
Abstract
The molecular and phenotypic irreversibility of mammalian cell differentiation was a fundamental principle of developmental biology at least until the 1980s, despite numerous reports dating back to the 1950s of the induction of pluripotency in amphibian cells by nuclear transfer (NT). Landmark reports in the 1980s and 1990s in sheep progressively challenged this dogmatic assumption; firstly, embryonic development of reconstructed embryos comprising whole (donor) blastomeres fused to enucleated oocytes, and famously, the cloning of Dolly from a terminally differentiated cell. Thus, the intrinsic ability of oocyte-derived factors to reverse the differentiated phenotype was confirmed. The concomitant elucidation of methods for human embryonic stem cell isolation and cultivation presented opportunities for therapeutic cell replacement strategies, particularly through NT of patient nuclei to enucleated oocytes for subsequent isolation of patient-specific (autologous), pluripotent cells from the resulting blastocysts. Associated logistical limitations of working with human oocytes, in addition to ethical and moral objections prompted exploration of alternative approaches to generate autologous stem cells for therapy, utilizing the full repertoire of factors characteristic of pluripotency, primarily through cell fusion and use of pluripotent cell extracts. Stunningly, in 2006, Japanese scientists described somatic cell reprogramming through delivery of four key factors (identified through a deductive approach from 24 candidate genes). Although less efficient than previous approaches, much of current stem cell research adopts this focused approach to cell reprogramming and (autologous) cell therapy. This chapter is a quasi-historical commentary of the various aforementioned approaches for the induction of pluripotency in lineage-committed cells, and introduces transcriptional and epigenetic changes occurring during reprogramming.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
2.1 Introduction
…reprogramming after transfer into the zygote is impossible in the mammalian embryo, either inher- ently or because of lack of time, whereas the amphibian nucleus probably has sufficient time to reprogram…
(McGrath and Solter 1984. Science) [8]
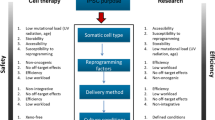
Despite the initial seminal work of Briggs and King [1, 2] in the 1950s, and subsequently by John Gurdon [3–7] and colleagues through the 1960s and 1970s, demonstrating the plasticity of a range of primitive and somatic cells in Xenopus and amphibia, the fundamental principle of mammalian developmental biology of molecular and phenotypic irreversibility of cellular differentiation persisted until the 1990s [8]. Three landmark studies in sheep challenged this dogmatic assumption, whereby ovine recipient (enucleated) oocytes supported embryonic development of donor nuclei from blastomeres, cultured cells and primary, adult mammary cells, respectively; the latter two studies resulting in both embryonic and fetal development, and live-born progeny [9–11]. With the concurrent development of methods to culture pluripotent stem cells isolated from donated, surplus IVF human blastocysts [12], new strategies for generation of patient-specific (autologous) stem cells for the treatment of human degenerative disorders were proposed and explored, namely (i) nuclear transfer of somatic cell nuclei to recipient oocytes for stem cell isolation from embryos, or (ii) direct reprogramming of somatic cell nuclei via cell fusion [13], application of cell extracts [14] or ‘induced pluripotency’ via direct delivery of key reprogramming factors (via numerous methods; Fig. 2.1) [15].

Diagrammatic representation of various methods to reprogram a somatic cell nucleus to pluripotency. ‘Wholistic Approach’ refers to exposure of somatic cells to a full repertoire of reprogramming factors (e.g. ooplasmic/cytoplasmic). Somatic cell nuclear transfer of somatic nuclei to oocytes generates cloned stem cells. Fusion of a somatic cell to a pluripotent stem cell via e.g. electrofusion reprograms the somatic cell to pluripotency. The “Minimalist/Defined Approach” refers to reprogramming of somatic cells with known or defined factors, in the absence of other factors. Genes such as Oct4 (O), Sox2 (S), Klf4 (K), cMyc (M), Nanog (N), Lin28 (L) have been shown to be key (although not exclusive) to this process [15]. Various chemical agents and microRNA constructs have been used as additions or substitutions to key reprogramming factors
In this chapter, we discuss the development of approaches to induce pluripotency in nuclei of lineage-committed somatic cells, and introduce the molecular changes that accompany this process, referred to as ‘[somatic] cell reprogramming’; namely, transcriptional changes (JAK-STAT & Wnt/Notch pathways), and epigenetic modifications (chromatin/histone modifications; expanded commentaries of each of these discussions can be found in Chaps. 10, 14 and 17 in this volume).
2.2 Early Nuclear Transfer Experiments in Amphibia (1950–1980): Questioning the Plasticity of the Committed
Much of the early nuclear transfer experiments were undertaken in amphibia. As early as 1952 [1], embryos comprising transferred donor nuclei from undifferentiated frog blastula to enucleated (frog; Rana pipiens) oocytes underwent normal embryonic development, prompting consideration of the potential of more committed donor nuclei to contribute to early embryonic development. Briggs and King (1953) [2] noted cleavage rates of constructed embryos, incorporating cells of the frog gastrula (cells undertaking commitment to one of three germ layers), were reduced in comparison to those constructed with frog blastula. Applying these results to more committed donor nuclei, blastulae and later stage (gut) donor cells supported tadpole development and even the generation of sexually mature frogs after 9–12 months transplantation [4], proving the ability to undertake complete reprogramming is not limited to primitive nuclei with lineage plasticity. Furthermore, the capacity of lineage-committed, Xenopus nuclei to regain pluripotency, and contribute to cleavage-stage embryonic development, is regained in the presence of ooplasmic factors of alternative Xenopus species (albeit, of the same Genus) [3]. Despite the Xenopus species X. tropicalis and X. laevis being incapable of generating a hybrid through natural reproductive means (or artificial fertilization), X. tropicalis ooplasm (enucleated) was able to successfully reprogram vegetal hemisphere/endoderm donor nuclei from X. laevis. During reprogramming, recipient ooplasm elicits structural and functional changes to transferred somatic cell nuclei. Following transfer to Xenopus oocytes, mid-blastula nuclei (mitotically active cells, active in DNA synthesis but with little RNA synthesis) resume RNA synthesis, and from 30 min post-injection, cease DNA synthesis and cell division [5]. Ooplasmic factors of recipient (maturing) eggs also rapidly reverse the hypo-proliferative nature of brain nuclei (0.5–3 h post transfer; [5]). Interestingly, cleavage rates of transfers using cultured cells from numerous organs of tadpole (stage 40) and adult Xenopus (kidney, heart, lung, testis, and skin) were similar, even adult donors compared to tadpole stage cells, highlighting the plasticity of nuclei from numerous anatomical sources [6]. Histology proved differentiation of donor nuclei to all cells of the developing tadpoles. Extending these results, transfer of in vitro cultured somatic Xenopus nuclei to Pleurodeles waltlii newt oocytes demonstrated reactivation of previously inactive genes and repression of somatic gene profile [7].
2.3 Generation of (Mouse and Human) Embryonic Stem Cell lines: Realizing the Therapeutic Potential
Blastocyst-derived, embryonic stem cells (ESC) are the ‘natural’ counterpart of induced pluripotent cell populations, and since they originally derive from totipotent blastomeres and not lineage-committed cell sources, they do not strictly satisfying the criteria of ‘induced pluripotency’. However, it is fitting a brief historical perspective of ESC isolation and maintenance be presented here, as it was this seminal work by Evans, Kaufman, Martin and Thomson [16–20] in the 1980s and 1990s that highlighted in vitro conditions required to maintain ESC, knowledge that was imperative for subsequent development of induced pluripotency technologies. Furthermore, the molecular, epigenetic and functional characteristics of ESC remain the ‘gold standard’ to which induced pluripotent cells are compared.
Careful consideration of ideal stage of isolation (epiblast of early post-implantation) and in vitro culture conditions led to establishment of the first mouse ESC lines [16, 17]. Evans and Kaufman (1981) [16] plated hatched murine blastocysts (129 mouse strain) in culture dishes, from which inner cell mass (ICM) was isolated and re-plated on inactivated feeders. Expanded lines maintained normal karyotype and displayed differentiation potential in vitro and in vivo. Soon after, additional mouse ESC lines were isolated and cultured in media pre-conditioned on embryonal carcinoma cell culture [17]. Since then, numerous mouse ESC lines have been established around the world, a feat that has fostered development of technologies for generation of transgenic experimental animals.
The obvious progression from derivation of mouse ESC lines was translation to other species. Bongso et al. (1994) [18] were the first to describe isolation of ICM from donated, human (IVF) blastocysts. Although grown on epithelial feeder layers and displaying stem cell morphology and expressing alkaline phosphatase (AP), this Singaporean group failed to maintain a human ES cell (hESC) beyond 2 passages. Interestingly, the establishment of stable hESC lines expressing stem cell markers, possessing differentiation potential and capable of long term growth was achieved some 4 years later [12], and only after pluripotent lines were established in two species of non-human primate [19, 20]. Subsequently, methods for human ICM isolation have been optimized, including a multi-step procedure using day 8 blastocysts (rather than the usual day 5–7) [21]. Statistically significant improvements in yield of human pluripotent cells were reported using this multi-step approach. hESC lines have also been derived from morula (pre-blastocyst) stage embryos, with approximately 17 % of original explants forming stable pluripotent lines; an efficiency similar to blastocyst derived lines [22]. Furthermore, establishment of stable hESC lines from microsurgical removal of single blastomeres circumvents the inherent embryo destruction when isolated from ICM cultures [23].
The ethical implications of hESC isolation (i.e. with implied embryo destruction), acquired knowledge of favorable in vitro ESC culture conditions, and continuing need for autologous stem cells for therapy, collectively drove exploration of alternative sources of stem cells for therapy. Despite SCNT-derived, pluripotent cells being reported in non-human primate (rhesus macaque) [24], derivation of stable hESC lines by SCNT still elude us. Alternative, more directed approaches to human somatic cell reprogramming, that circumvent embryo destruction associated with SCNT and ESC isolation, have evolved with varying success and efficiency. We outline mammalian SCNT, and alternative approaches, in the coming sections.
