
Abstract
The study of the mechanisms underlying the spread of cancer to sites of bone metastasis have benefitted greatly from recent advances in the high-throughput analysis of biomolecules using modern “omic” techniques. Omic-based profiling can provide both qualitative and quantitative data about the expression of key biomolecules within body fluids, tissues and sub-cellular compartments within both healthy and disease states. Individual omic platforms which analyse DNA-sequences (genomics), mRNA (transcriptomics), proteins (proteomics) and metabolites (metabolomics) have provided key information relating to the biological alterations which occur as a result of cancer spread to bone. Application of omic-techniques to both patient derived samples and animal models of bone metastasis have identified molecules which could serve as diagnostic and prognostic biomarkers of disease development. Biomarkers identified by omic techniques also offer the potential to assist in making cancer treatment decisions. Biomarkers identified by omic techniques require extensive validation in large patient cohorts and across multiple institutions before their adoption within clinical practice. The large number of potential biomarkers which have already been identified within pre-clinical omic-based studies in the field of bone metastatic cancer provides considerable promise for the future of both cancer detection and treatment.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction: The Promise of “Omics” in Bone Metastasis
Bone metastasis occurs in greater than 70 % of patients with advanced breast and prostate cancer and multiple myeloma. The consequent skeletal complications, which include pathological fracture, bone pain, spinal cord compression and hypercalcaemia represent a major cause of morbidity and loss of quality of life [1, 2]. Prediction of patients at high risk of developing bone metastases as well as early diagnosis would enable more timely and effective interventions aimed at prevention or treatment of bone metastases. Markers of cancer development and metastatic spread have historically been discovered by immunological profiling of tissues and body fluids (for instance the elucidation of serum prostate specific antigen-PSA [3]). Scientific developments such as the sequencing of the complete human genome (complete sequence published in 2003), combined with high speed computing and other technological developments within analytical chemistry have ushered in the era of large scale qualitative and quantitative analysis of biomolecules (“omics”-technologies). These high-throughput platforms for biomolecular analysis offer exciting prospects of discovering new and improved markers for cancer metastasis to bone, as well as the identification of pivotal molecules within cancer development and spread which may serves as future drug targets.
2 Molecular Profiling: Genomics, Proteomics, Metabolomics
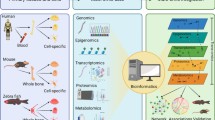
The term “omic technologies” refers to a series of techniques and methodological platforms which each aim to characterize biomolecules using approaches with a degree of generic applicability to a given type of biomolecule. In the process by which biological information flows from DNA (gene sequences and non-transcribed regulatory elements), through to transcription of mRNA, translation into proteins (and their associated post-translational modifications) and the eventual effect of protein expression upon metabolite levels within the cell, “omics” technologies embrace the fields of: genomics (DNA), functional genomics (mRNA), proteomics (proteins) and metabolomics (metabolites) respectively (see Fig. 7.1). Each of these fields of “omic” research includes a wide variety of potential techniques and a thorough description of all the methods available for omic-research is beyond the scope of this chapter, however a general overview will be given. The information which each method can provide can include: (1) identification of the molecules involved, (2) quantification of the amount of biomolecules present within defined biological states/systems-quantitative omics approaches, (3) characterization of the molecular interactions between biomolecules and (4) identification and quantification of the molecular alterations which can diversify a given biomolecule into numerous isoforms. The information from omic-studies can provide several useful outputs of clinical utility including mechanistic insight into the development of disease and/or candidate biomarkers. The official NIH definition of a biomarker is “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention” [4]. The discovery of biomarkers and of mechanistic insights into disease development are by no means mutually exclusive and key mechanistic players may indeed be biomarkers of disease.

“Omics” strategies within biomarker discovery and biological research-an overview: “Omic”-approaches apply molecular characterization methodologies to biomolecules within the flow of biological information from DNA, through to mRNA, protein and eventually alterations within cellular metabolites (“Cellular process”). Different classes of biomolecules are analysed within: genomics (for DNA sequence analysis), functional genomics (for mRNA analysis), proteomics (for protein analysis) and metabolomics (for the analysis of metabolites). The individual omic procedures each encompass a wide-range of different techniques which can produce different types of data pertinent to disease aetiology, prediction of clinical outcomes and guidance of treatment options. Genomic analysis (using techniques such as next-generation sequencing) can identify key genes mutated within disease, germline mutations which predispose towards disease or disease-associated single nucleotide polymorphisms (SNPs). Functional genomic profiling determines the level of gene-transcripts and in some cases provides useful fingerprints for molecular profiling of tumours enabling patient-centred treatment decisions for personalized medicine. Proteomics and metabolomics identify alterations in proteins and metabolites. The data arising from proteomics can be both qualitative (presence/absence of proteins) or quantitative depending upon the technique being used. Metabolomics provides quantitative information about the levels of metabolites within disease and this information can be used to supplement data from other molecular profiling strategies to provide an improved patient-diagnostic/prognostic/treatment-oriented decision tool to aid disease management. In addition to biomarker discovery all of these platforms have the potential to discover key molecules involved in disease which could function as drug targets
2.1 Genomic Analysis
2.1.1 Methodology
Genomic techniques involve sequencing of DNA, the determination of gene sequences, base-substitution mutations within genes, sequencing and identification of gene fusions, and the detection of duplications and deletions of key areas of the genome and their relation to disease states. Genomic platforms have evolved to allow the sequencing of whole genomes (using paired end sequencing) [5] and the technology has developed to enable genomic sequencing from single cells [6]. In addition to sequence alterations cancers can also display gene copy number alterations. Normal cells are diploid containing two copies of every gene (one on each chromosome pair-with the exception of sex-linked genes on the X and Y chromosomes in males). Within many cancers regions of chromosomes are duplicated resulting in genes having more than two copies per cell and sometimes entire chromosomes are duplicated (polyploidy). Cancers can also harbour deletions resulting in less than two copies of genes per cell, and this can also encompass loss of entire chromosomes (aneuploidy). Copy number alterations within genes can be detected by array-based comparative genomic hybridization (aCGH) which enables the detection of copy number alterations within genes and whole chromosomes [7]. Genomic techniques for molecular classification have begun to impact upon patient diagnosis and treatment. For instance within breast cancer the Mammaprint®, (Agendia, Irvine, CA, USA) microarray based kit [8], and the OncotypeDX®, (Genomic Health, Inc, Redwood City, CA, USA) PCR-based kits [9], are both approved for use in standard clinical treatment guidelines.
2.1.2 Applicability
Current state of the art methods for genomic analysis (i.e., next generation sequencing-NGS) require a cellular source of DNA which can be obtained from solid tumours or circulating cancer cells. Solid tumours are challenging due to their heterogeneity as well as the presence of normal, healthy cells within the tumour mass. For this reason most studies focus on tumours with >60 % tumour nuclei present [10]. For diffuse tumour-types, such as pancreas and prostate cancer, laser-capture microdissection (LCM) can be employed, however this approach is challenging due to the low yields of genomic DNA (<100 ng). Genomic sequencing has the potential to reveal mechanistic aspects of cancer development including the identification of somatic mutations predictive of poor disease outcome (e.g., in acute myeloid leukaemia-[11]), identifying the clonal origin and development of malignancy [12] and determining treatment options [13].
2.2 Functional Genomic Analysis
2.2.1 Methodology
Functional genomic methodologies study the products of gene expression, principally mRNA transcripts as well as regulatory RNAs such as microRNAs [14]. The main technological platform used within functional genomics have been microarrays. Microarray surfaces present a series of short impregnated oligonucleotides printed onto their surface which will hybridize along the length of specific mRNAs. More recently exon arrrays have been developed which present oligonucleotides specific to individual exons within genes. As exons are specific DNA regions which encode protein domains, and as these exons are frequently shuffled together in differing orders during gene expression (a process termed “alternative splicing”)-exon arrays can provide information relevant to the expression of alternative protein isoforms. In addition to these array based methods deep-sequencing of mRNA (mRNA-seq) methods are becoming increasingly applied [15, 16]. By fluorescently tagging genetic material and using the principle of hybridization of complementary nucleic acid strands followed by the digital evaluation of fluorescent signals, microarrays allow the expression of tens of thousands of genes to be quantified simultaneously, and within pair-wise sample comparisons. Functional genomic studies can provide an assessment of the differentially expressed genes between two biological samples (e.g., healthy vs. cancer), as well as identifying alternative splicing events. This provides key mechanistic insights into the disease process as well as providing information relevant to patient stratification and guiding treatment options.
