. Theor Appl Gen 120:1–12
Beaver JS, Osorno JM (2009) Achievements and limitations of contemporary common bean breeding using conventional and molecular approaches. Euphytica 168:145–175
Becerra-Velásquez VL, Gepts P (1994) RFLP diversity in common bean (Phaseolus vulgaris L.). Genome 37:256–263
Beebe S, Skroch P, Tohme J et al (2000) Structure of genetic diversity among common bean landraces of middle-American origin based on correspondence analysis of RAPD. Crop Sci 40:264–273
Beebe S, Rengifo J, Gaitan E et al (2001) Diversity and origin of Andean landraces of common bean. Crop Sci 41:854–862
Bennett MD, Leitch IJ (1995) Nuclear DNA amounts in angiosperms. Ann Bot 76:113–116
Bennett MD, Leitch IJ (2010) Angiosperm DNA C-values database (release 7.0, Dec. 2010) http://www.kew.org/cvalues/
Bitocchi E, Nanni L, Bellucci E et al (2012) Mesoamerican origin of the common bean (Phaseolus vulgaris L.) is revealed by sequence data. Proc Natl Acad Sci U S A 109(14):E788–E796
Bitocchi E, Bellucci E, Giardini A et al (2013) Molecular analysis of the parallel domestication of the common bean in Mesoamerica and the Andes. New Phytol 197:300–313
Blair MW, Iriarte G, Beebe S (2006a) QTL analysis of yield traits in an advanced backcross population derived from a cultivated Andean x wild common bean (Phaseolus vulgaris L.) cross. Theor Appl Gen 112:1149–1163
Blair MW, Giraldo MC, Buendia HF et al (2006b) Microsatellite marker diversity in common bean (Phaseolus vulgaris L.). Theor Appl Gen 113:100–109
Blair MW, Diaz LM, Buendia HF et al (2009) Genetic diversity, seed size associations and population structure of a core collection of common beans (Phaseolus vulgaris L.). Theor Appl Genet 119:955–972
Blair MW, González LF, Kimani M et al (2010) Genetic diversity, inter-gene pool introgression and nutritional quality of common beans (Phaseolus vulgaris L.) from Central Africa. Theor Appl Gen 121:237–248
Blair MW, Fernandez AC, Ishitani M et al (2011) Construction and EST sequencing of full-length, drought stress cDNA libraries for common beans (Phaseolus vulgaris L.). BMC Plant Biol 11:171
Blair MW, Soler A, Cortés AJ (2012) Diversification and population structure in common beans (Phaseolus vulgaris L.). PLoS One 7(11):e49488
Blair MW, Cortés AJ, Penmetsa RV et al (2013) A high-throughput SNP marker system for parental polymorphism screening, and diversity analysis in common bean (Phaseolus vulgaris L.). Theor Appl Genet 126:535–548
Broughton WJ, Hernandez G, Blair M et al (2003) Beans (Phaseolus spp.)—model food legumes. Plant Soil 252:55–128
Burle ML, Fonseca JR, Kami JA et al (2010) Microsatellite diversity and genetic structure among common bean (Phaseolus vulgaris L.) landraces in Brazil, a secondary center of diversity. Theor Appl Genet 121:801–813
Carvalho LMJ, Correa MM, Pereira EJ et al (2012) Iron and zinc retention in common beans (Phaseolus vulgaris L.) after home cooking. Food Nut Res 56:15618
Chacón SMI, Pickersgill B, Debouck DG (2005) Domestication patterns in common bean (Phaseolus vulgaris L.) and the origin of the Mesoamerican and Andean cultivated races. Theor Appl Genet 110:432–444
Coates AG, Collins LS, Aubry MP et al (2004) The geology of the Darien, Panama, and the late Miocene—Pliocene collision of the Panama arc with north–western South America. Geol Soc Amer Bull 116:1327–1344
Cortés AJ, Chavarro MC, Blair MW (2011) SNP marker diversity in common bean (Phaseolus vulgaris L.). Theor Appl Genet 123:827–845
Cuppen E (2007) Genotyping by allele–specific amplification (KASPar). Cold Spring Harb Protocols, pp 172–173
Davey JW, Hohenlohe PA, Etter PD et al (2011) Genome–wide genetic marker discovery and genotyping using next–generation sequencing. Nat Rev Genet 12:499–510
Debouck DG, Toro O, Paredes OM et al (1993) Genetic diversity and ecological distribution of Phaseolus vulgaris in northwestern South America. Econ Bot 47:408–423
