
Abstract
Epigenetic states are orchestrated by several converging and reinforcing signals, including DNA methylation, histone modifications and non-coding RNAs. Growing evidence indicates that acquired epigenetic abnormalities participate with genetic alterations to cause cancer. In this review we describe recent advances in the field of cancer epigenomics and microRNAs (miRNAs) with special emphasis on renal cancer. We discuss whether epigenetic changes are the cause or consequence of cancer initiation and the use of epigenetic biomarkers and miRNAs for cancer diagnosis or prognosis. Finally we address the potential of epigenetic based anti-cancer therapeutic strategies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
4.1 Background
The term ‘epigentics’ was originally coined by Conrad Waddington in 1942 for the molecular mechanisms that convert genetic information into observable traits or phenotypes during development [1]. By contrast, Arthur Riggs et al. defined epigenetics as “the study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence” [2]. The term may be currently defined as the mechanisms that initiate and maintain heritable patterns of gene function and regulation without affecting the sequence of the genome [3]. The sum total of all epigenetic information is termed the ‘epigenome’ and comprises some of the instructions directing the genome to express genes at particular places and times [4, 5]. Unlike the genome, the epigenome is highly variable between cells and fluctuates in time according to conditions even within a single cell. Each of us has essentially one genome, however each cell type in each individual is believed to have a distinct epigenome that reflects its developmental state [6]. The epigenetic state of a cell is affected by developmental as well as environmental influences that may leave epigenetic traces which the cell remembers, referred to as cellular memory [7]. Thus the epigenome provides a crucial interface between the environment and the genome. Recent breakthroughs in the understanding of epigenetic mechanisms provide evidence that they are fundamental to the regulation of many cellular processes, including gene and microRNA expression, DNA-protein interactions, suppression of transposable element mobility, cellular differentiation, embroygenesis, X-chromosome inactivation and genomic imprinting [8]. The disruption of epigenetic changes underlies a wide variety of pathologies including cancer [9]. The cancer epigenome is characterized by global changes in DNA methylation including hypomethylation, promoter specific hypermethylation, histone modification, chromatin-modifying enzyme expression profiles and global dysregulation of non-coding microRNAs (miRNAs). These aberrations confer a selective growth advantage to neoplastic cells, apoptotic deficiency and uncontrolled cell proliferation, leading to cancer initiation and progression. For didactic purposes, epigenetic mechanisms may be grouped into DNA methylation, histone modification and remodeling and miRNAs. In this review, we will describe these mechanisms with an emphasis on alterations of the epigenome taking place in renal cancer.
4.2 DNA Methylation
Aberrant DNA methylation is the best characterized cancer-related epigenetic modification. DNA methylation occurs predominantly at the symmetrical dinucleotide CpG sites [10] that are scattered throughout the genome at a lower-than-expected frequency. However, in certain areas of the genome, a high concentration of CpG dinucleotides is found, and are referred to as “CpG islands” (CGIs) [11]. In a normal differentiated cell, CpG loci disseminated across the genome are highly methylated, whereas most promoter CGIs are protected from methylation inside their boundaries [11]. In general CGI methylation is associated with gene silencing. Gene silencing associated with CGI promoter methylation may be due to restricted access of transcription factors or binding of methylcytosine-binding proteins (MBD), which cooperate with DNMTs and histone deacetylases (HDACs) [12]. An important role in the regulation of gene expression has also been credited to low density CpG regions located in the vicinity of CpG islands, the so-called “CpG island shores” [13, 14]. These are sequences up to 2 kb distant from CpG islands, that are associated with gene expression. Remarkably, methylation patterns at CpG island shores are mostly tissue-specific and cancer-associated alterations in these patterns occur at sites that vary normally in tissue differentiation [14]. Differentially methylated CpG island shores are sufficient to distinguish between specific tissues and are conserved between human and mouse [13, 15]. Aberrations in DNA methylation include both global and gene-specific hypomethylation as well as gene-specific CpG island promoter hypermethylation [3, 16] (Fig. 4.1). Since global DNA hypomethylation and promoter-specific hypermethylation can be commonly observed in benign neoplasias and early-stage tumors, it is becoming apparent that epigenetic deregulation may precede the classical preliminary transforming events such as mutations in tumor suppressors, protooncogenes and genomic instability [17]. These aberrations have also have been considered to be the earliest events in the process of tumorigenesis [18]. The impact of gene-specific alterations in DNA methylation depends on the function of the affected gene and the type of alteration. Whereas promoter hypomethylation may cause activation of proto-oncogenes, hypermethylation induces silencing of cancer-related genes with tumor suppressive properties [18]. On the other hand, genome-wide hypomethylation may lead to genomic instability in repetitive sequences, especially at pericentromeric regions, predisposing to abnormal recombination, facilitating translocations, deletions, and chromosomal rearrangements [19–21].

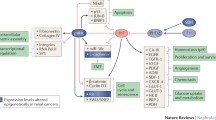
Two epigenetic pathways of transformation from normal cells to cancer cells. In normal cells, DNA is unmethylated in CpG islands, while in repeat sequences and CpG sparse regions, DNA is heavily methylated. When DNA is less methylated in repeat sequences and CpG sparse regions, cells are transformed (Cancer A with global hypomethylation). If DNA is heavily methylated in CpG islands, cells are also transformed (Cancer B with regional hypermethylation)
Renal cell carcinoma (RCC) is genetically and histopathologically a heterogeneous disorder. The most common subtype of RCC is clear cell RCC (ccRCC; approximately 75 %) and the next most frequent subtype is papillary RCC (pRCC; approximately 15 %) [22]. The most frequent genetic abnormality in ccRCC is inactivation of the von Hippel-Lindau (VHL) tumor suppressor gene [23] and promoter methylation of tumor suppressor geness (TSGs) is common in both subtypes of RCC. The VHL and p16 INK4a TSGs are inactivated by promoter hypermethylation in up to 20 % of clear cell [24] and 10 % of all RCC [25]. The RASSF1A and the Timp-3 genes are hypermethylated in 27–56 % [26] and 58–78 %, of primary RCCs respectively [26]. Table 4.1 provides an overview of the commonly methylated genes in renal cancer based on the published reports. A survey of published work in 2010 by Morris and Maher [36] has identified 58 genes that are methylated in RCC and 43 of these genes had a mean combined methylation/mutation rate of over 20 % (Ref. [36]). Cancer genome projects such as TCGA (http://cancergenome.nih.gov/l) and CAGEKID (http://www.icgc.org/icgc/cgp/65/812/817) have elected to define the mutational status and methylation profile of RCC. Hence large amount of data will be available to identify frequently methylated genes in RCC in the near future. Our group has also reported that various genes such as DNA mismatch repair genes [37], E-cadherin [38], gamma-catenin [39] and BTG3 [40] are silenced through promoter hypermethylation in renal cancer. We have also published extensively on the promoter methylation status of genes involved in the Wnt signaling pathway in renal cancer. Oncogenic activation of the Wnt pathway drives expression of genes that contribute to proliferation, survival and invasion. Inhibitors of this pathway can be divided into two functional classes, sFRP proteins that bind directly to Wnt and prevent its binding to frizzled receptor and the Dickkopt (DKK) proteins which bind to LRP component of the Wnt receptor complex. The sFRP-1, sFRP-2, sFRP-4, sFRP-5 and related Wif genes are all frequently methylated in RCC [41–44], as are the DKK genes [45, 46]. An interesting finding by our group is that sFRP1 is unmethylated/hypomethylated and thus over-expressed in metastatic renal tumors [47] compared to primary tumors where in its expression is attenuated by promoter hypermethylation [41]. Another study from our group by Yamamura et al. [48] challenged the Wnt inhibitory role of sFRP2 and reported that overexpression of sFRP2 activates the canonical Wnt pathway, promoting cell growth through diverse signaling cascades in renal cancer cells [48].