2.2.2 Applicability
Functional genomic screens profile cellular mRNA (the “transcriptome”) and thus require either tumour derived cells, circulating cancer cells within the blood, or released microsomes (small membranous vesicles reported recently to contain miRNAs [17]). The clinical value of functional genomic data is illustrated by the array of gene signature detection assays available to provide prognosis/prediction tools for breast cancer treatment [18]. Gene expression signatures can provide complementary information to histological tumour grade and patient health status to aid prediction of survival outcomes. Functional genomic profiling can also provide results aiding treatment decisions, e.g., the response to lenalidomide within del(5q) myelodysplastic syndromes (MDS) [19].
2.3 Proteomic Analysis
2.3.1 Methodology
A complete description of proteomic methods is beyond the scope of this book chapter however a review of these methods is provided within [20–22]. There are many different methodological platforms for proteomic analysis, and these can provide information including (i) identifying the proteins present within a biological sample, (ii) providing quantification of protein levels (and comparison of these levels amongst multiple samples), (iii) identification of protein-protein interactions (“interactomics”), (iv) identification of important regulatory post-translational modifications to proteins (e.g., phosphorylation), (v) profile temporal alterations in the levels of proteins within a biological system and (vi) identifying organelle and cellular localization. Whilst the number of proteomic hardware platforms and analytical strategies is great all methods use one of two different approaches: (a) “Top-down” proteomics-in which whole proteins and naturally occurring peptides are analysed and (b) “Bottom-up” proteomics-in which the proteins within a biological sample are digested into peptides in vitro using proteases (typically trypsin) and the resulting peptides analysed. Top-down proteomics is useful for identifying the range of Post-Translational Modifications (PTMs) within proteins-chemical modifications to the protein structure that are not part of the DNA encoded amino acid sequence (such as phosphorylation, glycosylation, ubiquitination) and alternative splicing/proteolytic isoforms within a sample, whilst bottom-up proteomics can generate larger data sets more rapidly due to the relative ease of identifying small peptides. Within biomarker discovery, proteomics has the advantage of identifying altered proteins, the class of molecules which are the target of almost all drug therapies. Furthermore altered protein expression cannot be inferred from genomic or functional genomic data sets.
2.3.2 Applicability
Proteomic approaches can be applied to tissue/cell-extracts, biological fluids (serum/plasma/urine) and more recently to tissue sections themselves. Each individual sample type provides its own unique challenges-e.g., within serum/plasma the high level of a few major protein components makes detecting disease-specific proteins/peptides more difficult, a limitation partially overcome by using immunodepletion [23]. Proteomic data sets can provide mechanistic insights into disease processes as well as providing diagnostic, prognostic and treatment-decision orientated information to guide cancer management.
2.4 Metabolomic Analysis
2.4.1 Methodology
Metabolomic methods enable the identification and quantification of metabolites (e.g., salts, lipids, steroids, sugars, hydrocarbons and salts) within body fluids as well as tissues. Metabolomic studies involve metabolite extraction followed by separation of the metabolites and their identification. The separation of metabolites can be performed using liquid chromatography-LC [24] or gas-chromatography-GC [25], and metabolite identification can be performed using either mass-spectrometry (MS) or nuclear magnetic resonance (NMR) [26]. The advantage of using MS within metabolomics is sensitivity, whilst NMR provides relatively low sensitivity but high reproducibility. Metabolic alterations are a frequent phenomena within cancers via cellular alterations such as the Warburg effect-(increased glycolytic flux within cancers [27]) and the reverse-Warburg effect [28]. Other key metabolic alterations observed within cancer include: hypoxia, increased synthesis of proteins, fatty acids and nucleotides, altered de novo fatty acid synthesis and alterations within lipid metabolism. Metabolomic data can be combined with proteomic data to provide a more detailed diagnostic fingerprint of cancer development, thus increasing the specificity of cancer diagnosis [29]. One potential advantage of metabolomic alterations within disease monitoring arises from the fact that metabolic alterations are already being used within diagnostic/therapeutic tests-for instance mass-spectrometry is frequently used for measuring inborn errors of fatty acid and amino acid metabolism within newborn babies [30].
2.4.2 Applicability
Metabolomic analysis within cancer diagnosis currently faces some of the same hurdles and challenges as proteomics. Although body fluids can be analysed by both MS-based and NMR-based metabolomics, and solid tissue samples are also applicable (by magic-angle NMR) several key challenges remain. In order to reliably detect disease states the normal range and variability of metabolite levels requires an improved definition, and sample preparation procedures need a greater degree of standardization to enable comparison between studies [31]. Sample preparation for LC-MS based metabolomics using solvent extraction also faces the limitation that each individual procedure samples only a sub-fraction of the entire metabolites present. Despite these limitations, metabolomics will provide biomarker signatures enhancing the diagnostic and prognostic utility of biomarkers discovered using other omic-platforms.
3 “Omic”-Strategies Within Bone-Metastatic Cancer
A summary of individual studies relating to cancers that metastasise to bone will now be presented. An overview of selected “omic”-biomarker studies is provided within Table 7.1.
4 Bone Metastasis in Multiple-Myeloma
4.1 Multiple Myeloma: Role of Epigenetic Regulation Within Bone Metastasis Revealed by Proteomic Profiling
The term “epigenetics” refers to a series of heritable modifications within the genome that do not consist of DNA-sequence alterations. Several forms of epigenetic modification have been identified within cancer including: (a) methylation of gene-promoter regions resulting in gene-silencing (“DNA-methylation”) [32]; (b) post-translational modification of the histone-components that bind DNA within the nucleus “histone modification” [33]; (c) repositioning of the nucleosomes to different DNA regions (“nucleosomal repositioning”) [34] and (d) the regulation of gene expression by short 18–25 nucleotide micro-RNAs (“miRNA”) [35].
Several miRNAS have been discovered which play a role in the developmental pathway of multiple myeloma from normal plasma cells through to MGUS and MM, including miR-21, miR-106b-25 cluster, miR181a and b, miR-32 and mIR-17-92 cluster [36]. miR-21 has received particular attention as a micro-RNA frequently over-expressed in a wide range of cancers including numerous solid tumours (hepatocellular carcinomas, gastric cancer, cervical carcinoma, ovarian carcinoma, head and neck cancers and papillary thyroid carcinomas) as well as leukemic cancers and thus a miRNA which functions as a classical oncogenic miRNA or “OncomiR” [37]. Quantitative reverse-transcription-polymerase chain reaction (qRT-PCR) enables the amplification of mRNA transcripts into cDNA with incorporation of fluorescent groups and the ability to monitor the rate of fluorescence-incorporation in real time. The rate of incorporation of the fluorescent signal is proportional to the amount of mRNA in the sample enabling as estimation of the relative level of different mRNA transcripts and this approach can be applied to miRNAs as well. In a PCR-based study of myeloma cells it was observed that miR-21 levels were increased when these cells were cultured in the presence of bone marrow stromal cells (BMSCs) [38]. The miR-21-induced alterations in protein expression occurring within MM cells were profiled by selective knockdown of miR-21 expression following transfection with a locked nucleic acid anti-miR-21 oligonucleotide (LNA-21) and in the control experiment transfection with a control oligonucleotide (LNA-cont) (see Fig. 7.2). SILAC-labelling of cells transfected with LNA-miR-21 and LNA-cont enabled the quantitative estimation of the global proteomic alterations occurring in response to the action of miR-21. Several proteins were identified as potential miR-21-targets including the Protein Inhibitor of activated STAT3 (PIAS3)-a negative regulator of Signal Transducer and Activator of Transcription 3 (STAT3) activity [39]. Constitutive STAT3 signalling has been strongly implicated in the development of MM [40] and PIAS3 has been demonstrated to negatively regulate IL-6-mediated STAT3-signalling within MM cells [41]. SILAC-based comparison of a MM-cell line before and after H1-parvovirus-mediated reversion of the malignant phenotype identified 379 proteins which were either increased or decreased during cell-reversion with STAT3 being the most significantly down-regulated, further pointing to a role of STAT3 in MM-progression [42].