2.4 (Mammalian) Somatic Cell Reprogramming, and the Autologous Therapeutic Cell: Reacquiring Plasticity
2.4.1 Somatic Cell Nuclear Transfer (SCNT)
As outlined in Sect. 2.2, the process of somatic cell nuclear transfer (SCNT) typically involves the removal of maternal chromosomes from an oocyte (‘enucleation’) followed by insertion of the donor cell nucleus, or the fusion of intact somatic cell or nucleus, to the enucleated oocyte. Embryonic development is artificially triggered, by inducing an increase in intracellular calcium, and can continue to pre-implantation stages in vitro. If transferred to a recipient animal, embryonic implantation and continued fetal development can give rise to a live-born, cloned animal. This process is referred to as reproductive cloning. Alternatively, cells of the inner cell mass of transferred blastocysts (in vitro) can be isolated and cultured, giving rise to nuclear transfer embryonic stem cells (ntESC); this process is referred to as therapeutic cloning. The resulting ntESC can be used as a tool for biomedical research or as a source of cells for transplantation back to the somatic cell donor. Since the ntESC are genetically identical to the donor at the genomic DNA level, transplanted cells are unlikely to be rejected by the host immune system. It is important to note that the mitochondrial DNA in clones and ntESC is predominantly inherited from the oocyte.
Despite documented success in deriving nuclear transfer offspring in amphibia, it was widely accepted that the molecular events characterizing mammalian cell differentiation prevented it from re-attaining totipotency and contributing to cloned offspring [8]. This dogmatic assumption was challenged by Willadsen (1996) [9] and Wilmut et al. (1997) [11], who generated viable embryos through nuclear transfer of 8- or 16-cell blastomeres, or live-born sheep from mammary epithelial cells, to enucleated ovine oocytes, respectively. Later, numerous cell types were shown to contribute to embryonic and fetal development, and live offspring [25]. SCNT/cloning has now been performed in a number of mammals, including non-human primates [24]. However, the biggest impact being in its translation to agricultural species. Conceptually, reproductive cloning could create multiple clones of animals with highly valued or desirable traits, including cows with high milk production or bulls that breed offspring with high quality meat [26]. Importantly, the biological properties and nutritional value of milk and meat obtained from cloned individuals does not differ from non-cloned animals [27, 28], and thus is considered safe for human consumption [29, 30].
The application of therapeutic cloning to the treatment of human, degenerative disease is even more promising. Generation of (cloned) pluripotent stem cells from somatic cells of a diseased individual, in combination with corrective gene therapy in vitro, has the potential to treat diseases through autologous cellular transplantation, not withstanding logistical and fiscal considerations. The first proof of principle study on the therapeutic application of SCNT derived cell lines was reported in 2002, whereby ntESC derived from Rag2 −/− mice were differentiated into hemato-poietic stem cells (HSC) in vitro for transplantation after correction of the characteristic Rag2 recombinase gene mutation by targeted homologous recombination [31].
However, there are a number of technical hurdles that need to be addressed before SCNT can be a viable source of pluripotent cells for human cell therapy. SCNT is a very resource intensive procedure requiring a large number of oocytes [32], and there are only a handful of reports of human SCNT embryos reaching the blastocyst stage [33–35]. To date, diploid ntESC lines have not been established. Recently, ntESC were isolated following SCNT to an intact oocyte (i.e. non-enucleated), resulting in a triploid embryo [36]. Interestingly, the triploid ESC isolated from the blastocysts maintained various characteristics of pluripotent cells and could differentiate into cells of all three germ layers.
Most importantly, the process of SCNT demonstrated that adult cells which are programmed to express a subset of specific genes as a differentiated cell, can be reprogrammed or de-differentiated to give rise to an organism karyotypically identical to the donor cell. Due to the ethical issues surrounding SCNT and technical difficulties in translating this to humans, alternate methods of somatic cell reprogramming have been explored including cell fusion, treating cells with cell extracts and viral induction of pluripotency by defined factors.
2.4.2 Cell Fusion
Fusion of interfacing plasma membranes of two cells can be achieved through chemically through treatment with polyethylene glycol (PEG), electrofused or through viral induction [37]. As enucleation does not precede fusion (as for SCNT), the resulting cell hybrids are tetraploid (since nuclear membranes commonly undergo intracytoplasmic fusion) and in most cases the phenotype of the less-differentiated fusion partner dominates the phenotype of the more-differentiated partner. Reprogramming of somatic cells by cell fusion involves the hybridization of a pluripotent cell to a somatic cell, resulting in a tetraploid cell hybrid.
In 1976, Miller and Ruddle first demonstrated that he phenotype of the pluripotent cell dominates following somatic cell fusion [13]. Mouse embryonic carcinoma (EC):thymocytes cell hybrids exhibited characteristics of the pluripotent EC cell and could form teratomas containing derivatives of the three germ layers. Other hybrids have adopted a number of pluripotency properties of embryonic germ cells and embryonic stem cells, and shown to modify gene expression profiles of the somatic cell hybrid partner [38–42]. ES-somatic cell hybrids display most properties of pluripotent stem cells; however, since they are tetraploid and have genomic DNA from both parental cells, their use for autologous cell replacement therapy is limited.
Conversion of a tetraploid (pluripotent) hybrid to a reprogrammed diploid cell, carrying the donor (autologous) somatic cell genome, conceptually generates a therapeutically relevant cell. One approach of excluding the ESC-derived DNA genome following reprogramming is maintaining a ‘heterokaryon’ (preventing fusion of the nuclear membranes) for the duration of reprogramming, followed by subsequent enucleation of the ES nucleus [43, 44]. Although a viable cell remains after extrusion of the ESC-derived nucleus, this approach resulted in only partial reprogramming of the somatic nucleus [44]. An alternative approach involves the removal of the specific ESC derived chromosomes responsible for self recognition in the ES-somatic cell hybrid resulting in a hybrid aneuploid cell that is immune matched to the somatic cell [45]. Despite these novel approaches, more advances in the cell fusion field are required before it can be considered a viable alternate method for generating autologous pluripotent stem cells for clinical applications.
2.4.3 Cell Extracts
In contrast to fusing intact pluripotent cells to somatic cells, methods of exposing differentiated cells or nuclei to cell extracts from totipotent and pluripotent cells have been devised as a means of somatic cell reprogramming. This technique involves the reversible membrane permeabilisation of differentiated cells, using the chemical streptolysin-O (SLO; a member of the family of cholesterol-dependent cytolysins), followed by exposure to ‘reprogramming’ cell extracts. The lesions induced in the plasma membrane by SLO are resealed upon application of Ca2+ [46]. Earlier reprogramming studies using cell extracts showed that the incubation of somatic cells in Xenopus egg extracts resulted in remodeling of chromatin and changes to gene expression [14, 47, 48]. The first demonstration of reprogramming of differentiated cells by exposure to mammalian cell extracts was performed with HEK293T (human embryonic kidney) cells exposed to stimulated T-cell extracts resulting in direct reprogramming toward a lymphoid-specific phenotype [49]. Furthermore, it was demonstrated that cell extract based reprogramming involves transcriptional changes in addition to ATP-dependent chromatin remodeling [49, 50].
The first reprogramming studies using pluripotent stem cell extracts showed that both HEK293T cells and immortalized NIH/3T3 mouse fibroblast cells acquired characteristics of pluripotent stem cells when exposed to extracts made from a pluripotent human carcinoma cell line and cultured [51]. The treated cells were partially reprogrammed and formed colonies, which were alkaline phosphatase (AP) positive and expressed pluripotency markers. They had also deactivated differentiation markers and had undergone epigenetic changes at the promoters of a number of pluripotent gene loci [51, 52]. More recently, human fetal fibroblasts have been shown to form hESC-like colonies when treated with a combination of chromatin inhibitors and hESC extracts [53]. Partial reprogramming was reported when the somatic cells were pre-treated with the epigenetic modifiers 5-aza-2′-deoxycytosine and Trichostatin A prior to exposure to hESC extracts. Treated cells were shown to upregulate a number of pluripotency genes and change morphology and growth characteristics. Although reprogramming to a complete pluripotent state was not achieved, the cells could be trans-differentiated into neurons under differentiation conditions [53].
To date cell extract exposure has been shown to partially reprogram the treated cells toward an embryonic state, predominantly in transformed and immortalized cell lines. However, it is noteworthy that survival and persistence of pluripotent stem cells in fusion preparations, having donated cell extract, constitute a potential source of contamination in subsequent analysis [54].
2.5 Induction of Pluripotency (Post-2006) Identification of the Critical Reprogramming Factors for Direct Induction of Pluripotency
A major turning point in international stem cell research came in 2006 with the generation of ‘induced pluripotent stem cells’ (iPSC), the significance of this discovery was recognized by the joint-award of the Nobel prize for Physiology or Medicine in 2012 to Shinya Yamanaka to its discoverer. Ectopic expression of just four transcription factors resets the transcriptional profile and epigenetic state of the host cell to one resembling an ESC [15]. The most widely used set of reprogramming factors, Oct4, Sox2, Klf4 and c-Myc, was identified initially by screening 24 pre-selected factors by Takahashi and Yamanaka [15]. Since this discovery, research papers characterizing properties of iPSC have flood scientific literature: murine (and subsequently human) iPSC can be generated without the oncogenic factor cMyc [55, 56], are germ line competent [57], and contribute all cell types to fertile offspring via the tetraploid complementation assay [58–60]. Human iPSC can be generated using the same set of factors [61, 62] or an alternative set of 4 factors, namely Oct4, Sox2, Nanog and Lin28 [63], suggesting that Oct4 and Sox2 are essential whereas Nanog, Klf4, Lin28 and c-Myc are alternative reprogramming supporting factors. iPSC can be generated without permanent integration of transgenes [64–67] and be generated from diseased patient cells [68–71]. Like ESC, they are capable of differentiating into multiple cell types of the germ layers, including heart, blood, islet, nerve, liver, and muscle [72–79]. Therefore, the potential benefits of iPSC for regenerative medicine are immense, for example, skin biopsies (notably, terminally differentiated readily accessible cells) could be taken from patients of degenerative disease or injury for conversion to pluripotent iPSC before their directed differentiation to the cell type of interest. Transplantation of the differentiated progeny to the afflicted organ or tissue could manage or cure disease/condition, and since the original donor cell came from the patient, the risk of immune rejection of the grafted cells is minimal.