Delgado-Salinas A, Turley T, Richman A et al (1999) Phylogenetic analysis of the cultivated and wild species of Phaseolus (Fabaceae). Syst Bot 24:438–460
Delgado-Salinas A, Bibler R, Lavin M (2006) Phylogeny of the genus Phaseolus (Leguminosae): a recent diversification in an ancient landscape. Syst Bot 31:779–791
Desiderio F, Bitocchi E, Bellucci E et al (2013) Chloroplast microsatellite diversity in Phaseolus vulgaris. Front Plant Sci 3:312
Dìaz LM, Blair MW (2006) Race structure within the Mesoamerican gene pool of common bean (Phaseolus vulgaris L.) as determined by microsatellite markers. Theor Appl Genet 114:143–154
Ebert D, Peakall R (2009) Chloroplast simple sequence repeats (cpSSRs): technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol Ecol Resour 9:673–690
Escribano MR, Santalla M, Casquero PA et al (1998) Patterns of genetic diversity in landraces of common bean (Phaseolus vulgaris L.) from Galicia. Plant Breed 117:49–56
Freyre R, Ríos R, Guzmán L et al (1996) Ecogeographic distribution of Phaseolus spp. (Fabaceae) in Bolivia. Econ Bot 50:195–215
Freyre R, Skroch P, Geffroy V et al (1998) Towards an integrated linkage map of common bean. 4. Development of a core map and alignment of RFLP maps. Theor Appl Genet 97:847–856
Freytag GF, Debouck DG (1996) Phaseolus costaricensis, a new wild bean species (Phaseolinae, Leguminosae) from Costa Rica and Panama, central America. Novon 6:157–163
Freytag GF, Debouck DG (2002) Taxonomy, distribution, and ecology of the genus Phaseolus (Leguminosae–Papilionoideae) in North America, Mexico and central America. Botanical Research Institute of Texas, Ft. Worth
Galeano CH, Fernandez AC, Gomez M et al (2009) Single strand conformation polymorphism based SNP and indel markers for genetic mapping and synteny analysis of common bean (Phaseolus vulgaris L.). BMC Genomics 10:629
Galeano C, Cortés A, Fernández A et al (2012) Gene–based single nucleotide polymorphism markers for genetic and association mapping in common bean. BMC Genet 13(1):48
Gepts P, Bliss FA (1985) F1 hybrid weakness in the common bean: differential geographic origin suggests two gene pools in cultivated bean germplasm. J Hered 76:447–450
Gepts P, Osborn TC, Rashka K et al (1986) Phaseolin–protein variability in wild forms and landraces of the common bean Phaseolus vulgaris evidence for multiple centers of domestication. Econ Bot 40:451–468
Gepts P, Bliss FA (1988) Dissemination pathways of common bean (Phaseolus vulgaris, Fabaceae) deduced from phaseolin electrophoretic variability. II Europe and Africa. Econ Bot 42:86–104
Gepts P, Kmiecik K, Pereira P et al (1988) Dissemination pathways of common bean (Phaseolus vulgaris, Fabaceae) deduced from phaseolin electrophoretic variability. I. The Americas. Econ Bot 42:73–85
Gepts P, Debouck DG (1991) Origin, domestication, and evolution of the common bean, Phaseolus vulgaris. In: Voysest O, Van Schoonhoven A (eds.) Common beans: research for crop improvement. CAB, Oxon, UK, pp 7–53
Gepts P (1998) Origin and evolution of common bean, past event and recent trends. J Am Soc Hortic Sci 33:1124–1130
Gepts P, Papa R, Coulibaly S et al (1999) Wild legume diversity and domestication – insights from molecular methods. In Vaughan D (ed), Wild legumes, Proc. 7th MAFF International Workshop on Genetic Resources. National Institute of Agrobiological Resources, Tsukuba, Japan, pp 19–31
Gepts P, Papa R (2002). Evolution during domestication. In: Encyclopedia of Life Sciences 1–7 LONDON: Nature Publishing Group. Macmillan Publishers Ltd (UK)
Gepts P, Aragão F, de Barros E et al (2008) Genomics of Phaseolus beans, a major source of dietary protein and micronutrients in the Tropics. In: Moore PH, Ming R (eds) Genomics of Tropical Crop Plants. Springer, Berlin, pp 113–143
Gioia T, Logozzo G, Attene G et al (2013) Evidence for introduction bottleneck and extensive inter-gene pool (Mesoamerica x Andes) hybridization in the European common bean (Phaseolus vulgaris L.) germplasm Plos ONE (in press)