Proteomic analysis of multiple myeloma cells identifies a regulatory network stimulating cancer cell proliferation: Multiple myeloma cells within the bone microenvironment are in contact with bone marrow stromal cells (BMSCs). The interaction with BMSCs promotes MM-cells to secrete several autocrine and paracrine factors including SDF1α, VEGF-A and IL-6. Contact with BMSCs also increases the level of miR-21 within bone-resident MM-cells. One of the targets of miR-21 action is the gene for PIAS-3. PIAS-3 decreases cell-proliferation by dephosphorylating STAT-3 downstream of the IL-6 receptor (IL-6-R). Contact with BMSCs thus activates MM-cell proliferation both by stimulating the release of proliferative autocrine and paracrine cytokines and growth factors from MM cells, but also by inhibiting a growth-inhibitory pathway acting via PIAS-3 and STAT3
In addition to a role in the regulation of miRNA-21 levels, binding or MM-cells to BMSCs also increases the secretion of cytokines such as stromal-derived factor-1α (SDF-1α), vascular endothelial growth factor (VEGF) and interleukin-6 (IL-6) which promote cell-survival, migration and angiogenesis. The bone environment thus promotes multiple myeloma cell survival via a number of mechanisms including cell-cell contact and receptor mediated signalling as well as epigenetic modification within metastatic MM cells themselves (see Fig. 7.2).
Functional genomic studies combined with SILAC-labelling and proteomic analysis have thus identified a key epigenetic switch responsible for the adaptation of multiple myeloma cells to growth within the bone metastatic niche. This work also identifies the IL-6/STAT3 signalling pathway as a potential drug target within multiple myeloma.
4.2 Functional Genomic Profiling Identifies a Gene-Signature Predictive of Dependence Upon the Bone-Microenvironment
The survival and proliferation of MM cells within the bone microenvironment is promoted by a number of autocrine and paracrine signalling systems which enhance tumour cell proliferation and inhibit tumour cell apoptosis. Several members of the tumour necrosis factor (TNF) family have been reported to be elevated within the serum of patients with MM, including B-cell Activating Factor (BAFF), and A Proliferation-Inducing Ligand (APRIL) [43, 44]. BAFF and APRIL are produced within the bone microenvironment with APRIL being a significant factor released by osteoclasts [45]. Several receptors for BAFF and APRIL have been identified within malignant plasma cells including the receptors TAC1 (Transmembrane activator and calcium modulator and Cyclophilin Ligand Interactor) and BCMA (B-Cell Maturation Antigen).
Gene expression profiling of purified MM cells from patients across a range of clinical grades, followed by hierarchical clustering identified two sub-groups of patients, a TACIhigh subgroup and a TACIlow subgroup with a 659-gene signature differentially expressed between them [45]. TACIhigh MM cells displayed a gene signature more similar to that of mature plasma cells, with a preponderance of up-regulated transcripts encoding autocrine/paracrine signalling components and receptors responsible for interaction with the extracellular matrix (ECM) and bone microenvironment. In contrast TACIlow MM cells express a gene signature with a preponderance of cell-cycle genes resembling the profile of plasmablastic cells. Treatment of purified MM-cells with BAFF/APRIL did not alter the expression pattern of the signature genes within the TACIhigh / TACIlow signatures suggesting that these transcriptional profiles may arise from exposure to the bone microenvironment and not be directly regulated BAFF/APRIL transcripts themselves. TACIlow patients have a higher proportion of advanced stage III MM cases, more frequent bone lesions and a decreased haemoglobin level and an overall worse prognosis than TACIhigh patients. In particular the TACIhigh/low status of patients did not correlate with other known clinical parameters and risk factors including levels of β2m/LDH or CRP, suggesting that TACI may well be an independent prognostic factor for outcome within MM. Stratification of MM cases into TACIhigh/low subclasses could also aid treatment decisions as many therapeutic agents target components of the bone microenvironment and autocrine/paracrine signalling components responsible for tumour cell survival.
4.3 Phosphoproteomic Profiling to Identify Key Signalling Components Within Multiple Myeloma Bone Metastases
Altered cellular signalling within MM could represent a potential target for future drug discovery. Several signalling networks involving tyrosine-phosphorylation are altered within multiple myelomas. A subgroup of MM cases harbour the t(4;14) chromosomal translocation which results in the activation of the fibroblast growth factor receptor-3 (FGFR3) [46] and a role for activation of FGFR3 has been identified within a variety of cancers including bladder, colon and cervical cancers as well as skeletal dysplasia’s [47]. Signalling via FGFR3 occurs via a similar mechanism to many receptor tyrosine kinases (RTKs), in which ligand binding to the extracellular domain of the receptor triggers receptor activation and autophosphorylation of key tyrosine-residues within the cytoplasmic domain of the receptor. These phosphorylated sites can act as docking sites for key-signalling proteins which contain src-homology-2 (SH2) and protein-tyrosine-binding (PTB)-binding domains [48, 49]. The activated receptor tyrosine kinases can also phosphorylate other proteins in a signalling cascade. Phosphoproteomic identification of key proteins involved in FGFR3 signalling has been facilitated by use of an FGFR3-inhibitor PD173074, as well as by stimulatory treatment of MM cells with FGF1 and the pan-tyrosine-phosphatase inhibitor orthovanandate. Isolation of phosphotyrosine-containing peptides from the MM-cell line KMS11 treated with PD173074, or with FGF1+orthovanandate, followed by label-free quantification identified a series of protein phosphorylation sites which were increased by FGF1-treatment and inhibited by PD173074-treatment [50]. These candidate FGFR3-mediated targets included proteins within cell-signalling cascades (Ribosomal S6 Kinase 2-RSK2, proteins involved in endocytosis which may regulate FGFR3 signalling, cytoskelatal proteins and proteins which regulate growth factor signalling to MM cells) [50]. This phosphoproteomic study identified key proteins responsible for the FGFR3-mediated growth of multiple-myeloma. Targets such as RSK-2 may also be potential drug targets within multiple myeloma.
5 Prostate Cancer Metastasis to Bone
5.1 Metabolomic Alterations Within Prostate Cancer Metastasis to Bone
Metabolic alterations accompanying prostate cancer metastasis to bone could potentially be utilized to aid the prognosis of prostate cancer metastatic spread enabling more rapid application of drug treatments. Several metabolomic studies have been performed within prostate cancer including: (a) a reduction in citrate concentrations within primary prostate tumours compared to benign prostatic hyperplasia (BPH) or normal prostate tissues [51], as well as (b) a 1H-NMR study which demonstrated statistically significant altered ratios of citrate/lactate, citrate/total choline, phosphocholine/total creatinine, choline/total creatinine, alanine/total creatinine, phosphoethanolamine/total phosphate, phosphocholine/total phosphate and glycerophosphoethanolamine/total phosphate within prostate cancer tissue samples compared to BPH samples [52]. In contrast here have been few metabolomic studies of prostate cancer metastasis to bone.
To date there have been a few metabolomic studies within prostate cancer metastasis. Using gas-chromatography-MS (GC-MS) Sreekumar et al. [53] identified elevated levels of sarcosine (an N-methylated derivative of the amino acid glycine) as being elevated in prostate cancer invasion. Within this study it was observed that reduction in the level of sarcosine (by knock-down of glycine n-methyltransferase) attenuated the invasive potential of prostate cancer cell lines. Similarly increasing the level of sarcosine (by knock down of the sarcosine degrading enzyme sarcosine dehydrogenase) increased the invasive potential of prostate endothelial cells [53].