Despite the therapeutic potential, technical and logistic issues surround iPSC technology. Efficiency of cell reprogramming remains low, attributable in part by incomplete transcriptional and epigenetic reprogramming, and actual/risk of mutagenesis during conversion makes the differentiated cells possibly oncogenic. If iPSC are to achieve therapeutic relevance, methods that ensure complete differentiation of all cells within a pool of iPSC are required to negate the possible tumourgenicity of rare pluripotent cells once transplanted. Also the time taken to convert somatic cells to clinically relevant and regulatory body approved therapeutic cells may prevent autologous use of the cells per se, rather cells matched to potential populations may require to be banked. Therefore, research is currently underway that address these, and other iPSC-related problems; some of which are discussed here.
2.5.1 Improvement of Efficiency, Quality and Purity of iPSCs Production
In Yamanaka’s landmark study, the efficiency of reprogramming mouse embryonic fibroblasts (MEF) into iPSC was reportedly 0.01–0.1 % [15]. Despite prolonged expression of the Yamanaka’s four factors, only a small percentage of cells achieve full reprogramming. In contrast, reprogramming by somatic cell nuclear transfer and cell fusion is quicker and more efficient [44, 80], indicating that additional key reprogramming factors may yet to be identified. Previous studies showed that in differentiated mouse ESC, COUP-TFs silences the Oct4 locus by binding to RAREoct, a composite RA responsive element in the Oct4 promoter [81, 82]. RA receptors (RARs) and members of Nr5a steroid hormone receptor family (Lrh-1) form heterodimers, which compete with COUP-TFs for RAREoct and maintain Oct4 expression [83]. By adding Rarg (RAR-γ) and Lrh-1 to the Yamanaka reprogramming cocktail, Liu and co-workers report 100-fold improvements in reprogramming efficiency [84]. Key indicators of iPSC reprogramming, namely, activation of Oct4 and Rex1 genes, were observed from as few as 3–4 days of (six) factor induction, a temporal indicator comparable to somatic cell nuclear transfer [85] or cell fusion [86].
One central question related to the molecular mechanisms of iPSC formation is how transcriptionally restrictive chromatin at loci of inactive pluripotent genes (e.g. Oct4, Sox2, and Nanog) in somatic cell is relaxed upon exogenous factor induction to permit resumption of expression. Chemical agents that relieve the restrictive conformation of heterochromatin, such as inhibitors of histone deacetylation and DNA methylation, increase the efficiency of iPSC generation [87, 88], indicating that modifications to chromatin structure in somatic cells is key to full reprogramming. Knockdown of p53 in B cells shortens cell cycle length results in relief of repressive heterochromatin conformation during DNA synthesis, greater access of reprogramming factors to previously inaccessible genomic loci and therefore reduces the time required to form iPSC twofold [89]. MyoD, a transcription factor for skeletal myogenesis, can recruit various transcription factors and chromatin remodeling proteins to its target genes more efficiently than Oct4, leading to activation of suppressed genes embedded in repressive chromatin [90]. Hirai and co-workers discovered that fusing a fragment transactivation domain (TAD) of MyoD to Oct4 (M3O) improves the iPSC reprogramming process [91]. Transduction of TAD-Oct4 with Sox2, Klf4, and cMyc to fibroblasts effectively remodeled patterns of DNA methylation, chromatin accessibility, histone modifications, and protein binding at pluripotency genes, raising the efficiency of mouse/human iPSC generation more than 50-fold in comparison to the Yamanaka four factors (OSKM). The resultant human iPSC colonies appeared in around 5 days, in contrast to two weeks with OSKM, and the purity of the iPSC was much higher with the M3O-SKM gene introduction (98 % of the colonies) compared with OSKM (5 %).
2.5.2 Epigenetic Characteristics of iPSC
Although iPSC exhibit many of the morphological and molecular characteristics of ESC, a number of recent reports have questioned the extent to which reprogrammed iPSC adopt an epigenetic signature characteristic of their ESC counterparts. iPSC appear to retain an ‘epigenetic memory’ of the donor tissue from which they were derived and exhibit somatic genome-wide messenger RNA and microRNA expression patterns, thus questioning their differentiation potential. Although sharing pluripotency status assessed by various criteria, iPSC derived from fetal fibroblasts, neonatal fibroblasts, adipose stem cells, and keratinocytes differ in expression profiles of core sets of donor genes [92]. Expression profiles in fetal fibroblasts-derived iPSC bore closer resemblance to human ESC followed by adipose, neonatal fibroblasts, and keratinocyte-derived iPSC.
Overall, iPSC and ESC share a well-defined core pluripotency network, although select core genes are often hypo-expressed in iPSC. George Daley’s lab found iPSC harbor residual DNA methylation signatures characteristic of their somatic tissue of origin, which could be reset by differentiation and serial reprogramming, or by treatment of iPSC with chromatin-modifying drugs (TSA and AZA) [93]. The DNA methylation of nuclear transfer-derived pluripotent stem cells resembles classical ESC than iPSC. However, other studies report resetting of epigenetic memory and cell function upon continuous passaging, suggesting that complete reprogramming is a gradual process that continues beyond the acquisition of a bona fide iPSC state assessed by activation of endogenous pluripotency genes, viral transgenes-independent growth and the ability to differentiate into cell types of all three germ layers [94]. Guenther et al. compared both global chromatin structure and gene expression profiles of a panel of human iPSC and ESC [95]. Genome-wide maps of nucleosomes with histone H3Kme3 and H3K27me3 modifications indicate that there is little difference between ESC and iPSC with respect to these marks. Gene expression profiles confirm that the transcriptional programs of ESC and iPSC show very few consistent differences. Importantly, the observed differences in these cell lines did not discriminate iPSC from ESC [95].
2.5.3 Generation of iPSC for Clinical Applications
Numerous modifications to the original, retroviral-based Oct4, Sox2, Klf4 and cMyc method have been reported since its original description with the aim to improve reprogramming efficiency or create iPSC for clinical application [15, 87, 88, 96, 97]. Retroviral or lentiviral vector-mediated transduction of reprogramming genes involves random integration of exogenous DNA into the genome of the recipient cells, representing a preventative obstacle to therapeutic use of the cells and their derivatives. iPSC can be obtained with removable PiggyBac transposons or episomal systems [65, 98–100], but these approaches are either (i) at least temporarily mutagenic, or (ii) still require delivery of exogenous DNA construct to the nuclear compartment of target cells, thus increasing risk of genomic recombination or insertional mutagenesis. Sendai virus, an RNA genome, has been used to deliver transgenes, but undesirable in the therapeutic arena given the required purging of reprogrammed cells of replicating virus [101, 102]. iPSC have been generated with recombinant proteins of Oct4, Sox2, Klf4 and cMyc incorporating cell-penetrating peptide moieties [103, 104] and synthetic modified mRNA of the four factors [105]. A recent study showed reprogramming of mouse and human cells to pluripotency by direct transfection of a combination of mir-200c plus mir-302 and mir-369 family microRNAs (miRNAs), albeit at considerably lower efficiency [106].
iPSC appear to share much of the differentiation potential characteristic of ESC. Recently, iPSC (from murine and human cell sources) have been differentiated into functional SM/C-2.6+ skeletal cells [107], IFN-γ expressing T-cells [108], human hematopoietic precursors [109], coagulation factor-expressing, liver-like cells [110], and hepatocyte- and cardiomyocyte-like cells [111], to name a few. The considerable health and economic burden of neurological disease has prompted development of protocols for directed differentiation to neural cell (progenitor and mature) and related lineages (e.g. glial cells; [112–119]). These cells enable immediate study of neurological disease development in vitro, and represent a potential transplantable pool of cells for the treatment of human neurological disease in vivo. Interestingly, Kim et al. (2011) [115] report transient induction of Oct4/Sox2/Klf4/cMyc, in combination with FGF2, FGF4 and EGFP-containing media, (trans) differentiate mouse fibroblasts to expandable neural progenitor cells and glia. Mature cells expressed numerous neuronal markers, could be induced to generate action potentials and formed functional synapses. Secreted protein from chick dorsal root ganglia in culture also directs differentiation of murine iPSC to motor and sensory neurones [116]. iPSC technology enabled the identification/study of novel/known molecular pathways mis-regulated in diseased neural cells, through neural differentiation from human fibroblast donor cells, as well as in vitro screening of proposed corrective measures for efficacy (genetic Parkinson’s disease and schizophrenia fibroblast donors; [113, 117]).
Whilst the propensity to differentiate to oligodendrocyte precursor cells (marker A2B5+) from a pluripotent phenotype is comparable between mouse ESC and iPSC, rates of differentiation directly to mature oligodendrocytes (marker O4+) differ markedly (24 % ESC v 2.4 % iPSC; [112]). It is noteworthy that the differentiation protocols employed in this study were optimized to differentiation of ESC; subtle differences between iPSC/ESC phenotypes may account for this discrepancy and specific, iPSC-related protocols may need to be devised for improvements in differentiated cell yield to be realized. In a three-step protocol, (i) induction of mouse iPSC for (ii) differentiation to an intermediate oligodendrocyte precursor cell yielded (iii) functional (i.e. myelinating) oligodendrocytes in vitro when co-cultured with dorsal root ganglial neurones [114]. It will be interesting if these results can be recapitulated in a human system.
Despite these impressive results, concerns surround pluripotent iPSC retaining a ‘memory’ of their original, differentiated donor cell state, and thus a propensity to spontaneously differentiate to their original phenotype [120, 121]. Partial DNA methylation in low passage iPSC, permits re-activation of the original somatic cell-related transcriptional profile [121]. This has obvious implications for cell transplantation in vivo.