4.3 Chromatin Remodeling and Histone Modifications
The coiling of DNA around nucleosome particles is the basis for organization of eukaryotic genomes. Each nucleosome encompasses ~147 bp of DNA wrapped around an octamer of histone proteins. The core histones H2A, H2B, H3 and H4 bind together (two H2A-H2B dimers and one H3-H4 tetramer) to form the nucleosome. The core histones are small basic proteins containing a globular domain and a flexible charged NH2 terminus known as the histone tail [49]. Regulation of gene expression occurs through posttranslational covalent modifications of the histone tails including acetylation, methylation, phosphorylation, ubiquitination, sumoylation, proline isomerization, and ADP ribosylation [49, 50]. Generally certain histone modifications such as acetylation or phosphorylation are thought to change chromatin structure by altering the net positive charge of the histone proteins, thereby making the underlying DNA sequence accessible [51]. Alternatively, histone modifications can be recognized by specific protein domains (e.g., bromodomains, Tudor domains, chromodomains), which in turn might enforce or stabilize the recruitment of additional factors [52, 53]. Posttranslational modifications to histone tails govern the structural status of chromatin and the resulting transcriptional status of genes within a particular locus. These modifications are reversible and controlled by a group of enzymes including histone acetyltransferases (HATs) and deacetylases (HDACs), methyltransferases (HMTs), demethylases (HDMs), kinases, phosphatases, ubiquitin ligases and deubiquitinases, SUMO ligases and proteases which add and remove these modifications [8, 49]. In relation to transcriptional state, the human genome can be roughly divided into two distinct chromatin conformation states: euchromatin, which has an open structure and is transcriptionally active and heterochromatin, which is densely compacted and transcriptionally inert [54]. Euchromatin is characterized by high levels of acetylation and trimethylated H3K4, H3K36 and H3K79. In contrast heterochromatin is characterized by low levels of acetylation and high levels of H3K9, H3K27 and H4K20 methylation [8, 54]. The notion of heterochromatin as transcriptionally inactive has been challenged by the discovery of numerous noncoding RNAs (ncRNAs) derived from heterochromatic loci [55]. Well-known examples of this phenomenon in humans are the ncRNAs XIST and HOTAIR [56, 57]. Histone modifications are predictive for gene expression as actively transcribed genes are characterized by enriched levels of H3K4me3, H3K27ac, H2BK5-azacytidine (H2BK5ac) and H4K20me1 in the promoter and H3K79me1 and H4K20me1 along the gene [58]. Therefore histone modifications influence chromatin structure which plays an important role in gene regulation and carcinogenesis (Fig. 4.2).

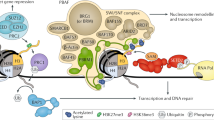
DNA methylation status in the promoter and the related chromatin structure. In normal cells, the CpG sites adjacent to transcription start site are unmethylated. The transcriptional machinery is activated by the binding of transcriptional factor (TFs) and co-acting factors (CAs) in this region. The gene promoter shown on the upper left is transcriptionally active. In upstream and downstream regions, DNA is methylated by DNA methyltransferases (DNMTs). In these regions, methylcytosine-binding proteins (MBPs) that bind to methylated CpG sites recruit histone deacetylases (HDACs) and histone methyltransferases to form a complex. Left bottom shows the related chromatin structure around the transcriptionally active, unmethylated promoter. The lysine residues in the tails of histone H3 are acetylated (acK). Lysine 4 is methylated (mK4) and lysine 9 is unmethylated (K9). These changes contribute to open and relaxed conformation of the chromatin allowing key components of the transcription apparatus accessible to the promoter. In the upstream and downstream regions, the lysine residues are deacetylated (K), demethylated (K4) and methylated (mK9) respectively and the chromatin structure have closed and dense conformation. In cancer cells, shown in the upper right, DNA methylation spreads toward the promoter regions near the transcriptional start site, resulting in transcriptional silencing. These events result in closed and dense chromatin conformation making it difficult for the key components of gene transcription apparatus to bind to the promoter
Genome-wide studies have revealed that various combinations of histone modifications in a specific genomic region can lead to a more ‘open’ or ‘closed’ chromatin structure resulting in the activation or repression of gene expression. Disruption of normal patterns of histone modifications is a hallmark of cancer [12, 59]. One of the most characteristic examples is the global reduction of H4K20 trimethylation (H4K20me3) and H4K16 acetylation (H4K16Ac), along with DNA hypomethylation, at repeat sequences in many primary tumors [12]. Furthermore, genes encoding for histone-modifying enzymes have been also reported to be mutated in ccRCC [60]. Mutated genes have been implicated in chromatin regulation through nucleosome repositioning and histone tail modification. PBRM1, which was found to be mutated in nearly 40 % of human RCCs [61, 62], is a component of the Polybromo BRG1-associated factor complex (PBAF, SWI/SNF-B). PBAF, like SWI/SNF, functions as a nucleosome remodeler and was shown to be involved in transcriptional regulation (24–26). Less common mutations were also identified in two methyltransferases, SETD2 and MLL2, and two demethylases, UTX (KDM6A) and JARID1C [KDM5C Ref. [60]]. Deletion of chromosome 3p is a common finding in ccRCC associated with the loss of VHL at 3p25 and can also affect SETD2 and PBRM1, which are located at 3p21 [63]. SETD2 mediates the trimethylation of H3K36 [64], a histone mark that is placed during transcription and may be important for maintaining faithful transcription [65], whereas MLL2 mediates H3K4me3, a mark associated with active transcription. UTX demethylates H3K27me3 [66, 67], a histone mark associated with repressed chromatin. Of interest, UTX associates with MLL2 [68], suggesting that demethylation of repressive modification is associated with transcriptional activation. The hypoxia response pathway has been shown to have a direct effect on histone modification. HIF upregulation is a feature of ccRCC and it was shown to activate several chromatin demethylases, including JMJD1A (KDM3A), JMJD2B (KDM4B), JMJD2C (KDM4C), and JARID1B (KDM5B), all of which are directly targeted by HIF [69–71]. Indeed, both JMJD1A and JMJD2B were found to be elevated in a RCC cell line with loss of VHL function [70], and the expression of JMJD1A was reported to be higher in RCC cancer tissue surrounding blood vessels, suggesting that JMJD1A is involved in tumor angiogenesis [72]. Reexpression of VHL in VHL-deficient cell lines increased H3K4me3 levels associated with decreasing levels of JARID1C, a target of HIF2a [62]. Silencing of JARID1C in VHL-deficient tumor cells augmented tumor growth in a xenograft mouse model, suggesting that JARID1C acts as a tumor suppressor. In contrast, hypoxia may increase methylation through HIF-independent mechanisms. Like HIF prolyl hydroxylase (PHD, EGLN3), histone demethylases are members of the dioxygenase superfamily, which requires oxygen as well as iron and 2-oxoglutarate for activity [73, 74]. In a manner analogous to stabilization of HIF via decreased hydroxylation, hypoxia was shown to suppress JARID1A (KDM5A) activity, resulting in increased H3K4me3 levels [75]. This suggests the hypothesis that loss of demethylases (and, by analogy, increased histone methylation) is part of a hypoxia phenotype that is selected for in RCC. This hypoxia phenotype, which is mimicked by VHL loss, would also be mimicked by loss of histone demethylase activity, which is a high-frequency event in RCC. Chromatin organization also influences HIF function. Studies of HIF induced under conditions of hypoxia showed preferential targeting of HIF to previously nucleosome depleted chromatin regions [76]. Moreover, the coexpression of SWI/SNF components BRG1, BAF170, and BAF57 augmented HIF activity from an HIF responsive reporter [77]. The extent to which mutations of epigenetic regulators influence chromatin or HIF targeting remains unknown.
4.4 MicroRNAs
MicroRNAs are small, non-protein-coding sequences thought to regulate >90 % of human genes by targeted repression of gene transcription and translation [78]. These endogenous, silencing RNAs have been shown to play important roles in development and differentiation [79, 80], cellular stress responses [81], and growing evidence has strongly implicated the involvement of miRNAs in carcinogenesis [82–84]. Specific subsets of miRNAs have also been shown to be dysregulated in various solid tumors [85, 86]. Due to their tremendous regulatory potential and tissue-specific and disease-specific expression patterns [87, 88], there is increasing evidence that miRNA expression profiles could be indicative of disease risk.