Metabolomic profiling of normal-bone and prostate cancer derived bone metastases by GC-MS identified a panel of 71 metabolite peaks of which 34 were identifiable [54]. Validation of this data set was also performed by GC-MS analysis of plasma samples from prostate cancer patients with and without bone metastases as well as plasma samples from patients with benign prostate disease. In addition metabolomic profiling of both malignant and benign prostate tissue was also performed and the results also indicated increased cholesterol levels within bone metastatic prostate cancer [54]. A key metabolite observed to alter within bone metastatic prostate cancer was cholesterol, with statistically significant higher levels of cholesterol within prostate cancer bone metastases than from bone metastases derived from other forms of cancer. Increased immunostaining for the low-density lipoprotein receptor (LDL-R) as well as the scavenger receptor class B type I receptor (SR-B1) suggested an increased potential for bone metastatic prostate cancer cells to take up cholesterol containing lipoproteins. In addition increased immunostaining for 3-hydroxy-3-methyl-glutaryl-CoA-reductase (HMG-CoA-Reductase) was observed in osteoblasts situated adjacent to the metastatic prostate cancer cells [54].
This panel of metabolites identified in advanced, metastatic prostate cancer may enable the earlier detection of cancer spread to bone (particularly when using high-sensitivity methods such as LC-MS). In combination with proteomic biomarker profiles this may facilitate the high-sensitivity, high specificity detection of malignant spread to bone.
5.2 Transcriptomic Alterations Within Bone-Metastatic Prostate Cancer Cells
Functional genomic studies of the altered gene expression profiles within bone metastatic prostate cancers have attempted to identify master transcriptional regulators of bone colonization. Several transcription factors have been implicated in osteoblastogenesis including the Runx-transcription factor family member Runx2 [55–57]. Runx2 transcriptional activity has been associated with expression of key-bone proteins including bone sialoprotein [58], MMP9 [59] and Runx2 expression induces the mineralization of prostate cancer cell-lines [60]. Gene expression analyses have identified a panel of genes which are Runx2 targets including: genes mediating anti-apoptotic protection of prostate cancer cells e.g., survivin and Bcl2 [61, 62], increases in prostate cancer cell survival via elevated expression of BMP7 [63, 64], as well as known genes involved in epithelial-mesenchymal transition (EMT), invasiveness, degradation of the extracellular matrix, bone breakdown and angiogenesis [59, 65–67] and osteoclast differentiation [59, 65, 66]. The combined effect of these transcriptional alterations is to promote prostate cancer growth within and adaptation to the bone environment (see Fig. 7.3).

Transcriptomic profiling identifies a key transcriptional regulator within bone-metastatic prostate cancer: Transcriptomic profiling has identified the Runx2 transcription factor as a key transcriptional regulator involved in prostate cancer metastasis to bone. Bone is a rich source of hyaluronan, a non-protein-containing glycosaminoglycan which binds to the receptor CD44 on metastatic prostate cancer cells triggering the phosphorylation of the SMAD5-transcriptional coactivator. Osteoprotogerin (OPN) which is secreted by metastatic prostate cancer cells, binds to the cell surface receptor αvβ3-integrin triggering phosphorylation of the transcription factor Runx2. The complex of phospho-SMAD5 and phospho-Runx2 can then activate the transcription and protein expression from genes involved in numerous aspects of prostate cancer metastasis to bone including: cell survival, increased bone resorption, extracellular matrix (ECM) degradation increased cell motility and osteoclast (OC) activation
The molecular events which trigger Runx2 expression and activation when prostate cancer cells metastasize to bone are a subject of intensive research. Recent studies revealed a role for SMAD5 phosphorylation within the signal transduction pathway leading to Runx2 activation [68]. Transcriptional activation of Runx2 with resultant increased RANKL production by metastatic prostate cancer cells requires phosphorylation of both SMAD5 and Runx2. SMAD5 phosphorylation increases when hyaluronan (a major component of the ECM within bone) binds to the cell surface receptor CD44 on prostate cancer cells [69]. Runx2 phosphorylation was observed to require ligation of the cell surface receptor αvβ3-integrin [68], and αvβ3-integrin has been demonstrated to bind osteopontin, a signalling component secreted by prostate cancer cells [70]. Increased bone resorption functions in concert with oestrogen receptor (ER) signalling to regulate Runx2 [71, 72] and there is evidence that Runx2 expression itself may be driven by a switch in the oestrogen receptor expression profile from the ERβ1 isoform (which suppresses Runx2 expression) to the ERβ2 isoform (which enhances Runx2 expression) [73] (See Fig. 7.3).
The transcriptomic profiling of bone-metastatic prostate cancer cells identifies a gene signature indicative of Runx2 transcriptional activation within bone metastases. Runx2 may therefore be a key target for therapies (including miRNA-mediated gene therapies) aiming to reduce prostate cancer spread to bone.
5.3 Serum Diagnostic Markers for Prostate Cancer Metastasis to Bone
Diagnostic markers for prostate cancer metastasis to bone are urgently required to supplement current assessment procedures which typically involve isotope bone scanning (reviewed in [74]). Serum/plasma represents a potentially invaluable sample source for biomarker discovery as it can be obtained non-invasively. In the time course of prostate cancer development the failure of anti-androgen therapy initially presents as a biochemical failure characterized by rising serum prostate-specific antigen (PSA)-levels [75]. This biochemical failure predates the development of detectable bone metastases and metastasis-associated symptoms by a median time of approximately 6-months [75]. Thus there is a window of time during which serum/plasma biochemical alterations predate the development of clinical symptoms of cancer-spread to bone. Earlier detection of bone micrometastases may enable more effective targeting of bone-directed therapies to target prostate cancer spread.
Proteomic profiling of prostate cancer serum samples using 4-plex iTRAQ was performed using 4 different groups of serum pools: (i) benign prostatic hyperplasia (BPH) samples, (ii) localised prostate cancer with no evidence of progression, (iii) localized prostate cancer with biochemical evidence of progression and (iv) serum from patients with confirmed bone metastases [76]. Of 122 proteins identified and quantified within this study 25 proteins were significantly differentially expressed between progressing vs. non-progressing cancer samples and 23 proteins were significantly differentially expressed between bone-metastatic and progressing samples. Within the 23 metastasis associated proteins eukaryotic translation elongation factor 1 alpha 1 (eEF1A1) was further validated by immunostaining of tissue microarrays and observed to be elevated within osteoblasts within close proximity to bone-metastases [76]. Low molecular weight-peptide-based biomarkers of prostate cancer metastasis to bone have also been identified by SELDI-TOF-MS, resulting in the identification of a series of serum amyloid protein A (SAA) isoforms with statistically significant elevated expression within serum from bone metastatic prostate cancer patients compared to prostate cancer patients without bone metastases, a result confirmed by immunoprecipitation assays [77].
“Bottom-up” proteomic analysis of prostate cancer serum samples, and characterization of the low-MW serum peptidome has thus identified potential early diagnostic markers for prostate cancer metastasis to bone.
6 Breast Cancer Metastasis to Bone
6.1 Breast Cancer Bone Metastasis: Transcriptional Profiling Reveals a Key Role for Transforming Growth-Factor-β (TGFβ)/Bone Morphogenetic Protein (BMP)-Signalling
Breast cancer primary tumours have been subject to extensive gene expression analysis using both commercially available microarray chips (i.e., Affymetrix) as well as using custom made chips, and the gene expression data from these studies are publicly available (via gene expression omnibus). These gene expression databases represent a potentially rich source of information for identifying key mediators of breast cancer development, relapse and metastatic spread. In a recent statistical analysis of these data sets a subset of genes were identified which correlated with the risk of relapse. Members of this gene family subset displayed either increased or decreased expression levels correlating with risk of relapse across a panel comprising hundreds of breast cancer samples representing all stages of development and subtypes of breast cancer [78] and gene ontology analysis identified key members of this relapse- and metastasis-related gene family to be transforming growth factor-β (TGFβ) family cytokines and a key TGFβ-family member antagonist-Noggin [78, 79].