Also, the non-immunogenicity of pluripotent ESC transplanted to allogenic recipients (leading to teratoma formation) raised hope that autologous iPSC transplantation would have applications in clinical medicine. However, Zhao et al. (2011) [122] recently questioned the clinical applicability of iPSC. Although ESC derived from B6 embryos circumvent the immune response in B6 recipients, leading to teratoma formation, transplantation of reprogrammed iPSC to syngeneic hosts initiates T-cell-mediated infiltration and necrosis (Zhao et al. 2011) [122]. The behavioral differences between transplanted ESC and iPSC to syngeneic hosts are perhaps attributable to gene expression profile differences. These concerning and unresolved issues highlight the need for caution when considering transferring current cell induction and differentiation protocols from bench to bedside.
In addition to being a cellular source for transplantation therapy, human iPSC also have great potential for disease modeling and drug development. Human iPSC can be generated from a variety of diseased individuals, display similar differentiation capacity to control iPSC derived from normal individuals [68, 70], and (diseased) iPSC-derived cells (e.g., motor neurons, cardiomyocytes) recapitulate disease-specific effects in vitro [69, 123–125]. iPSC technology enabled modeling of diseases such as dyskeratosis congenital and Friedreich’s ataxia, leading to identification and exploration of novel therapeutic strategies [126, 127]. The ability to derive iPSC and create disease models lacking high quality or appropriate animal models will facilitate disease biology studies and drug discovery in the future, notably, for diseases that are complex or polygenic and are not easily recapitulated by gene modifications in mice, and personalized medicine.
2.6 Transcriptional and Epigenetic Changes During Reprogramming
Wholesale changes to the transcriptional and epigenetic architecture of somatic cells must precede, and are a feature of, full adoption of the pluripotent phenotype. Molecular signatures of ESC provide a yardstick to which induced pluripotency is assessed in somatic cell reprogramming. In this section, we discuss the known transcriptional and epigenetic profile characteristics of ESC, and highlight their counterparts in the process, and full adoption, of (induced) pluripotency in somatic cells.
2.6.1 Changes to Transcriptional Profile During Reprogramming
Here, we provide a brief preview of transcriptional changes relating to two well-characterized gene families during reprogramming; (i) the Wnt/NOTCH Pathway and (ii) the JAK-STAT Pathway. We hope this provides an introduction to more comprehensive appraisals of these important gene families in Chaps. 10 and 14 of this volume, respectively.
2.6.1.1 Wnt/NOTCH Pathway
2.6.1.1.1 And Wnt Signaling Pathway
The membrane-bound Frizzled and LRP5/6 (low-density-lipoprotein (LDL)-receptor like protein 5 or 6) heterodimer forms the dual receptor complex for the canonical Wnt signaling pathway after binding with Wnt proteins [128–131]. Upon binding of Wnt ligand to the trans-membrane receptor, dual pathways are activated; (i) the cytosolic scaffolding phosphoprotein Disheveled is phosphorylated via CK1 activated by Wnt signaling [132] (ii) the function of serine threonine kinases glycogen synthase-3(GSK3-β) is deactivated through binding of Axin GSK3-β complex to LRP5/6 receptor co-factor. Both mechanisms lead to activation of the β-catenin; (i) phosphorylation of Disheveled prevents phosphorylation of β-catenin by the recruitment and binding of Axin GSK3-β complex to phosphorylated LRP5/6 via the PPPSP motif [133, 134] and (ii) deactivation of GSK3-β suppresses its ability to phosphorylate β-catenin, phosphorylated β-catenin is targeted for degradation by the 26S proteasome. Accumulation of cytosolic β-catenin results in its translocation to the nuclear compartment and binding of lymphoid enhancer factor (LEF)/T cell factor3 (TCF3) transcription factors to regulate expression of target genes [135]. The subsequent recruitment of specific co-factors to the β-catenin/Tcf3 heterodimer, from a range of possible co-binders, dictates activating or repressive functions on gene expression.
The Wnt signaling pathway contributes to the maintenance of pluripotency in mouse and human ESC [136–141] as well as the self-renewal of undifferentiated adult stem cells in multiple tissues [138]. Application of Wnt1 or Wnt3a to culture media, or over-expression in feeder cells, enhances proliferation of hESC and maintains pluripotency [140]. Inhibition of GSK-3 maintains the undifferentiated phenotype and sustains expression of pluripotent state-specific transcription factors, Oct-3/4, Rex-1 and Nanog [135, 140]. Indeed, chemical inhibition of GSK-3 by CHIR99021 permits generation and maintenance of rat ESC [141]. In ESC, the Tcf3 (± β-catenin) and Oct4/Sox2/Nanog, combine to maintain protein levels within desirable thresholds and thus regulate the balance between pluripotency and differentiation [142, 143]. To illustrate, Tcf3 co-precipitates with Oct4 and/or Nanog in genome-wide ChIP-ChIP experiments [142], and occupies the Myc promoter. Tcf3 is a key player in the regulation of Nanog expression, maintaining mRNA and proteins levels, and regulating promoter activity through binding to regulatory elements [144]. Furthermore, Nanog levels are elevated in Tcf3 null ESC. Wnts also act synergistically with LIF through the JAK/STAT pathway, the former increases STAT3 mRNA levels while the latter phosphorylates it [145].
The effect of Wnt signaling in pluripotent ESC can be applied to a cell reprogramming setting. Wnt3a enhances somatic cell reprogramming by cell fusion [145]. Wnt3a-containing conditioned medium can substitute for exogenous cMyc in Oct4-Sox2-Klf4 mediated reprogramming, and improve reprogramming efficiency by as much as 20-fold [146]. Reprogramming of neural progenitor cells (NPCs) through exogenous expression of Oct4 and Klf4 alone is facilitated by the knock down of Tcf3, suggesting that Tcf3 represses β catenin activated genes, which are relevant in the efficient formation of iPSC. Similar successes can be recapitulated in cell fusion of NPCs with Tcf3 null ESC, resulting in large epigenetic changes in the genome permitting endogenous Oct4 to bind previously inaccessible target promoters [147].
2.6.1.1.2 Notch Signaling Pathway
The Notch pathway is involved in differentiation processes and lineage fate in fetal and postnatal development, as well as in adult self-renewing organs [148]. Five mouse/human Jagged and Delta proteins (Jagged1, Jagged2, Delta-like (Dll)-1, -3 & -4) represent ligands for Notch receptors (Notch-1 to -4 in mouse/human) [148, 149]. The ligand–receptor interaction at the cell surface (of neighboring cells) leads to the proteolytic cleavage of the Notch receptor, leaving a membrane-bound cleavage product, referred to as Notch Extracellular Truncation (NEXT). The intracellular portion of NEXT is further cleaved by cytosolic γ secretase to the Notch intracellular domain (NICD), which translocates to the nucleus [150, 151]. Notch fragment NICD binds CBF/CSL in the nucleus and along with MAM(mastermind)/Lag3(mammals) forms a transcriptionally active ternary complex which recruits general transcription factors CBP/p300 and PCAF, promoting chromatin acetylation and increased expression of Notch target genes [152, 153], such as Hes/Hey [154], bHLH like Id family of proteins, Sox9, Pax6, Lineage specific transcription factors (LSTFs) [155]. Here we focus on pluripotent stem cells and Notch signaling in that context.
Notch signaling activates Hes/Hey transcription, which leads to repression of Hes/Hey target genes such as tissue specific transcriptional activators, thereby preventing differentiation [154]. Furthermore, CHiP-Seq mapping of 13 transcription factors has revealed that core pluripotency transcription factors associated with Oct4, Sox2 and Nanog as well as signaling effectors Smad1 and STAT3 co-localize with enhancer- associated transcriptional co-activator p300 at non-promoter region [156].
In the nervous system, Notch signaling influences balance between progenitors and its differentiating progeny corroborated by evidence in which forced expression of Notch-1C promotes neurogenesis [157–159]. Furthermore Jag1 exposure to hematopoietic stem cells increases the proportion of stem cells as opposed to differentiating cells [160]. Disruption of Notch1 signaling in ESC results in commitment to mesodermal lineage through upregulation of mesodermal and cardiac markers [161]. Thus, Notch regulates lineage commitment, particularly relevant to mesodermal and neuro-ectodermal fates [161].
This may suggests that Notch does not appear to be a general inhibitor of ESC differentiation but functions as a regulator of cell fate decisions in multipotent ESC that must choose between mesodermal and neuro-ectodermal fates [161]. Although maintenance of pluripotency is not affected by Notch signaling, whether or not it affects induction of pluripotency from somatic cells is yet to be established.
2.6.1.2 JAK-STAT Pathway
Binding of the Leukemia Inhibitory Factor (LIF) to its receptor triggers three intracellular cascades: the JAK/STAT3 (Janus kinase/signal transducer and activator of transcription 3), the PI3K (phosphoinositide 3-kinase)/AKT and the SHP2 [SH2 (Src homology 2) domain-containing tyrosine phosphatase 2]/MAPK (mitogen-activated protein kinase) pathways [162, 164]. The JAK/STAT pathway is of particular interest due to its pleiotropic nature and its regulation of proliferation, differentiation, cell migration, apoptosis cell renewal and pluripotency [165]. Herein we focus on the mains events during the transduction of LIF signal trough JAK/STAT3 cascade on mouse ES and iPSC in vitro (Fig. 2.2); more comprehensive reviews can be found elsewhere [163].

Simplified schematic LIF-dependent JAK1/STAT3 signaling in pluripotent stem cells. I The LIF receptor comprises two glycoproteins (gp130 and gp190/LIFRβ). JAK (intracellular transductor) and cytosolic STAT3 (second messenger). II JAK1 is phosphorylated after LIF ligand binds the receptor. III gp130 and STAT3 are sequentially phosphorylated IV After dimerization of STAT3 the homodimer is released V STAT3 dimer translocates to the nucleus trough the nuclear pore complex NPC; translocation is mediated by subunits importin-α3 or importin-α6 and subunit importin β. VI STAT3 mediates the expression of several target genes related to self renewal and stemness and inhibition of mesoderm and endoderm differentiation. VII SOCS3 is also upregulated under STAT3 signaling. SOCS3 binds the phosphorylated JAK and mediates its degradation. The latter provides a mechanism of negative feedback for attenuation and regulation of STAT3 signaling
Several activators of the JAK/STAT pathway have been identified to date including, the growth hormone, erythropoietin, interferons and interleukin family members. Following development of methods for murine ESC isolation and culture, persistent activation of the JAK/STAT pathway was found to be essential for the maintenance of pluripotency, with LIF driven JAK1/STAT3 activation [167–168]. The considerable expression of cytokines including LIF from initial feeder (e.g. Buffalo rat liver BRL epithelial cell line) shown to be responsible for repressing differentiation in murine ES in vitro [169]. Disruption of JAK1/STAT3 signaling promotes differentiation of ESC [170], additionally, constitutive STAT3 expression, using a fusion protein composed of STAT3 and the estrogen receptor is sufficient to maintain ES in an undifferentiated state [167].