DNA hypermethylation of CpG sites within CpG islands is known to lead to the inactivation of many tumor-suppressive miRNAs [89–91]. One of the most common causes of tumor-suppressor miRNA loss is silencing of their primary transcripts by CpG-island hypermethylation [92–96]. The DNA methylation profile of tumors is useful to define tumor type, clinical prognosis and treatment response [19, 20]. Epigenetic silencing of miRNAs is also involved in the acquisition of an invasive phenotype and the development of metastasis [93]. Dysregulation of miRNA expression seems to be pivotal for RCC development and progression. Table 4.2 presents a list of miRNAs that are dysregulated in RCC. Depletion of tumor suppressor genes or upregulation of oncogenes has also been correlated with dysregulated expression of miRNAs in RCC. Our group has reported that several tumor suppressor miRNAs such as miR-1826 [102], miR-708 [100], miR-205 [104], miR-584 [105] are attenuated in RCC, where as oncogenic miR-21 was overexpressed [108]. There are controversial reports about the status of miRNA-34a in RCC. One study reported that inactivation of miR-34a correlates with its methylation status as they found methylation frequency of 58 % in RCC [34]. “Whereas in contrast, Liu et al. [109] reported increased levels of mir-34a caused loss of function of tumor suppressor SFRP1 [which again is a controversial tumor suppressor in RCC [47]] indicating its oncogenic potential” [109]. However no functional analysis was performed in either study. A recent study by our group investigated the functional effects of miR-34a in RCC [103]. It reported that overexpression of miR-34a inhibited cell invasion and suppressed the assembly and function of the c-Myc complex that activates or elongates transcription, indicating a tumor suppressor role in RCC [103]. Matching patterns between deregulated miRNAs and chromosomal aberrations have been reported in ccRCC [110]. On the other hand, miRNA deregulation might serve as an alternative mechanism for gene expression alterations due to chromosomal aberrations. This is well illustrated by the miR-204/211 family. Gain of chromosome 3q is a common finding in papillaryRCC that leads to upregulation of several genes including C3orf58, CCDC50, DTX3L, PLD1, TRIM59, ECT2, RAP2B, and SERP1 that are targeted by miR-204/211 [Ref. [109]], whereas in ccRCC, miR-204/211 downregulation might be the mechanism causing upregulation of the same set of genes, since 3q gain is rare in ccRCC [109].
4.5 Interplay Between Epigenetic Factors
There is interplay between histone modifications and DNA methylation and the best example is the relationship between DNMT3L and H3K4. DNMT3L specifically interacts with histone H3 tails, inducing de novo DNA methylation by recruitment of DNMT3A, however this interaction is strongly inhibited by H3K4me. Furthermore, several histone methyltransferases have also been reported to direct DNA methylation to specific genomic targets by recruiting DNMTs [111, 112], helping in this way to set the silenced state established by the repressive histone marks. Moreover, histone methyltransferases and demethylases can also modulate the stability of DNMT proteins, thereby regulating DNA methylation levels [113, 114]. On the other hand, DNA methylation can also direct histone modifications. For instance, methylated DNA mediates H3K9me through MeCP2 recruitment [115]. MicroRNAs are also known to target the components of epigenetic machinery such as DNMTs, HDACs and polycomb genes [116]. Whereas, miRNAs may be affected by epigenetic changes, such as methylation of the CGIs and accompanying changes in histone modifications. miR-127 has been found to be attenuated in cancer cells by promoter hypermethylation and by a decrease in acetyl-H3 and methyl-H3K4 [95]. Genome-wide analysis of different cancer types has shown that global expression of miRNAs is influenced by DNA methylation and histone modifications [117].
4.6 Epigenetic and miRNA Biomarkers
Methylated DNA sequences provide attractive options for biomarkers for cancer detection and prognosis including RCC [118]. The last decade has provided an extensive map of the aberrant DNA methylation events occurring in cancer cells, particularly for the hypermethylated CpG islands of tumor suppressor genes (TSG) [19]. Consequently a myriad of DNA methylation-based biomarkers of many types of human neoplasias have been reported. Different RCC subtypes seem to display different gene sets deregulated by promoter hypermethylation [26], and a gene panel (CDH1, PTGS2, and RASSF2) identifying most frequent RCC subtypes in tissue samples has been evaluated [119]. The epigenomic data have helped highlight the unique profile of aberrant DNA methylation that defines each tumor type [120]. Epigenetic biomarkers are of particular interest as non-invasive biomarkers since methylated DNA can be detected from tumor cells sloughed into urine or blood. This has been shown with a three-gene panel (APC, RARβ2, RASSF1A) which detected RCC with high specificity and sensitivity [41, 121]. Moreover, RASSF1A promoter methylation might also prove useful for tumor surveillance/monitoring of RCC cancer patients [122]. Methylation of the Wnt pathway genes SFRP1, SFRP2, SFRP4, SFRP5, DKK3 and WIF1 have been detected in the serum of patients with corresponding tumor methylation and the frequency of methylation in serum correlated with increased grade and stage [41]. Therefore the detection of RCC-associated TSG methylation by analysis of serum or urine samples could have potential for early detection of RCC and for distinguishing benign and malignant renal cancers. Promoter hypermethylation of some genes has been associated with clinical and pathological features of tumor aggressiveness and also with prognostic relevance. Aberrant promoter methylation of APAF1, DAPK1 and GREM1 [123] has been associated with aggressive forms of RCC. Moreover, promoter methylation of APAF1, DAPK1 [124], JUP [39], PTEN [125], UCHL1 [126], DAL1-4.1B/EPB41L3 [127] BNC1 and COL14A1 [128] have been associated with poorer survival, and most of them (JUP, APAF1, DAPK1, PTEN, DAL1-4.1B, BNC1, and COL14A1) retained independent prognostic value in multivariate analysis [39, 124, 128]. Clearly it is important that there should be additional studies of potential methylated biomarkers in tumor tissues and urine and/or blood with the ultimate aim of producing a panel of biomarkers that will enable non-invasive detection, molecular staging and prediction of prognosis. As the number of potential methylated TSG biomarkers increases, it will be of great importance to assay these in a standardized manner in prospective studies to establish their clinical utility.
Genome-wide studies of histone modifications have been performed to characterize the chromatin of malignant cells by establishing the overall profile of histone modifications in cancer cells. Signatures of histone modifications patterns, such as trimethyl-H3K9, are associated with patient prognosis in acute myeloid leukemia [129]. Silencing of genes marked by trimethyl-H3K27 in the absence of DNA methylation has also been reported [130]. Several histone modifications have been associated with poor prognosis in RCC, including low H3K4me2, H3K18ac, and H3K9me2 [131]. H3K4me1–3 levels were also found to be inversely correlated with Fuhrman grade, stage, lymph node involvement and distant metastases, and an H3K4me score was an independent factor for RCC progression free survival [132]. Similar observations have been made for global H3Ac and H4Ac levels, as well as for H3K9Ac levels in RCCs treated with partial nephrectomy [133], whereas H3K18Ac levels were an independent predictor of RCC progression after surgery [134].