TGFβ has the ability to both inhibit as well as promote tumorigenesis depending upon the stage of cancer development [80–82]. The TGFβ-family of growth factors includes Bone morphogenetic Proteins (BMPs) which stimulate bone formation. Several BMP inhibitors have been identified which play diverse roles within developmental pathways, embryogenesis and cancer [83] including the BMP-antagonist Noggin. TGFβ-family cytokines play a variety of roles within breast cancer metastasis to bone in particular by altering the balance of bone formation and bone breakdown. Bone consists of mineralised extracellular matrix components, and numerous cell types including bone forming osteoblasts and bone-resorbing osteoclasts [84]. Osteoblasts secrete growth factors including Receptor of Activator of Nuclear Factor κB-Ligand (RANKL) which binds to the Receptor activator of Nuclear Factor-κB (RANK) on osteoclasts stimulating osteoclast maturation and bone degradation. Osteoblasts can also secrete osteoprotogerin (OPG) a soluble decoy receptor which inhibits RANKL function.
BMPs are members of the TGFβ-family of growth factors, a large family of growth factors with over 20 members with numerous diverse functions [85]. BMPs play key roles in bone-formation including the formation of the body-axis, and bone and cartilage formation [86]. Several BMP-family members promote bone formation by acting upon osteoblasts to increase their release of OPG and reduce the release of RANKL thereby inhibiting osteoclast mediated bone degradation. Within development BMP action is controlled by a series of secreted BMP-antagonists which also play key developmental roles [83]. Noggin is a key BMP-antagonist which is required for correct embryonic development [87] and gene-knockout studies have suggested that it plays a key role in skeletal development [88].
Mechanistic investigation of the role of noggin within breast cancer metastasis to bone revealed that high noggin-expression is strongly selected for within the bone environment (but not within metastases to the lung, liver or brain) [79]. Over-expression of noggin increased the growth rate of bone metastases within orthotopic mouse models as assessed by BioLuminescence imaging, furthermore shRNA-mediated gene silencing of noggin reduced the growth rate of bone metastases in the same study and modulation of noggin levels was observed to influence the ability of breast cancer cells to form tumourspheres-suggestive that noggin might also facilitate the re-initiation of metastases via inhibiting the differentiation of metastatic breast cancer cells [79]. In this way the BMP antagonist noggin may provide bone metastatic breast cancer cells with a double advantage for growth and colonization within the bone environment and be a potential drug-target for targeting of bone metastases.
6.2 Breast Cancer Adaptation to the Bone-Metastatic Environment: Patient-Matched Genomic/Proteomic Studies
The adaptation of metastatic cancer cells to the bone microenvironment is a key step within cancer dissemination and a potential source of therapeutic targets. There have been few studies on primary tumours and patient matched metastases. This is partly due to the long time frame -breast cancer bone metastases often present years after the resection of the primary tumour and also due to logistical challenges in obtaining bone metastasis biopsy material [89]. Despite these limitations there have been a few omic-profiling studies of patient-matched primary-tumour vs. bone metastasis samples as well as studies within mouse-model systems for bone metastasis (see Fig. 7.4).

Comparative molecular-profiling of primary tumours and matched bone metastases: Molecular profiling of patient matched primary tumours and bone metastases, as well as mouse model systems has the potential to identify functionally important molecules within bone metastasis. The common cellular-origin within the mouse-model, as well as the isogenic background for the patient-derived samples reduces the effect of inter-individual variability. This facilitates the identification of functional molecules within bone metastases. Functional genomic and proteomic studies have been conducted within such sample types identifying the up-regulation of osteoblastic differentiation genes, as well as the altered protein expression of cell-surface molecules such as Class-I HLA molecules and αvβ3-integrin
Genomic analysis of primary breast tumours and these tumours after their relapse to either brain or bone metastatic sites identified panels of genes which clustered together according to the site of metastasis [90]. In total 22 transcripts were differentially expressed between the primary tumour and bone metastases and hierarchical clustering revealed similarity between the bone metastases and the primary breast tumour. Gene expression analyses such as these offer the hope that a diagnostic signature could be profiled within a primary tumour which will predict the site of future metastases, thus aiding treatment decisions [90, 91].
Functional genomic profiling has also been applied to mouse models of metastatic breast cancer. Microarray analysis of a breast cancer cell line (MDA-MB-231) and a bone homing variant obtained by intra-cardiac injection (MDA-MB-231-B02) identified the upregulation of a panel of 11 mRNAs with known roles within osteoblastic differentiation, including the increased expression of the osteoblast specific differentiation protein cadherin-11 [92]. A separate study involving functional genomic profiling of MDA-MB-231 breast cancer cells and a mouse-bone homing variant identified a functionally significant role for vascular cell adhesion molecule-1 (VCAM-1) in the recruitment of osteoclast progenitors into the site of bone micrometastases [93]. Anti-VCAM-1 antibodies had demonstrable ability to inhibit the development of bone metastases in this study [89].
Proteomic profiling of paired primary tumour/bone metastasis samples focussing on cell-surface and secreted proteins identified proteins implicated in cell-cell communication, and autocrine and paracrine signalling events. Cell-membrane proteins are attractive potential targets for antibody-based therapies. Surface biotinylation (a technique which enriches for cell-membrane proteins) has been employed in studies to date. Isolation of biotinylated membrane proteins from the osteotropic cell-line MDA-MB-231-B02 (a bone homing variant of MDA-MB-231) revealed the upregulation of the cell-surface receptor αvβ3-integrin, and the down-regulation of class-I HLA molecules within the bone homing cells [94]. Proteomic analysis of a primary human breast tumour and a bone-metastasis from the same patient, with identification of surface biotinylated as well as glycosylated proteins, revealed a decreased expression of tumour suppressive α2β1-integrin within the bone metastasis [95]. Numerous proteins involved in cancer cell motility and tumour aggressiveness were identified in this study as being elevated in bone metastasis including activated leukocyte cell adhesion molecule (ALCAM/CD166), whilst Sushi-domain-containing protein-2 (SUSD2)-a known tumour suppressor-had reduced expression with the bone metastasis samples [95]. Addition of these differentially expressed proteins to the current breast cancer biomarkers oestrogen-receptor (ER) and HER2 may improve treatment decisions. Tumours are currently classified according to histological criteria as well as the presence of differing receptor expression levels such as for oestrogen-receptor and HER2. Measurement of the levels of the differential proteins identified in these studies and their inclusion within the classification criteria may enable a more accurate subdivision of tumour types according to aggresiveness and response to therapeutic interventions.
7 Bone Metastasis Biomarkers: From Pre-clinical “Omics” Screens to Clinical Application
The application of genomics, transcriptomics, proteomics and metabolomics to biomarker-discovery within bone-metastatic cancers has generated large quantities of data and numerous potential biomarkers for further development. Whilst these studies are very promising the application of “omic”-strategies to the field of bone metastatic cancer is relatively recent and few omic-insights have been pursued as far as clinical utility.
One of the key challenges in the development of clinical biomarkers is revealed by recent data regarding the high degree of heterogeneity of tumours. High throughput genomic sequencing within breast cancer has identified extensive inter-tumour heterogeneity, with each individual tumour containing multiple cell clones each with a different pattern of mutations [96]. The genomic sequencing of gene fusion products within breast cancer also reveals considerable inter-individual heterogeneity [97] and this diversity may partly be explained by defects in the apparatus responsible for mismatch repair leading to genomic instability [98]. There is therefore a diverse family of subtypes within each organ-specific cancer and this makes it unlikely that an individual biomarker will predict outcomes in all cases of that cancer. A consequence of this is that currently used individual markers can have high sensitivity but low specificity. Prostate specific antigen (PSA) within prostate cancer is just one example of a biomarker with high sensitivity but low specificity [99]. The requirement for high specificity to prevent false positive results and consequent patient stress (and unnecessary treatment costs) has driven the search for multiple biomarker panels which should have improved diagnostic ability. In this approach successful biomarker development must therefore aim to identify a series of molecules which are involved in the key steps within the disease process with sufficient diversity to represent the full spectrum of subtypes within that cancer. The requirement for biomarkers enabling early diagnosis is particularly acute. In the early stages of cancer development alterations in protein and metabolite levels are likely to be of small magnitude and therefore multi-marker panels may also provide a compound assessment of disease progression. The development of diagnostic/prognostic decision tools arising from “omics”-research thus frequently focuses upon multi-marker panels.