Glémin S, Bataillon T (2009) A comparative view of the evolution of grasses under domestication. New Phytol 183:273–290
Gupta PK, Rustgi S, Mir RR (2008) Array–based high–throughput DNA markers for crop improvement. Heredity 101:5–18
Hyten DL, Song Q, Fickus EW et al (2010) High–throughput SNP discovery and assay development in common bean. BMC Genomics 11:475
Johnson WC, Gepts P (1999) Segregation for performance in recombinant inbred populations resulting from inter–gene pool crosses of common bean (Phaseolus vulgaris L.). Euphytica 106:5–56
Johnson WC, Gepts P (2002) The role of epistasis in controlling seed yield and other agronomic traits in an Andean–Mesoamerican cross of common bean (Phaseolus vulgaris L.). Euphytica 125:69–79
Kalavacharla V, Liu Z, Meyers BC et al (2011) Identification and analysis of common bean (Phaseolus vulgaris L.) transcriptomes by massively parallel pyrosequencing. BMC Plant Biol 11:135
Kami J, Becerra–Velásquez V, Debouck DG et al (1995) Identification of presumed ancestral DNA sequences of phaseolin in Phaseolus vulgaris. Proc Natl Acad Sci U S A 92:1101–1104
Kelly JD, Gepts P, Miklas PN et al (2003) Tagging and mapping of genes and QTL and molecular–marker assisted selection for traits of economic importance in bean and cowpea. Field Crops Res 82:135–154
Koenig R, Gepts P (1989) Allozyme diversity in wild Phaseolus vulgaris further evidence for two major centers of diversity. Theor Appl Genet 78:809–817
Koinange EMK, Singh SP, Gepts P (1996) Genetic control of the domestication syndrome in common bean. Crop Sci 36:1037–1145
Kwak M, Kami JA, Gepts P (2006) Identification of the determinacy gene (Fin) and its evolution during domestication in common bean (Phaseolus vulgaris L.). In: Plant & Animal Genome XIV, poster 447. Abstract available at: http://www.intl-pag.org/14/abstracts/PAG14_P447.html
Kwak M, Velasco D, Gepts P (2008) Mapping homologous sequences for determinacy and photoperiod sensitivity in common bean (Phaseolus vulgaris). J Hered 99:283–291
Kwak M, Gepts P (2009) Structure of genetic diversity in the two major gene pools of common bean (Phaseolus vulgaris L., Fabaceae). Theor Appl Genet 118:979–992
Kwak M, Kami JA, Gepts P (2009) The putative Mesoamerican domestication center of Phaseolus vulgaris is located in the Lerma–Santiago basin of Mexico. Crop Sci 49:554–563
Lavin M, Herendeen PS, Wojjciechowski MF (2005) Evolutionary rate analysis of leguminosae implicates a rapid diversification of lineages during the tertiary. Syst Biol 54:575–594
Limongelli G, Laghetti G, Perrino P et al (1996) Variation of seed storage protein in landraces of common bean (Phaseolus vulgaris L.) from Basilicata, southern Italy. Plant Breed 119:513–516
Lioi L (1989) Geographical variation of phaseolin patterns in an old world collection of Phaseolus vulgaris. Seed Sci Technol 17:317–324
Logozzo G, Donnoli R, Macaluso L et al (2007) Analysis of the contribution of Mesoamerican and Andean gene pools to European common bean (Phaseolus vulgaris L.) germplasm and strategies to establish a core collection. Genet Resour Crop Ev 54:1763–1779
Mamidi S, Rossi M, Annam D et al (2011) Investigation of the domestication of common bean (Phaseolus vulgaris) using multilocus sequence data. Funct Plant Biol 38:953–967
Mardis ER (2008) The impact of next–generation sequencing technology on genetics. Trends Genet 24:133–141
Marshall HD, Newton C, Ritland K (2002) Chloroplast phylogeography and evolution of highly polymorphic microsatellites in lodgepolepine (Pinus contorta). Theor Appl Genet 104:367–378
Martin GB, Adams MW (1987a) Landraces of Phaseolus vulgaris (Fabaceae) in northern Malawi I. Regional variation. Econ Bot 41:190–203
Martin GB, Adams MW (1987b) Landraces of Phaseolus vulgaris (Fabaceae) in northern Malawi II. Generation and maintenance of variability. Econ Bot 41:204–215
McClean PE, Lee RK, Miklas PN (2004) Sequence diversity analysis of dihydroflavonol 4–reductase intron 1 in common bean. Genome 47:266–280