The use of genome wide approaches has enabled the production of miRNA fingerprints in a range of tumors and the identification of new potential biomarkers to distinguish tumor tissue from its normal counterpart. From a clinical point of view, miRNAs have great potential as diagnostic and therapeutic agents. Owing to the tissue specificity of miRNAs, they have become a useful tool for defining the origin of tumors in poorly differentiated cancers [135]. Prognosis and survival of patients depends on the cancer stage at diagnosis and miRNA signatures have been reported to be useful tools for early diagnosis of cancer [136, 137]. Differential miRNA expression patterns between neoplastic and non-neoplastic renal tissues, as well as among different renal tumor subtypes have been described. Discrimination between ccRCC and normal kidney tissue have been described with a panel of nine miRs (miR-21, miR-34a, miR-142-3p, miR-155, miR-185, miR-200c, miR-210, miR-224, and miR-592) [Ref. [138]], a combination of miR-141 and miR-155 [139] or by differential expression of miR-92a, miR-210, and miR-200c [140]. For a more clinical perspective with the aim of supporting diagnosis, a stepwise decision tree was created to differentiate between kidney cancer subtypes and oncocytoma, depending on miRNA signatures. This method is valuable in small biopsy samples and in cases where morphological assessment is not sufficient for diagnosis [141]. Unsupervised hierarchical cluster analysis of miRNA microarray data showed that tumors derived from the proximal and distal nephrons can be distinguished by their miRNA profile [140]. The differential expression patterns of miRNAs can also be used to subclassify renal cancer. In ccRCC 23 miRNAs are differentially expressed (let-7e, let-7f, let-7g, miR10b, miR-124, miR-126, miR-138, miR-140-5p, miR-142-5p, miR-144, miR-184, miR-200c, miR-203, miR-206, miR-210, miR-218, miR-27a, miR-27b, miR-335, miR-373, miR-378, miR-92a, miR-98. However, some miRNAs are characteristic of sporadic ccRCC (let-7c, let-7d, miR-1, miR-100, miR-10a, miR-148b, miR-191, miR-199a-3p, miR-19a, miR-215, miR-29b, miR-30c, miR-363, miR-9) and others of hereditary RCC (let-7a, miR-125a-5p, miR-125b, miR-143, miR-146b-5p, miR-15b, miR-17, miR-193a-5p, miR-193b, miR-196a, miR-20b, miR-214, miR-23b, miR-32, miR-372) [61]. miRNA levels in sera of RCC patients and healthy controls, identified miR-1233 as a promising biomarker for RCC detection and monitoring [142]. Altered levels of miRNA might also provide prognostic information. Whereas miR-155 and miR-21 expression in ccRCC tumors has been found to correlate with tumor size [143], higher miR-210 levels were found in tumors displaying higher Fuhrman grade [140]. In ccRCC, overexpression of miR-32, miR-210, miR-21, and miR-18a correlated with poor survival [143, 144]. Lower miR-106b levels were associated with metastatic disease and poorer relapse-free survival [145]. High miR-210 expression was also found in tumors with lymph node metastasis [140], suggesting unique miRNA signatures in metastatic RCC, distinct from those of primary tumors [146]. Khalla et al. [147] compared distant metastases with primary tumors and found a distinct miRNA signature in metastases. Some of the primary tumor samples clustered together with the distant metastasis, suggesting that these primary tumors have a metastasis-specific signature [147]. Because miRNAs can be easily detected and quantified in blood, serum assays based on metastasis-associated miRNAs may be of value. In addition, Lin et al. [148] identified 12 SNPs in miRNA-related genes that are significantly associated with recurrence or survival and found a cumulative effect of multiple SNPs with recurrence. Taken together, additional studies in large patient cohorts are necessary to validate the potential use of miRNAs as diagnostic/prognostic biomarkers.
4.7 Epigenetics as Consequence or Cause of Cancer Initiation
Cancers are caused by accumulative mutations in the genes [149]. Mutations cause rearrangements of large chromosomal regions, which confer the cells with growth advantage under selection pressure due to abnormal expression of oncogenes [149, 150]. The clonal expansion of the mutated cells leads to genomic instability and global demethylation, while the cell machinery progressively shuts down the anti-survival genes by hypermethylation. Thus mutations cause genomic instability, which precedes methylation changes. By contrast, congenital disorders such as ICF syndrome and Rett syndrome involve genes that encode the methylation machinery of the cell such as DNMT3B (ICF syndrome) and MECP2 (Rett syndrome), but these disorders do not predispose to cancer. Thus, epigenetic changes were thought to be a consequence of altered gene expression rather than causal [151]. Further, activation of tumor suppressor genes by 5-aza-2′-deoxycytidine or DNMT1 knockout may not be stable, as has been shown for both MLH1 [152] and p16 [153], suggesting that the altered methylation might be a consequence rather than a cause of gene silencing. Thus a key barrier to the acceptance of epigenetic alterations as a cause rather than a consequence of cancer has been the lack of well-defined human pre-neoplastic disorders that are caused by epigenetic mutations. However the discovery of the mechanisms of Beckwith-Wiedemann syndrome (BWS) provides a good example of constitutional epigenetic alterations linked to cancer risk. BWS was shown to have various molecular causes, including loss of imprinting (LOI) of IGF2 [154] or point mutations in the CDKNIC [155] gene or epigenetic lesions in the nearby antisense RNA LIT1. Furthermore, cancer predisposition might be specifically associated with LOI of IGF2 and hypermethylation of H19 [156]. In a large registry of patients with BWS gain of methylation at H19, presumably resulting in biallelic expression of IGF2, was found to be specifically and statistically associated with cancer risk [157]. BWS leads to an 800 fold increased risk of embryonal tumors such as Wilm’s tumor of the kidney and rhabdomyosarcoma [158]. LOI of IGF2 is specifically associated with increased cancer risk in children with BWS. Thus the epigenetic change precedes cancer and confers risk for cancer, a strong argument for causality. Another study showed that aberrant changes in the epigenome could indeed lead to cancers that do not display genomic instability [159]. Snf5 is a tumor suppressor gene and a core component of the chromatin remodeling complex SWI/SNF whose inactivation is detected in several types of tumors [160, 161], including the highly invasive malignant rhabdoid tumors (MRTs) [162]. Differing from most other tumors where the chromosomes are usually fragmented, MRTs often display an intact genome. The authors generated Snf5-deficient primary mouse embryonic fibroblasts and showed that tumors derived from these cells were diploid and the cancer phenotype was correlated with the expression of the cell cycle protein cyclin D1, which was epigenetically upregulated by SWI/SNF complexes [159]. An alternative approach to study the relationship between epigenetic changes and transformation is to study the epigenome of pre-cancerous cells. A series of studies on colon cancers found that global hypomethylation as well as regional gene promoter hypermethylation occur in pre-cancerous lesions or even benign colon polyps before they become malignant colon cancers [163–165]. Similar findings have been observed in breast cancers, where normal tissues surrounding the tumors have been detected with aberrant DNA methylation patterns [166, 167]. These observations of methylation patterns change in pre-cancerous cells suggest that the loss in methylation can be an early event that precedes malignancy. Experimental data in mice also support a causal role for epigenetic changes in cancer. When DNMT1 hypomorphs are crossed with Min (multiple intestinal neoplasia) mice with an Apc mutation, they show an increased frequency of intestinal neoplasia and liver cancers [168]. In addition, it has also been shown that global hypomethylation leads to elevated mutation rates [169], suggesting that epigenetic changes may initiate downstream oncogenetic pathways. Studying these model systems may therefore aid our understanding of how epigenetic processes contribute to the process of oncogenic malignancy.
4.8 Epigenetic Therapy
Given that epigenetic modifications are reversible, it seems likely that understanding and manipulating the epigenome may hold promise for preventing and treating common human diseases including cancer. Much attention has been focused on the quest for epigenetic drugs, which restore the normal epigenetic landscape in cancer cells by inhibiting enzymes of the epigenetic machineries. Understanding the mechanisms underlying the tumor suppressor gene silencing in cancer has promoted the idea of pharmacologically relieving the inhibitory effects of DNA methylation and chromatin remodeling on gene expression. Identification of frequently methylated RCC tumor suppressor genes has highlighted potential targets for therapeutic intervention. Decitabine, the clinical form of the demethylating agent 5-aza-2′-deoxycytidine, has been used in several clinical trials, and promising responses have been reported for hematological malignancies such as myelodysplastic syndrome [170, 171]. Various studies have tested DNMT inhibitors or HDAC inhibitors either alone or in combination with conventional chemotherapeutic agents in RCC cell lines with promising results [172–174] but clinical studies are required to conclusively demonstrate the therapeutic usefulness in RCC.