LIF is a “helical type 1”, interleukin (IL)-6-type family protein [162]. The LIF receptor comprises a heterodimer of (i) A common IL-6 family subunit (gp130) [171], and (ii) A low-affinity, LIF-specific subunit (gp190 or LIF receptor beta/LIFRβ) [170]. Upon formation of the ligand-receptor trimeric complex, numerous phosphorylation events characterize activation of the JAK/STAT pathway. The four members of the mammalian JAK family (JAK1, JAK2, JAK3 and TYK2) share seven regions of homology, named JH1 to JH7. They are functionally divided into three domains: amino-terminal region (N), a catalytically inactive kinase like (KL) domain and a tyrosine kinase (TK) domain [172]. Among the four JAK proteins, only JAK1 and JAK2 are involved to LIF signal as is suggested by knockout murine model experiments [173]. JAK1 is bound to gp130. Upon LIF mediated activation a reciprocal phosphorylation occurs between gp130 and JAK1. Firstly, phosphorylation of JAK1 at its TK domain (Tyr1022), followed by phosphorylation of four tyrosine residues on gp130, providing a docking site for the following component: STAT3 [163, 174, 175].
The seven isoforms of STAT share 6 conserved domains: an amino-terminal domain (NH2), a coiled-coil domain, the DNA binding domain (DBD), a linker domain, an SH2 domain, and a tyrosine activation domain and a carboxy-terminal transcriptional activation domain (TAD); the latter being conserved in function but not in sequence [173]. STAT3 is also phosphorylated by JAK at Tyr (705) [166], triggering formation of a STAT3 homodimer (STAT3h), through the N-terminal SH2 domain; STAT3h is subsequently released to the cytoplasm [176]. The translocation of STAT3h to the nuclear compartment is mediated by two importin protein family members, importin-α3 and importin-α6 [177, 178], Then STAT3h binds to the STAT3-related enhancer region harboring the consensus sequence TTCC(C/G)GGGAA on target genes [179].
Several genes have been described as targeted genes by STAT3 with a majority of them involved on the subsequent inhibition of mesoderm and endoderm differentiation [180]. To illustrate, STAT3 expression promotes self-renewal of ESC in part through transcriptional control and regulation of c-Myc and Klf4 [181, 182], as well as binding to the enhancer element of Nanog gene (in mice, [183]). Chromatin Immunoprecipitation (ChIP) experiments show co-localization of STAT3 at loci share with pluripotency regulators Nanog, Sox2, Oct4 and Smad1 [179]. Finally, among those genes which are targets of STAT3, is SOCS3, a regulator that attenuate LIF signaling by binding the phosphorylated JAK1 and mediates its proteosomal degradation after a previous conformational change by gp130 binding; this provides a negative feedback to the STAT3 signal [184, 185].
The dependence of LIF to maintenance of murine ESC pluripotency directly translates to the induced pluripotent phenotype [186, 187]. The JAK/STAT3 molecular cascade is activate during somatic cell reprogramming to pluripotent iPSC and in EpiSC reprogramming under Nanog or Klf4 over expression and specific conditions [188]. Additionally, the clonal yield is reduced if JAK/STAT3 cascade is inhibited [188].
iPSC generated from Neuronal Stem Cells (NSC) and embryonic fibroblast depends on JAK/STAT3 pathway to maintain a ground state. Furthermore, in the presence of LIF a 3–4-fold increase in the number of colonies was observed; an effect only attributable to JAK/STAT3 cascades [188]. This effect has been proved in experiments using chimeric receptors [188, 189]. Over expressing a JAK/STAT3 activating receptor GY118F, LIF independent and responsive to granulocyte colony stimulating factor (GCSF), was possible to reprogram EpiSc into chimera-competent iPSC [188]. In a similar approach, but using the expression of a tamoxifen inducible form of STAT3 (STAT3MER) was possible to retain a pluripotent phenotype in murine iPSC even in absence of LIF stimulus, but not once tamoxifen was removed from the media [189].
2.6.2 Epigenetic Changes During Reprogramming
Chromatin refers to the collective DNA/histone complexes within the nucleus of a cell, the modification of which regulates access of transcriptional machinery to genes and regulatory elements in the genome [190]. Epigenetic marks at specific loci throughout the genome of somatic and pluripotent stem cells are likely to differ markedly, and require considerable global remodeling when converting from the former to the latter cell type.
Analysis of the epigenetic state provides a meaningful way of determining the degree of reprogramming in iPSC. Pluripotent stem cells contain a characteristic chromatin signature, termed ‘bivalent domains’ [89]. These are regions enriched for repressive histone H3 lysine 27 trimethylation (H3K27me3) and simultaneously for histone H3 lysine 4 trimethylation (H3K4me3), an activating mark [191]. Bivalent domains are indicative of genes that remain in a poised state and iPSC are found to contain large numbers of bivalent domains. Consequently, pluripotent cells were found to contain a large number of bivalent domains compared with, for example, multi-potential neural progenitor cells (NPC) cells. Several of the murine iPSC studies have investigated a small number of representative loci for their chromatin and DNA methylation patterns [192–194].
In a more expanded survey of the iPSC epigenome, Maherali and colleagues (2008) [192] suggested that the epigenetic profile if iPSC closely mirrored their ESC counterparts, with 94.4 % of 957 ‘signature’ genes (defined as genes that have a different epigenetic state between MEF and ESC) being reset to an ESC state in the respective iPSC line. H3K4me3 pattern were also similar across all samples, indicating that reprogramming was largely associated with changes in H3K27me3 rather than H3K4me3 [192]. Applying ChIP-Seq to determine genome-wide chromatin maps in several iPSC lines that were derived in distinct ways; namely, (i) through drug selection using an Oct4–neomycin-resistance gene [194], (ii) through drug selection using a Nanog–neomycin-resistance gene [196], and (iii) by simply isolating reprogrammed cells through their morphological appearance [197], Mikkelsen et al. (2007) [198] found overall global levels of repressive H3K27me3 and the characteristic bivalent chromatin structure were restored across the different iPSC lines.
In addition to histone modifications, DNA can be modified directly through attachment of methyl groups to CpG islands [199]. DNA methylation is stable and heritable (yet reversible) and influences many biological processes, including gene regulation, genomic imprinting and X-chromosome inactivation. DNA methylation patterns are dynamic during early embryonic development and are essential for normal post-implantation development [199]. Overall DNA methylation levels remain stable during ES-cell differentiation. However, these marks are not static [200] and regulate chromatin structure and function in concert with histone modification. To illustrate, H3K4me3 and DNA methylation are considered mutually exclusive and rarely co-exist at a given loci [201]. The re-establishment of H3K4me3 and the associated loss of DNA methylation in particular at ‘ES-cell-associated transcript’ (ECAT) genes seem to be a crucial and potentially rate-limiting step during reprogramming [202]. At reprogramming, expression from ECAT’s in somatic cells is reinstated from a state of dormancy, with associated demethylation marks of promoter regions [203]. This illustrates the importance of demethylation of key genomic loci for complete reprogramming to be achieved. Consistent with this notion, application of DNA demethylation agent 5-azacytidine accelerated fourfold increase in the reprogramming of lineage-restricted cells to iPSC [209].
Despite similarities in overall epigenetic state of iPSC and ESC, iPSC line-to-line variation complicates the comparison. Application of different/fewer reprogramming factor combinations or methods (e.g. RNA, recombinant protein delivery), use of chemical substitutes as well as choice of target cell, and prolonged periods of culture is likely to alter the epigenetic state of the respective cells [201, 202]. In particular, imprinted genes can only be reset in the germ line and are unstable in murine ESC cultures [203], although apparently not in human ESC [204]. More studies are required to test these hypotheses and confirm whether epigenetic stability is cause for concern when considering iPSC for therapeutic applications.
2.7 Conclusion
Convincing the scientific community that the observed plasticity of amphibian somatic cell nuclei translates to the mammalian system took some 30 years. The cloning of Dolly was conclusive proof mammalian somatic cells can acquire totipotency in certain situations. The concurrent development of cell culture techniques for ESC facilitated application of the techniques to cultivate cloned, human ESC by SCNT; however this eventuality still eludes us. Since 2006, much of the stem cell research has adopted a direct approach for derivation of patient-specific stem cells for therapy, but numerous issues require addressing before their therapeutic potential is realized. However, the current state of iPSC allows a unique means for understanding development and disease and provides an unprecedented resource for drug discovery.