The key steps involved in the translation of pre-clinical biomarkers into clinical utility are briefly outlined below and summarised in Fig. 7.5 (biomarker development for clinical utility has been covered in detail in several excellent reviews including [100]).

“Omic”-strategies within cancer metastasis to bone: Workflow from the laboratory to clinical application: Omic strategies have the potential to impact upon patient diagnosis and treatment in several ways, most notably the development of new clinical tests for prognosis/diagnosis of disease as well as the discovery of new drug targets. (a) As the majority of “omic” discovery platforms are time-consuming and/or expensive initial discovery is usually performed in a small cohort of well-defined patients. (b) The results of this discovery phase can include potential disease biomarkers and/or drug targets. Validation of these biomarkers involves application of the potential predictive panels within class-prediction tests using a larger blinded panel of patients with or without the disease. This first validation phase frequently requires the development of high-throughput assays for the markers. (c) Further validation of the candidates discovered then proceeds through multi-centre testing of the biomarker(s) to ensure that the insight discovered by the original omics-based screen is applicable across multiple clinics and laboratories. Only when a biomarker panel or drug target has cleared these steps of development and received regulatory approval will the original omics-based discovery proceed to clinical applicability. Eventual clinical application depends upon health economic assessment and the new diagnostic marker is frequently combined with pre-existing markers to provide the final, improved patient-diagnostic tool
The translation of pre-clinical findings into improved early diagnosis tools for bone metastasis, as well as their incorporation into patient stratification nomograms and treatment option determinants involves a number of number of stages and challenges. Pre-clinical biomarker discovery using “omics”-technologies typically involves the use of time-consuming procedures and expensive technology platforms, and for this reason they usually involve small patient sets. The putative biomarker candidates resulting from these small scale discovery projects require confirmation in blinded validation cohorts. A significant proportion of candidates fail this validation step and this may be due to the small number of samples originally analysed, sample biases, or in some cases the lack of robust sample preparation procedures. Quantification of biomarkers within large patient sets frequently requires the development of high-throughput assays for use in clinical chemistry laboratories.
In order to provide an effective clinical test the putative biomarkers discovered within pre-clinical studies must have demonstrable reproducibility between institutions. A challenge here to date has been the lack of standardization within the platforms used to discover potential biomarkers in pre-clinical studies such that biomarker panels may not be reproducible over time within an institution or between institutions. Validation of biomarkers at this stage requires the ability of the biomarker panel to accurately predict which patients have disease (or the disease-stage in question) within large, population-based, multi-institutional blind test cohorts (see Fig. 7.5). Biomarker candidates and panel-based diagnostic/prognostic tools that prove their utility across multiple institutions using these high-throughput assays provide a suitable biological basis for the development of clinical test kits. Eventual application of the clinical products (test kits or pharmaceutical drugs) to the sphere of patient treatment requires regulatory approval and input from health-care professionals and health-economic advisors.
7.1 Genomics/Functional Genomics: Towards Clinical Applicability
Gene expression signatures have already made a significant contribution towards cancer treatment decisions and outcome prediction, as application of the 70-gene signature MammaPrint test and the 21-gene-signature OncoTypeDX kits within breast cancer illustrate [18]. There is evidence that as blood cells flow through tumour tissues signalling events modify the gene expression profiles of the blood cells. Whole RNA-based transcriptomics has recently identified gene expression signatures predictive of overall survival within castration-resistant prostate cancer [101, 102]. Therefore whole blood profiling of mRNA (and miRNA) expression levels within whole blood cells offers considerable promise for informing cancer treatment. These gene expression signatures may reflect the risk of bone metastasis as this is a major contributor towards the morbidity arising from these cancers. Gene expression profiling and correlation with overall survival does not always relate to bone metastasis however, as a recent study within breast cancer illustrates [103]. In the study of Rajski et al. 2012 MDA-MB-231 cells cultured in the presence of osteoblasts up-regulated two sets of genes, one set of interferon-response genes which strongly predicted overall survival, and another set of IL-6 related genes which did not significantly change overall survival but was associated with a shorter time to bone metastasis [103]. Genomic profiling and gene expression analysis thus holds out significant promise for the mechanistic elucidation, and clinical management of bone metastatic cancers. Large multi-centre trials with careful data analysis (including patient associated meta-data) has the potential to reveal key insights into bone metastasis.
7.2 Proteomic/Metabolomic Signatures of Disease: Towards Clinical Utility
There have been many pre-clinical proteomic/metabolomic-studies performed to date which have identified potential protein and metabolic alterations which occur within bone metastatic cancer. None of these observations have to date impacted upon the treatment of bone metastatic cancer in the clinic, though some of these putative biomarkers are progressing through downstream biomarker validation. This validation relies on quantitative measurement of the candidates discovered within preclinical studies in much larger patient cohorts and this requires the development of robust, quantitative assays. Proteomic biomarker validation to date has principally involved use of immunoassays (i.e., ELISA), however MS-based quantitative methods for assaying proteomic biomarkers such as multiple-reaction monitoring (MRM) are increasingly being used [104]. Despite the current early stage of translation of proteomic/metabolomic markers into the clinic, multi-marker panels composed of these candidates have considerable potential to impact upon patient treatment in bone metastatic cancer in the future, particularly when combined with existing diagnostic markers (such as PSA) and clinical observations.
8 Conclusions
Post-genomic technologies are relatively recent additions to the arsenal of techniques being applied to the diagnosis and treatment of cancers that metastasise to bone. To date these technologies have contributed considerable insights into the disease-mechanisms and potential drug targets for bone metastatic cancers. Continual refinement of the techniques involved, for instance improved sensitivity within NMR-based metabolomic studies, the improved accuracy of transcriptome analysis using techniques such as mRNA-seq, and the expansion of functional genomics to include recently identified non-coding regulatory RNAs (such as miRNAs) will further increase the utility of omic-strategies within bone metastasis in the foreseeable future.