4.9 Conclusions
Understanding the complexity of the epigenome and all the actors involved in modulating its interactions with genomic sequences is of fundamental importance in health and disease. Owing to the reversible and plastic nature of epigenetic alterations, these constitute an attractive target for novel therapeutic intervention. Studying epigenomic alterations and miRNAs provide opportunities for the development of innovative biomarkers to aid in disease detection, diagnosis, prognosis and prediction of response to therapy. Understanding the complex molecular mechanisms involved in epigenetics and miRNAs, may lead to more effective cancer treatments and promote the change from current cytotoxic therapies to more targeted control of malignant phenotypes.
References
Waddington CH (2012) The epigenotype. 1942. Int J Epidemiol 41(1):10–13
Russo VEA, Martienssen RA, Riggs AD (eds) (1996) Epigenetic mechanisms of gene regulation. Cold Spring Harbor Laboratory Press, Woodbury
Sandoval J, Esteller M (2012) Cancer epigenomics: beyond genomics. Curr Opin Genet Dev 22(1):50–55
Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9(6):465–476
Bird A (2007) Perceptions of epigenetics. Nature 447(7143):396–398
Murrell A, Rakyan VK, Beck S (2005) From genome to epigenome. Hum Mol Genet 14(Spec No 1):R3–R10
Zhang TY, Meaney MJ (2010) Epigenetics and the environmental regulation of the genome and its function. Annu Rev Psychol 61(439–66):C1–C3
Portela A, Esteller M (2010) Epigenetic modifications and human disease. Nat Biotechnol 28(10):1057–1068
Jones PA, Baylin SB (2007) The epigenomics of cancer. Cell 128(4):683–692
Bird AP, Wolffe AP (1999) Methylation-induced repression–belts, braces, and chromatin. Cell 99(5):451–454
Illingworth RS, Bird AP (2009) CpG islands – ‘a rough guide’. FEBS Lett 583(11):1713–1720
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G et al (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37(4):391–400
Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R et al (2009) Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet 41(12):1350–1353
Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P et al (2009) The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41(2):178–186
Ji H, Ehrlich LI, Seita J, Murakami P, Doi A, Lindau P et al (2010) Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 467(7313):338–342
Feinberg AP, Vogelstein B (1983) Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301(5895):89–92
Feinberg AP (2005) Cancer epigenetics is no Mickey Mouse. Cancer Cell 8(4):267–268
Feinberg AP, Ohlsson R, Henikoff S (2006) The epigenetic progenitor origin of human cancer. Nat Rev Genet 7(1):21–33
Esteller M (2008) Epigenetics in cancer. N Engl J Med 358(11):1148–1159
Rodriguez-Paredes M, Esteller M (2011) Cancer epigenetics reaches mainstream oncology. Nat Med 17(3):330–339
Eden A, Gaudet F, Waghmare A, Jaenisch R (2003) Chromosomal instability and tumors promoted by DNA hypomethylation. Science 300(5618):455
Mancini V, Battaglia M, Ditonno P, Palazzo S, Lastilla G, Montironi R et al (2008) Current insights in renal cell cancer pathology. Urol Oncol 26(3):225–238
Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML et al (1993) Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 260(5112):1317–1320
Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S et al (1994) Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci U S A 91(21):9700–9704
Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE et al (1995) Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res 55(20):4525–4530
Dulaimi E, Ibanez de Caceres I, Uzzo RG, Al-Saleem T, Greenberg RE, Polascik TJ et al (2004) Promoter hypermethylation profile of kidney cancer. Clin Cancer Res 10(12 Pt 1):3972–3979
Kondratov AG, Kvasha SM, Stoliar LA, Romanenko AM, Zgonnyk YM, Gordiyuk VV et al (2012) Alterations of the WNT7A Gene in Clear Cell Renal Cell Carcinomas. PLoS One 7(10):e47012
Ye YW, Jiang ZM, Li WH, Li ZS, Han YH, Sun L et al (2012) Down-regulation of TCF21 is associated with poor survival in clear cell renal cell carcinoma. Neoplasma 59(6):599–605
Ricketts CJ, Morris MR, Gentle D, Brown M, Wake N, Woodward ER et al (2012) Genome-wide CpG island methylation analysis implicates novel genes in the pathogenesis of renal cell carcinoma. Epigenetics 7(3):278–290
Peters I, Eggers H, Atschekzei F, Hennenlotter J, Waalkes S, Trankenschuh W et al (2012) GATA5 CpG island methylation in renal cell cancer: a potential biomarker for metastasis and disease progression. BJU Int 110(2 Pt 2):E144–E152
Kim WJ, Gersey Z, Daaka Y (2012) Rap1GAP regulates renal cell carcinoma invasion. Cancer Lett 320(1):65–71
Morris MR, Ricketts CJ, Gentle D, McRonald F, Carli N, Khalili H et al (2011) Genome-wide methylation analysis identifies epigenetically inactivated candidate tumour suppressor genes in renal cell carcinoma. Oncogene 30(12):1390–1401
Zhang Q, Ying J, Li J, Fan Y, Poon FF, Ng KM et al (2010) Aberrant promoter methylation of DLEC1, a critical 3p22 tumor suppressor for renal cell carcinoma, is associated with more advanced tumor stage. J Urol 184(2):731–737
Vogt M, Munding J, Gruner M, Liffers ST, Verdoodt B, Hauk J et al (2011) Frequent concomitant inactivation of miR-34a and miR-34b/c by CpG methylation in colorectal, pancreatic, mammary, ovarian, urothelial, and renal cell carcinomas and soft tissue sarcomas. Virchows Arch 458(3):313–322
Hildebrandt MA, Gu J, Lin J, Ye Y, Tan W, Tamboli P et al (2010) Hsa-miR-9 methylation status is associated with cancer development and metastatic recurrence in patients with clear cell renal cell carcinoma. Oncogene 29(42):5724–5728
Morris MR, Maher ER (2010) Epigenetics of renal cell carcinoma: the path towards new diagnostics and therapeutics. Genome Med 2(9):59
Deguchi M, Shiina H, Igawa M, Kaneuchi M, Nakajima K, Dahiya R (2003) DNA mismatch repair genes in renal cell carcinoma. J Urol 169(6):2365–2371
Nojima D, Nakajima K, Li LC, Franks J, Ribeiro-Filho L, Ishii N et al (2001) CpG methylation of promoter region inactivates E-cadherin gene in renal cell carcinoma. Mol Carcinog 32(1):19–27
Breault JE, Shiina H, Igawa M, Ribeiro-Filho LA, Deguchi M, Enokida H et al (2005) Methylation of the gamma-catenin gene is associated with poor prognosis of renal cell carcinoma. Clin Cancer Res 11(2 Pt 1):557–564
Majid S, Dar AA, Ahmad AE, Hirata H, Kawakami K, Shahryari V et al (2009) BTG3 tumor suppressor gene promoter demethylation, histone modification and cell cycle arrest by genistein in renal cancer. Carcinogenesis 30(4):662–670
Urakami S, Shiina H, Enokida H, Hirata H, Kawamoto K, Kawakami T et al (2006) Wnt antagonist family genes as biomarkers for diagnosis, staging, and prognosis of renal cell carcinoma using tumor and serum DNA. Clin Cancer Res 12(23):6989–6997
Kawakami K, Hirata H, Yamamura S, Kikuno N, Saini S, Majid S et al (2009) Functional significance of Wnt inhibitory factor-1 gene in kidney cancer. Cancer Res 69(22):8603–8610
Kawakami K, Yamamura S, Hirata H, Ueno K, Saini S, Majid S et al (2010) Secreted frizzled-related protein-5 is epigenetically downregulated and functions as a tumor suppressor in kidney cancer. Int J Cancer 128(3):541–550
Kawamoto K, Hirata H, Kikuno N, Tanaka Y, Nakagawa M, Dahiya R (2008) DNA methylation and histone modifications cause silencing of Wnt antagonist gene in human renal cell carcinoma cell lines. Int J Cancer 123(3):535–542
Hirata H, Hinoda Y, Nakajima K, Kawamoto K, Kikuno N, Kawakami K et al (2009) Wnt antagonist gene DKK2 is epigenetically silenced and inhibits renal cancer progression through apoptotic and cell cycle pathways. Clin Cancer Res 15(18):5678–5687