References
Briggs R, King TJ (1952) Transplantation of living nuclei from blastula cells into enucleated frogs’ eggs. PNAS 38:455–463
Briggs R, King TJ (1953) Factors affecting the transplantation of nuclei of frog embryonic cells. J Exp Zool 122:485–505
Gurdon JB (1962) The transplantation of nuclei between two species of Xenopus. Dev Biol 5:68–83
Gurdon JB (1962) Adult frogs derived from the nuclei of single somatic cells. Dev Biol 4:256–273
Gurdon JB (1968) Changes in somatic cell nuclei inserted into growing and maturing amphibian oocytes. J Embryol Exp Morphol 20(3):401–414
Laskey RA, Gurdon JB (1970) Genetic content of adult somatic cells tested by nuclear transplantation from cultured cells. Nature 228(5278):1332–1334
De Robertis EM, Gurdon JB (1977) Gene activation in somatic nuclei after injection into amphibian oocytes. PNAS 74(6):2470–2474
McGrath J, Solter D (1984) Inability of mouse blastomere nuclei transferred to enucleated zygotes to support development in vitro. Science 226:1317–1319
Willadsen SM (1986) Nuclear transplantation in sheep embryos. Nature 320:63–65
Campbell KH, McWhir J, Ritchie WA et al (1996) Sheep cloned by nuclear transfer from a cultured cell line. Nature 380:64–66
Wilmut I, Schnieke AE, McWhir J et al (1997) Viable offspring derived from fetal and adult mammalian cells. Nature 385:810–813
Thomson JA, Itskovitz-Eldor J, Shapiro SS et al (1998) Embryonic stem cell lines derived from human blastocysts. Science 282:1145–1147
Miller R, Ruddle FH (1976) Pluripotent teratocarcinoma-thymus somatic cell hybrids. Cell 9:45–55
Kikyo N, Wade PA, Guschin D et al (2000) Active remodeling of somatic nuclei in egg cytoplasm by the nucleosomal ATPase ISWI. Science 289:2360–2362
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676
Evans MJ, Kaufman MH (1981) Establishment in culture of pluripotential cells from mouse embryos. Nature 292(5819):154–156
Martin GR (1981) Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. PNAS 78(12):7634–7638
Bongso A, Fong CY, Ng SC et al (1994) Isolation and culture of inner cell mass cells from human blastocysts. Hum Reprod 9(11):2110–2117
Thomson JA, Kalishman J, Golos TG et al (1995) Isolation of a primate embryonic stem cell line. PNAS 92(17):7844–7848
Thomson JA, Kalishman J, Golos TG et al (1996) Pluripotent cell lines derived from common marmoset (Callithrix jacchus) blastocysts. Biol Reprod 55(2):254–259
Stojkovic M, Lako M, Stojkovic P et al (2004) Derivation of human embryonic stem cells from day-8 blastocysts recovered after three-step in vitro culture. Stem Cells 22:790–797
Strelchenko N, Verlinsky O, Kukharenko V et al (2004) Morula-derived human embryonic stem cells. Reprod Biomed Online 9:623–629
Klimanskaya I, Chung Y, Becker S et al (2006) Human embryonic stem cell lines derived from single blastomeres. Nature 444:481–485
Byrne JA, Pedersen DA, Clepper LL et al (2007) Producing primate embryonic stem cells by somatic cell nuclear transfer. Nature 450:497–502
Wilmut I, Beaujean N, de Sousa PA et al (2002) Somatic cell nuclear transfer. Nature 419(6907):583–586
Paterson L, DeSousa P, Ritchie W et al (2003) Application of reproductive biotechnology in animals: implications and potentials. Applications of reproductive cloning. Anim Reprod Sci 79:137–143
Takahashi S, Ito Y (2004) Evaluation of meat products from cloned cattle: biological and biochemical properties. Cloning Stem Cells 6:165–171
Tome D, Dubarry M, Fromentin G (2004) Nutritional value of milk and meat products derived from cloning. Cloning Stem Cells 6:172–177
Rudenko L, Matheson JC (2007) The US FDA and animal cloning: risk and regulatory approach. Theriogenology 67:198–206
Rudenko L, Matheson JC, Sundlof SF (2007) Animal cloning and the FDA – the risk assessment paradigm under public scrutiny. Nat Biotechnol 25:39–43
Rideout WM 3rd, Hochedlinger K, Kyba M et al (2002) Correction of a genetic defect by nuclear transplantation and combined cell and gene therapy. Cell 109:17–27
Sumer H, Liu J, Ta PA et al (2009) Somatic cell nuclear transfer: pros & cons. J Stem Cells 4:85–94
French AJ, Adams CA, Anderson LS et al (2008) Development of human cloned blastocysts following somatic cell nuclear transfer with adult fibroblasts. Stem Cells 26:485–493
Li J, Liu X, Wang H et al (2009) Human embryos derived by somatic cell nuclear transfer using an alternative enucleation approach. Cloning Stem Cells 11:39–50
Stojkovic M, Stojkovic P, Leary C et al (2005) Derivation of a human blastocyst after heterologous nuclear transfer to donated oocytes. Reprod Biomed Online 11:226–231
Noggle S, Fung HL, Gore A et al (2011) Human oocytes reprogram somatic cells to a pluripotent state. Nature 478:70–75
Lucas JJ, Terada N (2003) Cell fusion and plasticity. Cytotechnology 41:103–109
Tada M, Tada T, Lefebvre L et al (1997) Embryonic germ cells induce epigenetic reprogramming of somatic nucleus in hybrid cells. EMBO J 16:6510–6520
Tada M, Takahama Y, Abe K et al (2001) Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr Biol 11:1553–1558
Kimura H, Tada M, Nakatsuji N et al (2004) Histone code modifications on pluripotential nuclei of reprogrammed somatic cells. Mol Cell Biol 24:5710–5720
Cowan CA, Atienza J, Melton DA et al (2005) Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science 309:1369–1373
Sumer H, Nicholls C, Pinto AR et al (2010) Chromosomal and telomeric reprogramming following ES-somatic cell fusion. Chromosoma 119:9
Pralong D, Mrozik K, Occhiodoro F et al (2005) A novel method for somatic cell nuclear transfer to mouse embryonic stem cells. Cloning Stem Cells 7:265–271
Sumer H, Jones KL, Liu J et al (2009) Transcriptional changes in somatic cells recovered from embryonic stem-somatic heterokaryons. Stem Cells Dev 18:1361–1368
Matsumura H, Tad M, Otsuji T et al (2007) Targeted chromosome elimination from ES-somatic hybrid cells. Nat Methods 4:23–25
Walev I, Bhakdi SC, Hofmann F et al (2001) Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc Natl Acad Sci USA 98:3185–3190
Hansis C, Barreto G, Maltry N et al (2004) Nuclear reprogramming of human somatic cells by Xenopus egg extract requires BRG1. Curr Biol 14:1475–1480
Gonda K, Kikyo N (2006) Nuclear remodeling assay in Xenopus egg extract. Methods Mol Biol 348:247–258
Hakelien AM, Landsverk HB, Robl JM et al (2002) Reprogramming fibroblasts to express T-cell functions using cell extracts. Nat Biotechnol 20:460–466
Collas P (2003) Nuclear reprogramming in cell-free extracts. Philos Trans R Soc Lond B Biol Sci 358:1389–1395
Taranger CK, Noer A, Sorensen AL et al (2005) Induction of dedifferentiation, genomewide transcriptional programming, and epigenetic reprogramming by extracts of carcinoma and embryonic stem cells. Mol Biol Cell 16:5719–5735
Freberg CT, Dahl JA, Timoskainen S et al (2007) Epigenetic reprogramming of OCT4 and NANOG regulatory regions by embryonal carcinoma cell extract. Mol Biol Cell 18:1543–1553
Han J, Sachdev PS, Sidhu KS (2010) A combined epigenetic and non-genetic approach for reprogramming human somatic cells. PLoS One 5:e12297
Neri T, Monti M, Rebuzzini P et al (2007) Mouse fibroblasts are reprogrammed to Oct-4 and Rex-1 gene expression and alkaline phosphatase activity by embryonic stem cell extracts. Cloning Stem Cells 9:394–406
Wernig M, Meissner A, Cassady JP et al (2008) c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell 2:10–12
Nakagawa M, Koyanagi M, Tanabe K et al (2008) Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 26:101–106
Okita K, Ichisaka T, Yamanaka S (2007) Generation of germline-competent induced pluripotent stem cells. Nature 448:313–317
Boland MJ, Hazen JL, Nazor KL et al (2009) Adult mice generated from induced pluripotent stem cells. Nature 461:91–94
Kang L, Wu T, Tao Y et al (2011) Viable mice produced from three-factor induced pluripotent stem (iPS) cells through tetraploid complementation. Cell Res 21:546–549
Zhao XY, Li W, Lv Z (2009) iPS cells produce viable mice through tetraploid complementation. Nature 461:86–90
Takahashi K, Tanabe K, Ohnuki M et al (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872
Park IH, Zhao R, West JA et al (2008) Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451:141–146
Yu J, Vodyanik MA, Smuga-Otto K et al (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318:1917–1920
Carey BW, Markoulaki S, Hanna J et al (2009) Reprogramming of murine and human somatic cells using a single polycistronic vector. PNAS 106:157–162
Okita K, Nakagawa M, Hyenjong H et al (2008) Generation of mouse induced pluripotent stem cells without viral vectors. Science 322:949–953
Soldner F, Hockemeyer D, Beard C et al (2009) Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136:964–977
Stadtfeld M, Nagaya M, Utikal J et al (2008) Induced pluripotent stem cells generated without viral integration. Science 322:945–949
Dimos JT, Rodolfa KT, Niakan KK et al (2008) Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321:1218–1221
Ebert AD, Yu J, Rose FF Jr et al (2009) Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457:277–280
Park IH, Arora N, Huo H et al (2008) Disease-specific induced pluripotent stem cells. Cell 134:877–886
Raya A, Rodriguez-Piza I, Guenechea G et al (2009) Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature 460:53–59
Choi KD, Yu J, Smuga-Otto K et al (2009) Hematopoietic and endothelial differentiation of human induced pluripotent stem cells. Stem Cells 27:559–567
Schenke-Layland K, Rhodes KE, Angelis E et al (2008) Reprogrammed mouse fibroblasts differentiate into cells of the cardiovascular and hematopoietic lineages. Stem Cells 26:1537–1546
Zhang J, Wilson GF, Soerens AG et al (2009) Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res 104:e30–e41
Chambers SM, Fasano CA, Papapetrou EP et al (2009) Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 27:275–280