McClean PE, Lee RK (2007) Genetic architecture of chalcone isomerase non–coding regions in common bean (Phaseolus vulgaris L.). Genome 50:203–214
McClean PE, Lavin M, Gepts P et al (2008) Phaseolus vulgaris: a diploid model for soybean. In: Stacey G (eds) Soybean Genomics. Springer, Berlin, pp 55–78
McClean PE, Mamidi S, McConnell M et al (2010) Synteny mapping between common bean and soybean reveals extensive blocks of shared loci. BMC Genomics 11:184. http://www.biomedcentral.com/1471-2164/11/184
McClean PE, Jackson S, Schmutz J et al (2013) Progress toward a draft sequence of the common bean genome. Grains and Legumes (in press)
McConnell M, Mamidi S, Lee R et al (2010) Syntenic relationships among legumes revealed using a gene–based genetic linkage map of common bean (Phaseolus vulgaris L.). Theor Appl Genet 121:1103–1116
McCouch S (2004) Diversifying selection in plant breeding. PLoS Biol 2:e347
Melotto M, Monteiro–Vitorello CB, Bruschi AG et al (2005) Comparative bioinformatic analysis of genes expressed in common bean (Phaseolus vulgaris L.) seedlings. Genome 48:562–570
Miklas PN, Kelly JD, Beebe SE et al (2006) Common bean breeding for resistance against biotic and abiotic stresses: from classical to MAS breeding. Euphytica 147:105–131
Nanni L, Bitocchi E, Bellucci E et al (2011) Nucleotide diversity of a genomic sequence similar to SHATTERPROOF (PvSHP1) in domesticated and wild common bean (Phaseolus vulgaris L.). Theor Appl Genet 123:1341–1357
Papa R, Gepts P (2003) Asymmetry of gene flow and differential geographical structure of molecular diversity in wild and domesticated common bean (Phaseolus vulgaris L.) from Mesoamerica. Theor Appl Genet 106:239–250
Papa R, Acosta J, Delgado–Salinas A et al (2005) A genome–wide analysis of differentiation between wild and domesticated Phaseolus vulgaris from Mesoamerica. Theor Appl Genet 111:1147–1158
Papa R, Nanni L, Sicard D et al (2006) The evolution of genetic diversity in Phaseolus vulgaris L. In: Motley TJ, Zerega N, Cross H (eds) New Approaches to the Origins, Evolution and Conservation of Crops. Darwin’s Harvest. Columbia University Press, New York
Papa R, Bellucci E, Rossi M et al (2007) Tagging the signatures of domestication in common bean (Phaseolus vulgaris) by means of pooled DNA samples. Ann Bot 100:1039–1051
Piergiovanni AR, Cerbino D, Brandi M (2000a) The common bean populations from Basilicata (southern Italy). An evaluation of their variation. Genet Resour Crop Ev 47:489–495
Piergiovanni AR, Taranto G, Pignone D (2000b) Diversity among common bean populations from the Abruzzo region (central Italy): a preliminary inquiry. Genet Resour Crop Ev 47:467–470
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Provan J, Soranzo N, Wilson NJ et al (1999) A low mutation rate for chloroplast microsatellites. Genetics 153:943–947
Provan J, Powell W, Hollingsworth PM (2001) Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends Ecol Evol 16:142–147
Ramirez M, Graham MA, Blanco–Lopez L et al (2005) Sequencing and analysis of common bean ESTs. Building a foundation for functional genomics. Plant Physiol 137:1211–1227
Repinski S, Kwak M, Gepts P (2012) The common bean growth habit gene PvTFL1y is a functional homolog of Arabidopsis TFL1. Theor Appl Genet 124:1539–1547
Rossi M, Bitocchi E, Bellucci E et al (2009) Linkage disequilibrium and population structure in wild and domesticated populations of Phaseolus vulgaris L. Evol Appl 2:504–522
Santalla M, Rodiño AP, De Ron AM (2002) Allozyme evidence supporting southwester Europe as a secondary center of genetic diversity for common bean. Theor Appl Genet 104:934–944
Santalla M, Menéndez–Sevillano MC, Monteagudo AB et al (2004) Genetic diversity of Argentinean common bean and its evolution during domestication. Euphytica 135:75–87