Hirata H, Hinoda Y, Nakajima K, Kawamoto K, Kikuno N, Ueno K et al (2010) Wnt antagonist DKK1 acts as a tumor suppressor gene that induces apoptosis and inhibits proliferation in human renal cell carcinoma. Int J Cancer 128(8):1793–1803
Saini S, Liu J, Yamamura S, Majid S, Kawakami K, Hirata H et al (2009) Functional significance of secreted Frizzled-related protein 1 in metastatic renal cell carcinomas. Cancer Res 69(17):6815–6822
Yamamura S, Kawakami K, Hirata H, Ueno K, Saini S, Majid S et al (2010) Oncogenic functions of secreted Frizzled-related protein 2 in human renal cancer. Mol Cancer Ther 9(6):1680–1687
Kouzarides T (2007) Chromatin modifications and their function. Cell 128(4):693–705
Rando OJ, Chang HY (2009) Genome-wide views of chromatin structure. Annu Rev Biochem 78:245–271
Wolffe AP, Hayes JJ (1999) Chromatin disruption and modification. Nucleic Acids Res 27(3):711–720
Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ (2007) How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol 14(11):1025–1040
Gardner KE, Allis CD, Strahl BD (2011) Operating on chromatin, a colorful language where context matters. J Mol Biol 409(1):36–46
Li B, Carey M, Workman JL (2007) The role of chromatin during transcription. Cell 128(4):707–719
Zaratiegui M, Irvine DV, Martienssen RA (2007) Noncoding RNAs and gene silencing. Cell 128(4):763–776
Agrelo R, Wutz A (2010) X inactivation and disease. Semin Cell Dev Biol 21(2):194–200
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA et al (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129(7):1311–1323
Karlic R, Chung HR, Lasserre J, Vlahovicek K, Vingron M (2010) Histone modification levels are predictive for gene expression. Proc Natl Acad Sci U S A 107(7):2926–2931
Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M et al (2005) Global histone modification patterns predict risk of prostate cancer recurrence. Nature 435(7046):1262–1266
Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A et al (2010) Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 463(7279):360–363
Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P et al (2011) Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469(7331):539–542
Niu X, Zhang T, Liao L, Zhou L, Lindner DJ, Zhou M et al (2012) The von Hippel-Lindau tumor suppressor protein regulates gene expression and tumor growth through histone demethylase JARID1C. Oncogene 31(6):776–786
Duns G, van den Berg E, van Duivenbode I, Osinga J, Hollema H, Hofstra RM et al (2010) Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res 70(11):4287–4291
Sun XJ, Wei J, Wu XY, Hu M, Wang L, Wang HH et al (2005) Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J Biol Chem 280(42):35261–35271
Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK et al (2005) Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123(4):581–592
Hong S, Cho YW, Yu LR, Yu H, Veenstra TD, Ge K (2007) Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci U S A 104(47):18439–18444
Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J et al (2007) UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449(7163):731–734
Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D et al (2007) Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318(5849):447–450
Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS et al (2009) Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc Natl Acad Sci U S A 106(11):4260–4265
Beyer S, Kristensen MM, Jensen KS, Johansen JV, Staller P (2008) The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J Biol Chem 283(52):36542–36552
Krieg AJ, Rankin EB, Chan D, Razorenova O, Fernandez S, Giaccia AJ (2010) Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol Cell Biol 30(1):344–353
Guo X, Shi M, Sun L, Wang Y, Gui Y, Cai Z et al (2011) The expression of histone demethylase JMJD1A in renal cell carcinoma. Neoplasma 58(2):153–157
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M et al (2001) HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292(5516):464–468
Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ et al (2001) Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292(5516):468–472
Zhou X, Sun H, Chen H, Zavadil J, Kluz T, Arita A et al (2010) Hypoxia induces trimethylated H3 lysine 4 by inhibition of JARID1A demethylase. Cancer Res 70(10):4214–4221
Xia X, Kung AL (2009) Preferential binding of HIF-1 to transcriptionally active loci determines cell-type specific response to hypoxia. Genome Biol 10(10):R113
Kenneth NS, Mudie S, van Uden P, Rocha S (2009) SWI/SNF regulates the cellular response to hypoxia. J Biol Chem 284(7):4123–4131
Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM et al (2006) A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 126(6):1203–1217
Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV et al (2004) MicroRNA-143 regulates adipocyte differentiation. J Biol Chem 279(50):52361–52365
Hornstein E, Mansfield JH, Yekta S, Hu JK, Harfe BD, McManus MT et al (2005) The microRNA miR-196 acts upstream of Hoxb8 and Shh in limb development. Nature 438(7068):671–674
Leung AK, Sharp PA (2007) microRNAs: a safeguard against turmoil? Cell 130(4):581–585
Garzon R, Calin GA, Croce CM (2009) MicroRNAs in Cancer. Annu Rev Med 60:167–179
Shi XB, Tepper CG, White RW (2008) MicroRNAs and prostate cancer. J Cell Mol Med 12(5A):1456–1465
Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F et al (2006) A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 103(7):2257–2261
Michael MZ, O’Connor SM, van Holst Pellekaan NG, Young GP, James RJ (2003) Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res 1(12):882–891
Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M et al (2006) Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 9(3):189–198
Bargaje R, Hariharan M, Scaria V, Pillai B (2009) Consensus miRNA expression profiles derived from interplatform normalization of microarray data. RNA 16(1):16–25
Saunders MA, Lim LP (2009) (micro)Genomic medicine: microRNAs as therapeutics and biomarkers. RNA Biol 6(3):324–328
Chuang JC, Jones PA (2007) Epigenetics and microRNAs. Pediatr Res 61(5 Pt 2):24R–29R
Majid S, Dar AA, Saini S, Shahryari V, Arora S, Zaman MS et al (2012) MicroRNA-34b inhibits prostate cancer through demethylation, active chromatin modifications and AKT pathways. Clin Cancer Res. [Epub ahead of print]. PMID: 23147995
Majid S, Dar AA, Saini S, Arora S, Shahryari V, Zaman MS et al (2012) MicroRNA-23b represses proto-oncogene Src kinase, functions as a methylation-silenced tumor suppressor with diagnostic and prognostic significance in prostate cancer. Cancer Res. [Epub ahead of print]. PMID: 23074286
Huang YW, Liu JC, Deatherage DE, Luo J, Mutch DG, Goodfellow PJ et al (2009) Epigenetic repression of microRNA-129-2 leads to overexpression of SOX4 oncogene in endometrial cancer. Cancer Res 69(23):9038–9046
Lujambio A, Esteller M (2009) How epigenetics can explain human metastasis: a new role for microRNAs. Cell Cycle 8(3):377–382
Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setien F et al (2007) Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res 67(4):1424–1429
Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA et al (2006) Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 9(6):435–443
Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K, Shinomura Y et al (2008) Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res 68(11):4123–4132