Hirami Y, Osakada F, Takahashi K et al (2009) Generation of retinal cells from mouse and human induced pluripotent stem cells. Neurosci Lett 458:126–131
Chen YF, Tseng CY, Wang HW et al (2011) Rapid generation of mature hepatocyte-like cells from human induced pluripotent stem cells by an efficient three-step protocol. Hepatology 55:1193–1203
Takayama K, Inamura M, Kawabata K (2011) Efficient generation of functional hepatocytes from human embryonic stem cells and induced pluripotent stem cells by HNF4alpha transduction. Mol Ther 19:400–407
Inamura M, Kawabata K, Takayama K et al (2011) Efficient generation of hepatoblasts from human ES cells and iPS cells by transient overexpression of homeobox gene HEX. Mol Ther 19:400–407
Pasque V, Miyamoto K, Gurdon JB (2010) Efficiencies and mechanisms of nuclear reprogramming. Cold Spring Harb Symp Quant Biol 75:189–200
Ben-Shushan E, Sharir H, Pikarsky E et al (1995) A dynamic balance between ARP-1/COUP-TFII, EAR-3/COUP-TFI, and retinoic acid receptor:retinoid X receptor heterodimers regulates Oct-3/4 expression in embryonal carcinoma cells. Mol Cell Biol 15:1034–1048
Pikarsky E, Sharir H, Ben-Shushan E et al (1994) Retinoic acid represses Oct-3/4 gene expression through several retinoic acid-responsive elements located in the promoter-enhancer region. Mol Cell Biol 14:1026–1038
Barnea E, Bergman Y (2000) Synergy of SF1 and RAR in activation of Oct-3/4 promoter. J Biol Chem 275:6608–6619
Wang W, Yang J, Liu H et al (2011) Rapid and efficient reprogramming of somatic cells to induced pluripotent stem cells by retinoic acid receptor gamma and liver receptor homolog 1. PNAS 108:18283–18288
Boiani M, Gentile L, Gambles VV et al (2005) Variable reprogramming of the pluripotent stem cell marker Oct4 in mouse clones: distinct developmental potentials in different culture environments. Stem Cells 23:1089–1104
Do JT, Han DW, Gentile L et al (2007) Erasure of cellular memory by fusion with pluripotent cells. Stem Cells 25:1013–1020
Huangfu D, Osafune K, Maehr R et al (2008) Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol 26:1269–1275
Huangfu D, Maehr R, Guo W et al (2008) Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol 26:795–797
Hanna J, Markoulaki S, Schorderet P et al (2008) Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell 133:250–264
Choi J, Costa ML, Mermelstein CS et al (1990) MyoD converts primary dermal fibroblasts, chondroblasts, smooth muscle, and retinal pigmented epithelial cells into striated mononucleated myoblasts and multinucleated myotubes. PNAS 87:7988–7992
Hirai H, Tani T, Katoku-Kikyo N et al (2011) Radical acceleration of nuclear reprogramming by chromatin remodeling with the transactivation domain of MyoD. Stem Cells 29:1349–1361
Ghosh Z, Wilson KD, Wu Y et al (2010) Persistent donor cell gene expression among human induced pluripotent stem cells contributes to differences with human embryonic stem cells. PLoS One 5:e8975
Kim K, Doi A, Wen B et al (2010) Epigenetic memory in induced pluripotent stem cells. Nature 467:285–290
Polo JM, Liu S, Figueroa ME et al (2010) Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol 28:848–855
Guenther MG, Frampton GM, Soldner F et al (2010) Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell Stem Cell 7:249–257
Yoshida Y, Takahashi K, Okita K et al (2009) Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell 5:237–241
Esteban MA, Wang T, Qin B et al (2010) Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell 6:71–79
Kaji K, Norrby K, Paca A et al (2009) Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature 458:771–775
Woltjen K, Michael IP, Mohseni P et al (2009) piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 458:766–770
Jia F, Wilson KD, Sun N et al (2010) A nonviral minicircle vector for deriving human iPS cells. Nat Methods 7:197–199
Fusaki N, Ban H, Nishiyama A et al (2009) Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci 85:348–362
Seki T, Yuasa S, Oda M et al (2010) Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell 7:11–14
Kim D, Kim CH, Moon JI et al (2009) Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 4:472–476
Zhou H, Wu S, Joo JY et al (2009) Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 4:381–384
Warren L, Manos PD, Ahfeldt T et al (2010) Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7:618–630
Miyoshi N, Ishii H, Nagano H et al (2011) Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 8:633–638
Mizuno Y, Chang H, Umeda K et al (2010) Generation of skeletal muscle stem/progenitor cells from murine induced pluripotent stem cells. FASEB J 24(7):2245–2253
Wada H, Kojo S, Kusama C et al (2011) Successful differentiation to T cells, but unsuccessful B-cell generation, from B-cell-derived induced pluripotent stem cells. Int Immunol 23(1):65–74
Woods NB, Parker AS, Moraghebi R et al (2011) Efficient generation of hematopoietic precursors and progenitors from human pluripotent stem cell lines. Stem Cells 29(7):1158–1164
Kasuda S, Tatsumi K, Sakurai Y et al (2011) Expression of coagulation factors from murine induced pluripotent stem cell-derived liver cells. Blood Coagul Fibrinolysis 22(4):271–279
Si-Tayeb K, Noto FK, Sepac A et al (2010) Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev Biol 10:81
Tokumoto Y, Ogawa S, Nagamune T et al (2010) Comparison of efficiency of terminally differentiation of oligodendrocytes from induced pluripotent stem cells versus embryonic stem cells in vitro. J Biosci Bioeng 109(6):622–628
Brennand KJ, Simone A, Jou J et al (2011) Modelling schizophrenia using human induced pluripotent stem cells. Nature 479(7374):556
Czepiel M, Balasubramaniyan V, Schaafsma W et al (2011) Differentiation of induced pluripotent stem cells into functional oligodendrocytes. Glia 59(6):882–892
Kim J, Efe JA, Zhu S et al (2011) Direct reprogramming of mouse fibroblasts to neural progenitors. PNAS 108(19):7838–7843
Kitazawa A, Shimizu N (2010) Differentiation of mouse induced pluripotent stem cells into neurons using conditioned medium of dorsal root ganglia. N Biotechnol 28(4):326–333
Seibler P, Graziotto J, Jeong H et al (2011) Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci 31(16):5970–5976
Tucker BA, Park IH, Qi SD et al (2011) Transplantation of adult mouse iPS cell-derived photoreceptor precursors restores retinal structure and function in degenerative mice. PLoS One 29; 6(4):e18992
Zhou L, Wang W, Liu Y et al (2011) Differentiation of induced pluripotent stem cells of swine into rod photoreceptors and their integration into the retina. Stem Cells 29(6):972–980
Hu Q, Friedrich AM, Johnson LV et al (2010) Memory in induced pluripotent stem cells: reprogrammed human retinal-pigmented epithelial cells show tendency for spontaneous redifferentiation. Stem Cells 28(11):1981–1991
Ohi Y, Qin H, Hong C et al (2011) Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat Cell Biol 13(5):541–549
Zhao T, Zhang ZN, Rong Z et al (2011) Immunogenicity of induced pluripotent stem cells. Nature 474(7350):212–215
Itzhaki I, Rapoport S, Huber I et al (2011) Calcium handling in human induced pluripotent stem cell derived cardiomyocytes. PLoS One 6:e18037
Itzhaki I, Maizels L, Huber I et al (2011) Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471:225–229
Moretti A, Bellin M, Welling A et al (2010) Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med 363:1397–1409
Agarwal S, Loh YH, McLoughlin EM et al (2010) Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature 464:292–296
Liu J, Verma PJ, Evans-Galea M, Delatycki M, Michalska A et al (2011) Generation and function of induced-pluripotent stem cell lines from Friedreich ataxia patients. Stem Cell Rev Reports 7(3):703–713
Wehrli M, Dougan ST, Caldwell K, O’Keefe L, Schwartz S et al (2000) Arrow encodes an LDL-receptor-related protein essential for Wingless signaling. Nature 407:527–530
Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC (2000) An LDL-receptor-related protein mediates Wnt signaling in mice. Nature 407:535–538
Bhanot P, Brink M, Samos CH, Hsieh JC, Wang Y et al (1996) A new member of the frizzled family from Drosophila functions as a wingless receptor. Nature 382:225–230
Tamai K, Semenov M, Kato Y, Spokony R, Liu C et al (2000) LDL-receptor-related proteins in Wnt signal transduction. Nature 407:530–535
Bernatik O, Ganji RS, Dijksterhuis JP, Konik P, Cervenka I et al (2011) Sequential activation and Inactivation of Dishevelled in the Wnt/β-Catenin pathway by casein kinases. J Biol Chem 286:10396–10410
Zeng X, Huang H, Tamai K, Zhang X, Harada Y et al (2008) Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development 135:367–375
Tamai K, Zeng X, Liu C, Zhang X, Harada Y et al (2004) A mechanism for Wnt coreceptor activation. Mol Cell 13:149–156
Grigoryan T, Wend P, Klaus A, Birchmeier W (2008) Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev 22:2308–2341
Sato N, Meijer L, Skaltsounis L et al (2004) Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med 10:55–63
Grigoryan T, Wend P, Klaus A et al (2008) Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev 22:2308–2341
Ogawa K, Nishinakamura R, Iwamatsu Y et al (2006) Synergistic action of Wnt and LIF in maintaining pluripotency of mouse ES cells. Biochem Biophys Res Commun 343:159–166
Singla DK, Schneider DJ et al (2006) wnt3a but not wnt11 supports self-renewal of embryonic stem cells. Biochem Biophys Res Commun 345:789–795
Cai L, Ye Z, Zhou BY et al (2007) Promoting human embryonic stem cell renewal or differentiation by modulating Wnt signal and culture conditions. Cell Res 17:62–72
Silva J, Barrandon O, Nichols J et al (2008) Promotion of reprogramming to Ground STATe pluripotency by signal inhibition. PLoS Biol 6:e253
Cole MF, Johnstone SE, Newman JJ, Kagey MH, Young RA (2008) Tcf3 is an integral component of the core regulatory circuitry of embryonic stem cells. Genes Dev 22:746–755