Abbreviations
- APRIL:
-
A proliferation inducing ligand
- BAFF:
-
B-cell activating factor
- BCa:
-
Breast cancer
- cDNA:
-
Complementary DNA
- miRNA:
-
Micro-RNA
- MM:
-
Multiple myeloma
- mRNA:
-
Messenger RNA
- MS:
-
Mass spectrometry
- NMR:
-
Nuclear magnetic resonance
- PCa:
-
Prostate cancer
- TF:
-
Transcription factor
References
Mundy G (2002) Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer 2:584–593
Weilbaecher K, Guise T, Mccauley L (2011) Cancer to bone: a fatal attraction. Nat Rev Cancer 11:411–425
Ablin RJ, Soanes WA, Gonder MJ (1969) Immunologic studies of the prostate. A review. Int Surg 52:8–21
Strimbu K, Tavel JA (2010) What are biomarkers? Curr Opin HIV AIDS 5(6):463–466
Fullwood MJ, Wei CL, Liu ET et al (2009) Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res 19:521–532
Baslan T, Kendall J, Rodgers L et al (2012) Genome-wide copy number analysis of single cells. Nat Protoc 7:1024–1041
Ueno T, Emi M, Sato H et al (2012) Genome-wide copy number analysis in primary breast cancer. Expert Opin Ther Targets 16(Suppl 1):S31–S35
Slodkowska EA, Ross JS (2009) MammaPrint 70-gene signature: another milestone in personalized medical care for breast cancer patients. Expert Rev Mol Diagn 9:417–422
Malo TL, Lipkus I, Wilson T et al (2012) Treatment choices based on oncotype Dx in the breast oncology care setting. J Cancer Epidemiol 2012:941495
Mardis ER (2012) Genome sequencing and cancer. Curr Opin Genet Dev 22:245–250
Mardis ER, Ding L, Dooling DJ et al (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361:1058–1066
Ding L, Ley TJ, Larson DE et al (2012) Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481:506–510
Jones SJ, Laskin J, Li YY et al (2010) Evolution of an adenocarcinoma in response to selection by targeted kinase inhibitors. Genome Biol 11:R82
Cho WC (2010) MicroRNAs: potential biomarkers for cancer diagnosis, prognosis and targets for therapy. Int J Biochem Cell Biol 42:1273–1281
Cowell JK, Hawthorn L (2007) The application of microarray technology to the analysis of the cancer genome. Curr Mol Med 7:103–120
Marguerat S, Bahler J (2010) RNA-seq: from technology to biology. Cell Mol Life Sci 67:569–579
Ajit SK (2012) Circulating microRNAs as biomarkers, therapeutic targets, and signaling molecules. Sensors (Basel) 12:3359–3369
Hornberger J, Alvarado MD, Rebecca C et al (2012) Clinical validity/utility, change in practice patterns, and economic implications of risk stratifiers to predict outcomes for early-stage breast cancer: a systematic review. J Natl Cancer Inst 104:1068–1079
Bacher U, Kohlmann A, Haferlach T (2010) Gene expression profiling for diagnosis and therapy in acute leukaemia and other haematologic malignancies. Cancer Treat Rev 36:637–646
Aebersold R, Mann M (2003) Mass spectrometry-based proteomics. Nature 422:198–207
Liang S, Xu Z, Xu X et al (2012) Quantitative proteomics for cancer biomarker discovery. Comb Chem High Throughput Screen 15:221–231
Walther TC, Mann M (2010) Mass spectrometry-based proteomics in cell biology. J Cell Biol 190:491–500
Smith MP, Wood SL, Zougman A et al (2011) A systematic analysis of the effects of increasing degrees of serum immunodepletion in terms of depth of coverage and other key aspects in top-down and bottom-up proteomic analyses. Proteomics 11:2222–2235
Becker S, Kortz L, Helmschrodt C et al (2012) LC-MS-based metabolomics in the clinical laboratory. J Chromatogr B Analyt Technol Biomed Life Sci 883–884:68–75
Dunn WB, Broadhurst D, Begley P et al (2011) Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat Protoc 6:1060–1083
Malet-Martino M, Holzgrabe U (2011) NMR techniques in biomedical and pharmaceutical analysis. J Pharm Biomed Anal 55:1–15
Bensinger SJ, Christofk HR (2012) New aspects of the Warburg effect in cancer cell biology. Semin Cell Dev Biol 23:352–361
Martinez-Outschoorn UE, Pavlides S, Howell A et al (2011) Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol 43:1045–1051
Benjamin DI, Cravatt BF, Nomura DK (2012) Global profiling strategies for mapping dysregulated metabolic pathways in cancer. Cell Metab 16(5):565–577
Garg U, Dasouki M (2006) Expanded newborn screening of inherited metabolic disorders by tandem mass spectrometry: clinical and laboratory aspects. Clin Biochem 39:315–332
Van QN, Veenstra TD (2009) How close is the bench to the bedside? Metabolic profiling in cancer research. Genome Med 1:5
Baylin SB, Jones PA (2011) A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 11:726–734
Suganuma T, Workman JL (2011) Signals and combinatorial functions of histone modifications. Annu Rev Biochem 80:473–499
Valouev A, Johnson SM, Boyd SD et al (2011) Determinants of nucleosome organization in primary human cells. Nature 474:516–520
Rouhi A, Mager DL, Humphries RK et al (2008) MiRNAs, epigenetics, and cancer. Mamm Genome 19:517–525
Pichiorri F, Suh SS, Ladetto M et al (2008) MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc Natl Acad Sci U S A 105:12885–12890
Krichevsky AM, Gabriely G (2009) miR-21: a small multi-faceted RNA. J Cell Mol Med 13:39–53
Wang X, Li C, Ju S et al (2011) Myeloma cell adhesion to bone marrow stromal cells confers drug resistance by microRNA-21 up-regulation. Leuk Lymphoma 52:1991–1998
Xiong Q, Zhong Q, Zhang J et al (2012) Identification of novel miR-21 target proteins in multiple myeloma cells by quantitative proteomics. J Proteome Res 11:2078–2090
Catlett-Falcone R, Landowski TH, Oshiro MM et al (1999) Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 10:105–115
Wang LH, Yang XY, Mihalic K et al (2001) Activation of estrogen receptor blocks interleukin-6-inducible cell growth of human multiple myeloma involving molecular cross-talk between estrogen receptor and STAT3 mediated by co-regulator PIAS3. J Biol Chem 276:31839–31844
Ge F, Zhang L, Tao SC et al (2011) Quantitative proteomic analysis of tumor reversion in multiple myeloma cells. J Proteome Res 10:845–855
Moreaux J, Legouffe E, Jourdan E et al (2004) BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood 103:3148–3157
Novak AJ, Darce JR, Arendt BK et al (2004) Expression of BCMA, TACI, and BAFF-R in multiple myeloma: a mechanism for growth and survival. Blood 103:689–694
Moreaux J, Cremer FW, Reme T et al (2005) The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood 106:1021–1030
Paterson JL, Li Z, Wen XY et al (2004) Preclinical studies of fibroblast growth factor receptor 3 as a therapeutic target in multiple myeloma. Br J Haematol 124:595–603
Katoh M, Nakagama H (2013) FGF receptors: cancer biology and therapeutics. Med Res Rev 1–21
Olsen JV, Blagoev B, Gnad F et al (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127:635–648
Schulze WX, Deng L, Mann M (2005) Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol Syst Biol 1(2005):0008
St-Germain JR, Taylor P, Tong J et al (2009) Multiple myeloma phosphotyrosine proteomic profile associated with FGFR3 expression, ligand activation, and drug inhibition. Proc Natl Acad Sci U S A 106:20127–20132
Kurhanewicz J, Dahiya R, Macdonald JM et al (1993) Citrate alterations in primary and metastatic human prostatic adenocarcinomas: 1H magnetic resonance spectroscopy and biochemical study. Magn Reson Med 29:149–157
Cornel EB, Smits GA, Oosterhof GO et al (1993) Characterization of human prostate cancer, benign prostatic hyperplasia and normal prostate by in vitro 1H and 31P magnetic resonance spectroscopy. J Urol 150:2019–2024
Sreekumar A, Poisson LM, Rajendiran TM et al (2009) Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 457:910–914
Thysell E, Surowiec I, Hornberg E et al (2010) Metabolomic characterization of human prostate cancer bone metastases reveals increased levels of cholesterol. PLoS One 5:e14175
Ducy P, Zhang R, Geoffroy V et al (1997) Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89:747–754
Komori T, Yagi H, Nomura S et al (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89:755–764
Otto F, Thornell AP, Crompton T et al (1997) Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89:765–771
Barnes GL, Javed A, Waller SM et al (2003) Osteoblast-related transcription factors Runx2 (Cbfa1/AML3) and MSX2 mediate the expression of bone sialoprotein in human metastatic breast cancer cells. Cancer Res 63:2631–2637