Cui L, Zhou H, Zhao H, Zhou Y, Xu R, Xu X et al (2012) MicroRNA-99a induces G1-phase cell cycle arrest and suppresses tumorigenicity in renal cell carcinoma. BMC Cancer 12(1):546
Yamasaki T, Seki N, Yamada Y, Yoshino H, Hidaka H, Chiyomaru T et al (2012) Tumor suppressive microRNA138 contributes to cell migration and invasion through its targeting of vimentin in renal cell carcinoma. Int J Oncol 41(3):805–817
Mikhaylova O, Stratton Y, Hall D, Kellner E, Ehmer B, Drew AF et al (2012) VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell 21(4):532–546
Saini S, Yamamura S, Majid S, Shahryari V, Hirata H, Tanaka Y et al (2011) MicroRNA-708 induces apoptosis and suppresses tumorigenicity in renal cancer cells. Cancer Res 71(19):6208–6219
Kawakami K, Enokida H, Chiyomaru T, Tatarano S, Yoshino H, Kagara I et al (2012) The functional significance of miR-1 and miR-133a in renal cell carcinoma. Eur J Cancer 48(6):827–836
Hirata H, Hinoda Y, Ueno K, Nakajima K, Ishii N, Dahiya R (2012) MicroRNA-1826 directly targets beta-catenin (CTNNB1) and MEK1 (MAP2K1) in VHL-inactivated renal cancer. Carcinogenesis 33(3):501–508
Yamamura S, Saini S, Majid S, Hirata H, Ueno K, Chang I et al (2012) MicroRNA-34a suppresses malignant transformation by targeting c-Myc transcriptional complexes in human renal cell carcinoma. Carcinogenesis 33(2):294–300
Majid S, Saini S, Dar AA, Hirata H, Shahryari V, Tanaka Y et al (2011) MicroRNA-205 inhibits Src-mediated oncogenic pathways in renal cancer. Cancer Res 71(7):2611–2621
Ueno K, Hirata H, Shahryari V, Chen Y, Zaman MS, Singh K et al (2010) Tumour suppressor microRNA-584 directly targets oncogene Rock-1 and decreases invasion ability in human clear cell renal cell carcinoma. Br J Cancer 104(2):308–315
Liu W, Zabirnyk O, Wang H, Shiao YH, Nickerson ML, Khalil S et al (2010) miR-23b targets proline oxidase, a novel tumor suppressor protein in renal cancer. Oncogene 29(35):4914–4924
Dey N, Das F, Ghosh-Choudhury N, Mandal CC, Parekh DJ, Block K et al (2012) microRNA-21 governs TORC1 activation in renal cancer cell proliferation and invasion. PLoS One 7(6):e37366
Zaman MS, Shahryari V, Deng G, Thamminana S, Saini S, Majid S et al (2012) Up-regulation of microRNA-21 correlates with lower kidney cancer survival. PLoS One 7(2):e31060
Liu H, Brannon AR, Reddy AR, Alexe G, Seiler MW, Arreola A et al (2010) Identifying mRNA targets of microRNA dysregulated in cancer: with application to clear cell Renal Cell Carcinoma. BMC Syst Biol 4:51
Chow TF, Mankaruos M, Scorilas A, Youssef Y, Girgis A, Mossad S et al (2010) The miR-17-92 cluster is over expressed in and has an oncogenic effect on renal cell carcinoma. J Urol 183(2):743–751
Tachibana M, Matsumura Y, Fukuda M, Kimura H, Shinkai Y (2008) G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J 27(20):2681–2690
Zhao Q, Rank G, Tan YT, Li H, Moritz RL, Simpson RJ et al (2009) PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol 16(3):304–311
Esteve PO, Chin HG, Benner J, Feehery GR, Samaranayake M, Horwitz GA et al (2009) Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proc Natl Acad Sci U S A 106(13):5076–5081
Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J et al (2009) The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet 41(1):125–129
Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T (2003) The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem 278(6):4035–4040
Valeri N, Vannini I, Fanini F, Calore F, Adair B, Fabbri M (2009) Epigenetics, miRNAs, and human cancer: a new chapter in human gene regulation. Mamm Genome 20(9–10):573–580
Choudhry H, Catto JW (2011) Epigenetic regulation of microRNA expression in cancer. Methods Mol Biol 676:165–184
Cairns P (2007) Gene methylation and early detection of genitourinary cancer: the road ahead. Nat Rev Cancer 7(7):531–543
Costa VL, Henrique R, Ribeiro FR, Pinto M, Oliveira J, Lobo F et al (2007) Quantitative promoter methylation analysis of multiple cancer-related genes in renal cell tumors. BMC Cancer 7:133
Fernandez AF, Assenov Y, Martin-Subero JI, Balint B, Siebert R, Taniguchi H et al (2012) A DNA methylation fingerprint of 1628 human samples. Genome Res 22(2):407–419
Hoque MO, Begum S, Topaloglu O, Jeronimo C, Mambo E, Westra WH et al (2004) Quantitative detection of promoter hypermethylation of multiple genes in the tumor, urine, and serum DNA of patients with renal cancer. Cancer Res 64(15):5511–5517
Peters I, Rehmet K, Wilke N, Kuczyk MA, Hennenlotter J, Eilers T et al (2007) RASSF1A promoter methylation and expression analysis in normal and neoplastic kidney indicates a role in early tumorigenesis. Mol Cancer 6:49
van Vlodrop IJ, Baldewijns MM, Smits KM, Schouten LJ, van Neste L, van Criekinge W et al (2010) Prognostic significance of Gremlin1 (GREM1) promoter CpG island hypermethylation in clear cell renal cell carcinoma. Am J Pathol 176(2):575–584
Christoph F, Kempkensteffen C, Weikert S, Kollermann J, Krause H, Miller K et al (2006) Methylation of tumour suppressor genes APAF-1 and DAPK-1 and in vitro effects of demethylating agents in bladder and kidney cancer. Br J Cancer 95(12):1701–1707
Kim HL, Seligson D, Liu X, Janzen N, Bui MH, Yu H et al (2005) Using tumor markers to predict the survival of patients with metastatic renal cell carcinoma. J Urol 173(5):1496–1501
Kagara I, Enokida H, Kawakami K, Matsuda R, Toki K, Nishimura H et al (2008) CpG hypermethylation of the UCHL1 gene promoter is associated with pathogenesis and poor prognosis in renal cell carcinoma. J Urol 180(1):343–351
Yamada D, Kikuchi S, Williams YN, Sakurai-Yageta M, Masuda M, Maruyama T et al (2006) Promoter hypermethylation of the potential tumor suppressor DAL-1/4.1B gene in renal clear cell carcinoma. Int J Cancer 118(4):916–923
Morris MR, Ricketts C, Gentle D, Abdulrahman M, Clarke N, Brown M et al (2010) Identification of candidate tumour suppressor genes frequently methylated in renal cell carcinoma. Oncogene 29(14):2104–2117
Muller-Tidow C, Klein HU, Hascher A, Isken F, Tickenbrock L, Thoennissen N et al (2010) Profiling of histone H3 lysine 9 trimethylation levels predicts transcription factor activity and survival in acute myeloid leukemia. Blood 116(18):3564–3571
Kondo Y, Shen L, Cheng AS, Ahmed S, Boumber Y, Charo C et al (2008) Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet 40(6):741–750
Seligson DB, Horvath S, McBrian MA, Mah V, Yu H, Tze S et al (2009) Global levels of histone modifications predict prognosis in different cancers. Am J Pathol 174(5):1619–1628
Ellinger J, Kahl P, Mertens C, Rogenhofer S, Hauser S, Hartmann W et al (2010) Prognostic relevance of global histone H3 lysine 4 (H3K4) methylation in renal cell carcinoma. Int J Cancer 127(10):2360–2366
Minardi D, Lucarini G, Filosa A, Milanese G, Zizzi A, Di Primio R et al (2009) Prognostic role of global DNA-methylation and histone acetylation in pT1a clear cell renal carcinoma in partial nephrectomy specimens. J Cell Mol Med 13(8B):2115–2121
Mosashvilli D, Kahl P, Mertens C, Holzapfel S, Rogenhofer S, Hauser S et al (2010) Global histone acetylation levels: prognostic relevance in patients with renal cell carcinoma. Cancer Sci 101(12):2664–2669
Rosenfeld N, Aharonov R, Meiri E, Rosenwald S, Spector Y, Zepeniuk M et al (2008) MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol 26(4):462–469
Heinzelmann J, Henning B, Sanjmyatav J, Posorski N, Steiner T, Wunderlich H et al (2011) Specific miRNA signatures are associated with metastasis and poor prognosis in clear cell renal cell carcinoma. World J Urol 29(3):367–373