Tam W-L, Lim CY, Han J, Zhang J, Ang Y-S et al (2008) T-cell factor 3 regulates embryonic stem cell pluripotency and self-renewal by the transcriptional control of multiple lineage pathways. Stem Cells 26:2019–2031
Pereira L, Yi F, Merrill BJ (2006) Repression of Nanog gene transcription by Tcf3 limits embryonic stem cell self-renewal. Mol Cell Biol 26:7479–7491
Ogawa K, Nishinakamura R, Iwamatsu Y, Shimosato D, Niwa H (2006) Synergistic action of Wnt and LIF in maintaining pluripotency of mouse ES cells. Biochem Biophys Res Commun 343:159–166
Marson A, Foreman R, Chevalier B, Bilodeau S, Kahn M et al (2008) Wnt signaling promotes reprogramming of somatic cells to pluripotency. Cell Stem Cell 3:132–135
Lluis F, Ombrato L, Pedone E, Pepe S, Merrill BJ et al (2011) T-cell factor 3 (Tcf3) deletion increases somatic cell reprogramming by inducing epigenome modifications. Proc Natl Acad Sci USA 108:11912–11917
Artavanis-Tsakonas S, Rand MD, Lake RJ (1999) Notch signaling: cell fate control and signal integration in development. Science 284:770–776
Radtke F, Raj K (2003) The role of Notch in tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer 3:756–767
Bray SJ, Takada S, Harrison E, Shen S-C, Ferguson-Smith AC (2008) The atypical mammalian ligand Delta-like homologue 1 (Dlk1) can regulate Notch signaling in Drosophila. BMC Dev Biol 8:11
Kovall RA (2008) More complicated than it looks: assembly of Notch pathway transcription complexes. Oncogene 27:5099–5109
Fryer CJ, Lamar E, Turbachova I, Kintner C, Jones KA (2002) Mastermind mediates chromatin-specific transcription and turnover of the Notch enhancer complex. Genes Dev 16:1397–1411
Wallberg AE, Pedersen K, Lendahl U, Roeder RG (2002) p300 and PCAF act cooperatively to mediate transcriptional activation from chromatin templates by notch intracellular domains in vitro. Mol Cell Biol 22:7812–7819
Iso T, Kedes L, Hamamori Y (2003) HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol 194:237–255
Meier-Stiegen F, Schwanbeck R, Bernoth K, Martini S, Hieronymus T et al (2010) Activated Notch1 target genes during embryonic cell differentiation depend on the cellular context and include lineage determinants and inhibitors. PLoS One 5:e11481
Chen X, Xu H, Yuan P, Fang F, Huss M et al (2008) Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133:1106–1117
Henrique D, Adam J, Myat A, Chitnis A, Lewis J et al (1995) Expression of a delta homologue in prospective neurons in the chick. Nature 375:787–790
Henrique D, Hirsinger E, Adam J, Le Roux I, Pourquie O et al (1997) Maintenance of neuroepithelial progenitor cells by Delta-Notch signaling in the embryonic chick retina. Curr Biol 7:661–670
Lowell S, Benchoua A, Heavey B, Smith AG (2006) Notch promotes neural lineage entry by pluripotent embryonic stem cells. PLoS Biol 4
Jones P, May G, Healy L, Brown J, Hoyne G et al (1998) Stromal expression of Jagged 1 promotes colony formation by fetal hematopoietic progenitor cells. Blood 92:1505–1511
Nemir M, Croquelois A, Pedrazzini T, Radtke F (2006) Induction of cardiogenesis in embryonic stem cells via downregulation of Notch1 signaling. Circ Res 98:1471–1478
Heinrich PC, Behrmann I, Haan S et al (2003) Principles of interleukin (IL)-6-type cytokine signaling and its regulation. Biochem J 374:1–20
Hirai H, Karian P, Kikyo N (2011) Regulation of embryonic stem cell self-renewal and pluripotency by leukaemia inhibitory factor. Biochem J 438:11–23
Paling NR, Wheadon H, Bone HK et al (2004) Regulation of embryonic stem cell self-renewal by phosphoinositide 3-kinase-dependent signaling. J Biol Chem 279:48063–48070
Rawlings JS, Rosler KM, Harrison DA (2004) The JAK/STAT signaling pathway. J Cell Sci 117:1281–1283
Smith AG, Nichols J, Robertson M et al (1992) Differentiation inhibiting activity (DIA/LIF) and mouse development. Dev Biol 151:339–351
Matsuda T, Nakamura T, Nakao K et al (1999) STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO J 18:4261–4269
Williams RL, Hilton DJ, Pease S et al (1988) Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature 336:684–687
Smith AG, Hooper ML (1987) Buffalo rat liver cells produce a diffusible activity which inhibits the differentiation of murine embryonal carcinoma and embryonic stem cells. Dev Biol 121:1–9
Niwa H, Burdon T, Chambers I et al (1998) Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev 12:2048–2060
Yoshida K, Chambers I, Nichols J et al (1994) Maintenance of the pluripotential phenotype of embryonic stem cells through direct activation of gp130 signaling pathways. Mech Dev 45:163–171
Yeh TC, Pellegrini S (1999) The Janus kinase family of protein tyrosine kinases and their role in signaling. Cell Mol Life Sci 55:1523–1534
Kisseleva T, Bhattacharya S, Braunstein J et al (2002) Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 285:1–24
Stahl N, Farruggella TJ, Boulton TG et al (1995) Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science 267:1349–1353
Gerhartz C, Heesel B, Sasse J et al (1996) Differential activation of acute phase response factor/STAT3 and STAT1 via the cytoplasmic domain of the interleukin 6 signal transducer gp130. I. Definition of a novel phosphotyrosine motif mediating STAT1 activation. J Biol Chem 271:12991–12998
Ihle JN, Kerr IM (1995) Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet 11:69–74
Cimica V, Chen HC, Iyer JK et al (2011) Dynamics of the STAT3 transcription factor: nuclear import dependent on Ran and importin-beta1. PLoS One 6:e20188
Liu L, McBride KM, Reich NC (2005) STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc Natl Acad Sci USA 102:8150–8155
Chen X, Xu H, Yuan P et al (2008) Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133:1106–1117
Bourillot PY, Aksoy I, Schreiber V et al (2009) Novel STAT3 target genes exert distinct roles in the inhibition of mesoderm and endoderm differentiation in cooperation with Nanog. Stem Cells 27:1760–1771
Cartwright P, McLean C, Sheppard A et al (2005) LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 132:885–896
Kristensen DM, Kalisz M, Nielsen JH (2005) Cytokine signaling in embryonic stem cells. APMIS 113:756–772
Suzuki A, Raya A, Kawakami Y et al (2006) Nanog binds to Smad1 and blocks bone morphogenetic protein-induced differentiation of embryonic stem cells. Proc Natl Acad Sci USA 103:10294–10299
Boyle K, Zhang JG, Nicholson SE et al (2009) Deletion of the SOCS box of suppressor of cytokine signaling 3 (SOCS3) in embryonic stem cells reveals SOCS box-dependent regulation of JAK but not STAT phosphorylation. Cell Signal 21:394–404
Duval D, Reinhardt B, Kedinger C et al (2000) Role of suppressors of cytokine signaling (Socs) in leukemia inhibitory factor (LIF) -dependent embryonic stem cell survival. FASEB J 14:1577–1584
Xu J, Wang F, Tang Z et al (2010) Role of leukaemia inhibitory factor in the induction of pluripotent stem cells in mice. Cell Biol Int 34:791–797
Nishishita N, Ijiri H, Takenaka C et al (2011) The use of leukemia inhibitory factor immobilized on virus-derived polyhedra to support the proliferation of mouse embryonic and induced pluripotent stem cells. Biomaterials 32:3555–3563
Yang J, van Oosten AL, Theunissen TW et al (2010) Stat3 activation is limiting for reprogramming to ground state pluripotency. Cell Stem Cell 7:319–328
Graf U, Casanova EA, Cinelli P (2011) The role of the leukemia inhibitory factor (LIF) – pathway in derivation and maintenance of murine pluripotent stem cells. Genes 2:280–297
Brambrink T, Foreman R, Welstead G et al (2008) Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell 2:151–159
Wernig M, Legner C, Jacob H et al (2008) A drug-inducible transgenic system for direct reprogramming of multiple somatic cell types. Nat Biotechnol 26:916–924
Maherali N, Ahfeldt T, Rigamonti A et al (2008) A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell 3:340–345
Hockemeyer D, Soldener F, Cook EG et al (2008) A drug-inducible system for direct reprogramming of human somatic cells to pluripotency. Cell Stem Cell 3:346–353
Lowry WE, Richter L, Yachechko R et al (2008) Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci USA 105:2883–2888
Niwa H, Miyajaki J, Smith AG (2000) Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet 24:372–376
Bernstein BE, Meissner A, Lander ES (2007) The mammalian epigenome. Cell 128:669–681
Bernstein BE, Mikkelsen TS, Xie X et al (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125(315):326
Mikkelsen TS, Manching KU, Jaffe DB et al (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448:553–560
Meissner A, Mikkelson TS, Hongcang GU et al (2008) Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454:766–770
Meissner A, Wernig M, Jaenisch R (2007) Direct reprogramming of genetically unmodified fibroblasts into pluripotent stem cells. Nat Biotechnol 25:1177–1181
Rougier N, Bourchis D, Gomes DM et al (1998) Chromosome methylation patterns during mammalian preimplantation development. Genes Dev 12:2108–2113
Imamura M, Miura K, Iwabuchi K et al (2006) Transcriptional repression and DNA hypermethylation of a small set of ES cell marker genes in male germline stem cells. BMC Dev Biol 6:34
Jones PA, Wolkowich MJ, Rideout WM et al (1990) De novo methylation of the MyoD1 CpG island during the establishment of immortal cell lines. PNAS 87:6117–6121
Humpherys D, Eggan K, Akutsu H et al (2001) Epigenetic instability in ES cells and cloned mice. Science 293:95–97
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Heffernan, C. et al. (2013). Induction of Pluripotency. In: Hime, G., Abud, H. (eds) Transcriptional and Translational Regulation of Stem Cells. Advances in Experimental Medicine and Biology, vol 786. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6621-1_2
Download citation
DOI: https://doi.org/10.1007/978-94-007-6621-1_2
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6620-4
Online ISBN: 978-94-007-6621-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)