Pratap J, Javed A, Languino LR et al (2005) The Runx2 osteogenic transcription factor regulates matrix metalloproteinase 9 in bone metastatic cancer cells and controls cell invasion. Mol Cell Biol 25:8581–8591
Lin DL, Tarnowski CP, Zhang J et al (2001) Bone metastatic LNCaP-derivative C4-2B prostate cancer cell line mineralizes in vitro. Prostate 47:212–221
Altieri DC (2008) Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer 8:61–70
Lim M, Zhong C, Yang S et al (2010) Runx2 regulates survivin expression in prostate cancer cells. Lab Invest 90:222–233
Morrissey C, Brown LG, Pitts TE et al (2010) Bone morphogenetic protein 7 is expressed in prostate cancer metastases and its effects on prostate tumor cells depend on cell phenotype and the tumor microenvironment. Neoplasia 12:192–205
Yang S, Lim M, Pham LK et al (2006) Bone morphogenetic protein 7 protects prostate cancer cells from stress-induced apoptosis via both Smad and c-Jun NH2-terminal kinase pathways. Cancer Res 66:4285–4290
Akech J, Wixted JJ, Bedard K et al (2010) Runx2 association with progression of prostate cancer in patients: mechanisms mediating bone osteolysis and osteoblastic metastatic lesions. Oncogene 29:811–821
Baniwal SK, Khalid O, Gabet Y et al (2010) Runx2 transcriptome of prostate cancer cells: insights into invasiveness and bone metastasis. Mol Cancer 9:258
Pratap J, Lian JB, Stein GS (2011) Metastatic bone disease: role of transcription factors and future targets. Bone 48:30–36
Gupta A, Cao W, Chellaiah MA (2012) Integrin alphavbeta3 and CD44 pathways in metastatic prostate cancer cells support osteoclastogenesis via a Runx2/Smad 5/receptor activator of NF-kappaB ligand signaling axis. Mol Cancer 11:66
Cao JJ, Singleton PA, Majumdar S et al (2005) Hyaluronan increases RANKL expression in bone marrow stromal cells through CD44. J Bone Miner Res 20:30–40
Desai B, Rogers MJ, Chellaiah MA (2007) Mechanisms of osteopontin and CD44 as metastatic principles in prostate cancer cells. Mol Cancer 6:18
Baniwal SK, Khalid O, Sir D et al (2009) Repression of Runx2 by androgen receptor (AR) in osteoblasts and prostate cancer cells: AR binds Runx2 and abrogates its recruitment to DNA. Mol Endocrinol 23:1203–1214
Khalid O, Baniwal SK, Purcell DJ et al (2008) Modulation of Runx2 activity by estrogen receptor-alpha: implications for osteoporosis and breast cancer. Endocrinology 149:5984–5995
Dey P, Jonsson P, Hartman J et al (2012) Estrogen receptors beta1 and beta2 have opposing roles in regulating proliferation and bone metastasis genes in the prostate cancer cell line PC3. Mol Endocrinol 26(12):1991–2003
Hricak H, Choyke PL, Eberhardt SC et al (2007) Imaging prostate cancer: a multidisciplinary perspective. Radiology 243:28–53
Newling DW, Denis L, Vermeylen K (1993) Orchiectomy versus goserelin and flutamide in the treatment of newly diagnosed metastatic prostate cancer. Analysis of the criteria of evaluation used in the European Organization for Research and Treatment of Cancer–Genitourinary Group Study 3085. Cancer 72:3793–3798
Rehman I, Evans CA, Glen A et al (2012) iTRAQ identification of candidate serum biomarkers associated with metastatic progression of human prostate cancer. PLoS One 7:e30885
Le L, Chi K, Tyldesley S et al (2005) Identification of serum amyloid A as a biomarker to distinguish prostate cancer patients with bone lesions. Clin Chem 51:695–707
Morales M, Planet E, Arnal-Estape A et al (2011) Tumor-stroma interactions a trademark for metastasis. Breast 20(Suppl 3):S50–S55
Tarragona M, Pavlovic M, Arnal-Estape A et al (2012) Identification of NOG as a specific breast cancer bone metastasis-supporting gene. J Biol Chem 287:21346–21355
Massague J (2008) TGFbeta in cancer. Cell 134:215–230
Roberts AB, Anzano MA, Wakefield LM et al (1985) Type beta transforming growth factor: a bifunctional regulator of cellular growth. Proc Natl Acad Sci USA 82:119–123
Witz IP (2008) Yin-yang activities and vicious cycles in the tumor microenvironment. Cancer Res 68:9–13
Walsh DW, Godson C, Brazil DP et al (2010) Extracellular BMP-antagonist regulation in development and disease: tied up in knots. Trends Cell Biol 20:244–256
Logothetis CJ, Lin SH (2005) Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer 5:21–28
Wozney JM (2002) Overview of bone morphogenetic proteins. Spine 27:S2–S8, Phila Pa 1976
Canalis E, Economides AN, Gazzerro E (2003) Bone morphogenetic proteins, their antagonists, and the skeleton. Endocr Rev 24:218–235
Mcmahon JA, Takada S, Zimmerman LB et al (1998) Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube and somite. Genes Dev 12:1438–1452
Brunet LJ, Mcmahon JA, Mcmahon AP et al (1998) Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science 280:1455–1457
Aguirre-Ghiso JA (2007) Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 7:834–846
Klein A, Olendrowitz C, Schmutzler R et al (2009) Identification of brain- and bone-specific breast cancer metastasis genes. Cancer Lett 276:212–220
Weigelt B, Glas AM, Wessels LF et al (2003) Gene expression profiles of primary breast tumors maintained in distant metastases. Proc Natl Acad Sci USA 100:15901–15905
Bellahcene A, Bachelier R, Detry C et al (2007) Transcriptome analysis reveals an osteoblast-like phenotype for human osteotropic breast cancer cells. Breast Cancer Res Treat 101:135–148
Lu X, Mu E, Wei Y et al (2011) VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell 20:701–714
Kischel P, Guillonneau F, Dumont B et al (2008) Cell membrane proteomic analysis identifies proteins differentially expressed in osteotropic human breast cancer cells. Neoplasia 10:1014–1020
Dumont B, Castronovo V, Peulen O et al (2012) Differential proteomic analysis of a human breast tumor and its matched bone metastasis identifies cell membrane and extracellular proteins associated with bone metastasis. J Proteome Res 11:2247–2260
Navin N, Krasnitz A, Rodgers L et al (2010) Inferring tumor progression from genomic heterogeneity. Genome Res 20:68–80
Stephens PJ, Mcbride DJ, Lin ML et al (2009) Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 462:1005–1010
Martin SA, Mccabe N, Mullarkey M et al (2010) DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell 17:235–248
Lilja H (2008) Testing new PSA subforms to enhance the accuracy of predicting cancer risk and disease outcome in prostate cancer. Clin Chem 54:1248–1249
Henry NL, Hayes DF (2012) Cancer biomarkers. Mol Oncol 6:140–146
Olmos D, Brewer D, Clark J et al (2012) Prognostic value of blood mRNA expression signatures in castration-resistant prostate cancer: a prospective, two-stage study. Lancet Oncol 13:1114–1124
Ross RW, Galsky MD, Scher HI et al (2012) A whole-blood RNA transcript-based prognostic model in men with castration-resistant prostate cancer: a prospective study. Lancet Oncol 13:1105–1113
Rajski M, Vogel B, Baty F et al (2012) Global gene expression analysis of the interaction between cancer cells and osteoblasts to predict bone metastasis in breast cancer. PLoS One 7:e29743
Percy AJ, Chambers AG, Yang J et al (2012) Comparison of standard- and nano-flow liquid chromatography platforms for MRM-based quantitation of putative plasma biomarker proteins. Anal Bioanal Chem 404:1089–1101
Fuhler GM, Diks SH, Peppelenbosch MP et al (2011) Widespread deregulation of phosphorylation-based signaling pathways in multiple myeloma cells: opportunities for therapeutic intervention. Mol Med 17:790–798
Jin L, Zhang Y, Li H et al (2012) Differential secretome analysis reveals CST6 as a suppressor of breast cancer bone metastasis. Cell Res 22:1356–1373
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Wood, S.L., Brown, J.E. (2014). The Application of ‘Omics’ Techniques for Cancers That Metastasise to Bone: From Biological Mechanism to Biomarkers. In: Vassiliou, V., Chow, E., Kardamakis, D. (eds) Bone Metastases. Cancer Metastasis - Biology and Treatment, vol 21. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-7569-5_7
Download citation
DOI: https://doi.org/10.1007/978-94-007-7569-5_7
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-7568-8
Online ISBN: 978-94-007-7569-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)