Kosaka N, Iguchi H, Ochiya T (2010) Circulating microRNA in body fluid: a new potential biomarker for cancer diagnosis and prognosis. Cancer Sci 101(10):2087–2092
Juan D, Alexe G, Antes T, Liu H, Madabhushi A, Delisi C et al (2010) Identification of a microRNA panel for clear-cell kidney cancer. Urology 75(4):835–841
Jung M, Mollenkopf HJ, Grimm C, Wagner I, Albrecht M, Waller T et al (2009) MicroRNA profiling of clear cell renal cell cancer identifies a robust signature to define renal malignancy. J Cell Mol Med 13(9B):3918–3928
Valera VA, Walter BA, Linehan WM, Merino MJ (2011) Regulatory Effects of microRNA-92 (miR-92) on VHL Gene Expression and the Hypoxic Activation of miR-210 in Clear Cell Renal Cell Carcinoma. J Cancer 2:515–526
Youssef YM, White NM, Grigull J, Krizova A, Samy C, Mejia-Guerrero S et al (2011) Accurate molecular classification of kidney cancer subtypes using microRNA signature. Eur Urol 59(5):721–730
Wulfken LM, Moritz R, Ohlmann C, Holdenrieder S, Jung V, Becker F et al (2011) MicroRNAs in renal cell carcinoma: diagnostic implications of serum miR-1233 levels. PLoS One 6(9):e25787
Neal CS, Michael MZ, Rawlings LH, Van der Hoek MB, Gleadle JM (2010) The VHL-dependent regulation of microRNAs in renal cancer. BMC Med 8:64
Petillo D, Kort EJ, Anema J, Furge KA, Yang XJ, Teh BT (2009) MicroRNA profiling of human kidney cancer subtypes. Int J Oncol 35(1):109–114
Slaby O, Jancovicova J, Lakomy R, Svoboda M, Poprach A, Fabian P et al (2010) Expression of miRNA-106b in conventional renal cell carcinoma is a potential marker for prediction of early metastasis after nephrectomy. J Exp Clin Cancer Res 29:90
White NM, Khella HW, Grigull J, Adzovic S, Youssef YM, Honey RJ et al (2011) miRNA profiling in metastatic renal cell carcinoma reveals a tumour-suppressor effect for miR-215. Br J Cancer 105(11):1741–1749
Khella HW, White NM, Faragalla H, Gabril M, Boazak M, Dorian D et al (2012) Exploring the role of miRNAs in renal cell carcinoma progression and metastasis through bioinformatic and experimental analyses. Tumour Biol 33(1):131–140
Lin J, Horikawa Y, Tamboli P, Clague J, Wood CG, Wu X (2010) Genetic variations in microRNA-related genes are associated with survival and recurrence in patients with renal cell carcinoma. Carcinogenesis 31(10):1805–1812
Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194(4260):23–28
Lengauer C, Kinzler KW, Vogelstein B (1997) DNA methylation and genetic instability in colorectal cancer cells. Proc Natl Acad Sci U S A 94(6):2545–2550
Feinberg AP (2007) Phenotypic plasticity and the epigenetics of human disease. Nature 447(7143):433–440
Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S et al (1998) Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci U S A 95(15):8698–8702
Bachman KE, Park BH, Rhee I, Rajagopalan H, Herman JG, Baylin SB et al (2003) Histone modifications and silencing prior to DNA methylation of a tumor suppressor gene. Cancer Cell 3(1):89–95
Weksberg R, Shen DR, Fei YL, Song QL, Squire J (1993) Disruption of insulin-like growth factor 2 imprinting in Beckwith-Wiedemann syndrome. Nat Genet 5(2):143–150
Hatada I, Ohashi H, Fukushima Y, Kaneko Y, Inoue M, Komoto Y et al (1996) An imprinted gene p57KIP2 is mutated in Beckwith-Wiedemann syndrome. Nat Genet 14(2):171–173
Tycko B (1999) Genomic imprinting and cancer. Results Probl Cell Differ 25:133–169
DeBaun MR, Niemitz EL, McNeil DE, Brandenburg SA, Lee MP, Feinberg AP (2002) Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith-Wiedemann syndrome with cancer and birth defects. Am J Hum Genet 70(3):604–611
DeBaun MR, Tucker MA (1998) Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J Pediatr 132(3 Pt 1):398–400
McKenna ES, Sansam CG, Cho YJ, Greulich H, Evans JA, Thom CS et al (2008) Loss of the epigenetic tumor suppressor SNF5 leads to cancer without genomic instability. Mol Cell Biol 28(20):6223–6233
Kohashi K, Oda Y, Yamamoto H, Tamiya S, Oshiro Y, Izumi T et al (2008) SMARCB1/INI1 protein expression in round cell soft tissue sarcomas associated with chromosomal translocations involving EWS: a special reference to SMARCB1/INI1 negative variant extraskeletal myxoid chondrosarcoma. Am J Surg Pathol 32(8):1168–1174
Trobaugh-Lotrario AD, Tomlinson GE, Finegold MJ, Gore L, Feusner JH (2009) Small cell undifferentiated variant of hepatoblastoma: adverse clinical and molecular features similar to rhabdoid tumors. Pediatr Blood Cancer 52(3):328–334
Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B (1999) Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59(1):74–79
Feinberg AP, Gehrke CW, Kuo KC, Ehrlich M (1988) Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res 48(5):1159–1161
Finch PW, He X, Kelley MJ, Uren A, Schaudies RP, Popescu NC et al (1997) Purification and molecular cloning of a secreted, Frizzled-related antagonist of Wnt action. Proc Natl Acad Sci U S A 94(13):6770–6775
Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD et al (2004) Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet 36(4):417–422
Yan PS, Venkataramu C, Ibrahim A, Liu JC, Shen RZ, Diaz NM et al (2006) Mapping geographic zones of cancer risk with epigenetic biomarkers in normal breast tissue. Clin Cancer Res 12(22):6626–6636
Cheng AS, Culhane AC, Chan MW, Venkataramu CR, Ehrich M, Nasir A et al (2008) Epithelial progeny of estrogen-exposed breast progenitor cells display a cancer-like methylome. Cancer Res 68(6):1786–1796
Yamada Y, Jackson-Grusby L, Linhart H, Meissner A, Eden A, Lin H et al (2005) Opposing effects of DNA hypomethylation on intestinal and liver carcinogenesis. Proc Natl Acad Sci U S A 102(38):13580–13585
Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R (1998) DNA hypomethylation leads to elevated mutation rates. Nature 395(6697):89–93
Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R et al (2002) Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 20(10):2429–2440
Oki Y, Jelinek J, Shen L, Kantarjian HM, Issa JP (2008) Induction of hypomethylation and molecular response after decitabine therapy in patients with chronic myelomonocytic leukemia. Blood 111(4):2382–2384
Shang D, Liu Y, Xu X, Han T, Tian Y (2011) 5-aza-2′-deoxycytidine enhances susceptibility of renal cell carcinoma to paclitaxel by decreasing LEF1/phospho-beta-catenin expression. Cancer Lett 311(2):230–236
Takano Y, Iwata H, Yano Y, Miyazawa M, Virgona N, Sato H et al (2010) Up-regulation of connexin 32 gene by 5-aza-2′-deoxycytidine enhances vinblastine-induced cytotoxicity in human renal carcinoma cells via the activation of JNK signalling. Biochem Pharmacol 80(4):463–470
Mahalingam D, Medina EC, Esquivel JA 2nd, Espitia CM, Smith S, Oberheu K et al (2010) Vorinostat enhances the activity of temsirolimus in renal cell carcinoma through suppression of survivin levels. Clin Cancer Res 16(1):141–153
Acknowledgments
We thank Dr. Roger Erickson for his support and assistance with the preparation of the manuscript. This study was supported by National Center for Research Resources of the National Institutes of Health through Grant Number RO1CA138642, RO1CA130860, VA Merit Review grants, VA Program Project.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Majid, S., Saini, S., Deng, G., Dahiya, R. (2013). Epigenetics and MicroRNAs in Renal Cancer. In: Sarkar, F. (eds) Epigenetics and Cancer. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6612-9_4
Download citation
DOI: https://doi.org/10.1007/978-94-007-6612-9_4
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6611-2
Online ISBN: 978-94-007-6612-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)