Santalla M, De Ron AM, De La Fuente M (2010) Integration of genome and phenotypic scanning gives evidence of genetic structure in Mesoamerican common bean (Phaseolus vulgaris L.) landraces from the southwest of Europe. Theor Appl Genet 120:1635–1651
Schlueter JA, Dixon P, Granger C et al (2004) Mining the EST databases to determine evolutionary events in the legumes and grasses. Genome 47:868–876
Schmutz J, Cannon SB, Schlueter J et al (2010) Genome sequence of the paleopolyploid soybean. Nature 463:178–183
Sicard D, Nanni L, Porfiri O et al (2005) Genetic diversity of Phaseolus vulgaris L and P. coccineus L. landraces in central Italy. Plant Breed 124:464–472
Singh SP (2001) Broadening the genetic base of common bean cultivars. Crop sci 41:1659–1675
Singh SP, Nodari R, Gepts P (1991a) Genetic diversity in cultivated common bean. I. Allozymes. Crop Sci 31:19–23
Singh SP, Gutiérrez JA, Molina A et al (1991b) Genetic diversity in cultivated common bean. II. Marker–based analysis of morphological and agronomic traits. Crop Sci 31:23–29
Singh SP, Gepts P, Debouck DG (1991c) Races of common bean (Phaseolus vulgaris L., Fabaceae). Econ Bot 45:379–396
Stefanović S, Pfeil BE, Palmer JD et al (2009) Relationships among phaseoloid legumes based on sequences from eight chloroplast regions. Syst Bot 34:115–128
Tanksley SD, McCouch SR (1997) Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277:1063–1066
Thibivilliers S, Joshi T, Campbell KB et al (2009) Generation of Phaseolus vulgaris ESTs and investigation of their regulation upon uromyces appendiculatus infection. BMC Plant Biol 9:46
Thuillet AC, Bataillon T, Poirier S et al (2005) Estimation of long–term effective population sizes through the history of durum wheat using microsatellite data. Genetics 169:1589–1599
Tian J, Venkatachalam P, Liao H et al (2007) Molecular cloning and characterization of phosphorous starvation responsive genes in common bean (Phaseolus vulgaris L.). Planta 227:151–165
Tohme J, Gonzalez DO, Beebe S et al (1996) AFLP analysis of gene pools of a wild bean core collection. Crop Sci 36:1375–1384
Toro O, Tohme J, Debouck DG (1990) Wild bean (Phaseolus vulgaris L.): Description and distribution. Centro Internacional de Agricultura Tropical, Cali, Colombia
Tuberosa R, Graner A, Varshney RK (2011) Genomics of plant genetic resources: an introduction. Plant Genet Resour 9:151–154
Vähä JP, Primmer CR (2006) Efficiency of model–based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol Ecol 15:63–72
Heerwaarden J van, Doebley J, Briggs WH et al (2011) Genetic signals of origin, spread, and introgression in a large sample of maize landraces. Proc Natl Acad Sci U S A 108:1088–1092
Vera J, Wheat C, Fescemyer H et al (2008) Rapid transcriptome characterization for a non model organism using 454 pyrosequencing. Mol Ecol 17:1636–1647
Wortmann CS, Kirkby RA, Eledu CA et al (1998) Atlas of common bean (Phaseolus vulgaris L.) production in Africa. CIAT Pan–African Bean Research Alliance, vol 133
Yan JB, Yang XH, Shah T et al (2010) High–throughput SNP genotyping with the GoldenGate assay in maize. Mol Breed 25:441–451
Young ND, Cannon SB, Sato S et al (2005) Sequencing the genespaces of Medicago truncatula and Lotus japonicus. Plant Phys 137:1174–1181
Zhang X, Blair MW, Wang S (2008) Genetic diversity of Chinese common bean (Phaseolus vulgaris L.) landraces assessed with simple sequence repeats markers. Theor Appl Genet 117:629–640
Zheng ZJ (1997) Food legumes in China. China Agriculture Press, Beijing, pp 222–249
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Bellucci, E. et al. (2014). Genomics of Origin, Domestication and Evolution of Phaseolus vulgaris . In: Tuberosa, R., Graner, A., Frison, E. (eds) Genomics of Plant Genetic Resources. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-7572-5_20
Download citation
DOI: https://doi.org/10.1007/978-94-007-7572-5_20
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-7571-8
Online ISBN: 978-94-007-7572-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)