
Abstract
Angiogenesis, or new blood vessel formation, is necessary for the growth and progression of malignant tumors. Among the endogenous regulators of angiogenesis, catecholamines have recently drawn attention owing to the discovery that they have opposing roles in regulating tumor angiogenesis. Dopamine (DA), norepinephrine (NE), and epinephrine (E) are the members of the catecholamine family. DA suppresses tumor angiogenesis and hence inhibits tumor growth, whereas NE and E increase tumor growth by promoting angiogenesis in tumor tissues. Therefore, on the whole, catecholamines function as an angiogenic switch. These neurotransmitters act upon their target cells via specific receptors, exerting pro- or anti-angiogenic effects, and thus are excellent targets for the regulation of tumor angiogenesis by dopaminergic or adrenergic receptor agonists or antagonists.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Tumor Angiogenesis
- Angiogenic Switch
- Tumor Endothelial Cell
- Inhibit Tumor Angiogenesis
- Placenta Growth Factor
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Neovascularization occurs by two distinct mechanisms: angiogenesis, in which new blood vessels sprout from the pre-existing blood vessels, and vasculogenesis, in which new blood vessels are derived from the circulating bone marrow-derived endothelial progenitor cells (EPCs) [1–5]. This process of new blood vessel formation is essential not only in normal physiological situations (e.g., the menstrual cycle, implantation, embryogenesis, and wound-healing), but also in the growth and metastasis of malignant tumors [1–5].
In the normal physiological milieu, angiogenesis is tightly regulated by intricately balanced endogenous pro- and anti-angiogenic molecules [6–8]. However, in malignancy, this fine tuning of the balance between pro- and anti-angiogenic molecules is disabled by either the overexpression of pro-angiogenic molecules or the down-regulation of anti-angiogenic molecules, resulting in the activation of the angiogenic switch [6–8]. The overexpression of pro-angiogenic growth factors, such as vascular permeability factor/vascular endothelial growth factor (VPF/VEGF), fibroblast growth factor (FGF), interleukin-8 (IL-8), placenta growth factor (PlGF), transforming growth factor β (TGF-β), and platelet derived growth factor (PDGF) tilt the tumor microenvironment in favor of angiogenesis and allow the transition of an avascular dormant tumor into a growing vascular tumor mass [1–3]. Because targeting growth factor-induced angiogenesis has shown clinical promise, designing therapies to target tumor neovessels is of interest owing to the decreased toxicity of the approach, the minimal drug resistance, and the ability to increase the efficacies of anti-cancer drugs and radiation therapy [1–10].
Catecholamines are a group of neurotransmitters that includes dopamine (DA), norepinephrine (NE), and epinephrine (E) [11]. In addition to their conventional roles in the brain, these molecules also have important functions in the periphery [11]. Recent discoveries of a regulatory role for different catecholamines in tumor angiogenesis are of current interest from a clinical viewpoint for the development of anti-angiogenic drugs to treat cancer patients [12–14]. These newly identified roles for catecholamines also enable us to understand the biology of catecholamines in peripheral systems [13]. The available information regarding the roles of catecholamines in the regulation of tumor angiogenesis is discussed in this chapter.
NE and E are Endogenous Promoters of Tumor Angiogenesis
NE and E act on their target cells through α (α1 and α2) and β (β1 and β2) adrenoceptors [11]. Evidence indicates that exposure to chronic stress promotes tumor growth [12, 15] through the stress mediators NE and E [16, 17], and the up-regulation of tumor angiogenesis is suggested to be the underlying mechanism [13, 18]. In a model of orthotopically xenografted human ovarian tumors in nude mice, a tumor growth-promoting effect was observed in animals following exposure to chronic stress or treatment with the β-adrenergic agonist isoproterenol, and this effect was abrogated by the β-adrenergic antagonist propranolol [19]. Interestingly, this increase in tumor growth was associated with the up-regulation of VEGF in tumor tissues, which led to the induction of tumor angiogenesis [19]. However, inhibition of the VEGF pathway suppressed the tumor growth stimulatory effect of the β-adrenergic agonist [19]. In vitro studies also demonstrated the NE-mediated secretion of VEGF by ovarian carcinoma cells [20]. In addition, there are reports that indicate that NE, by acting on the adrenoceptors present in the tumor-associated macrophages (TAM), induces angiogenesis by stimulating the production of matrix metalloproteinase 9 (MMP-9) [21].
In addition to the NE-mediated increase in the expression of the pro-angiogenic cytokine VEGF, studies have also indicated that in different tumor cells bearing the β-adrenoceptor (such as melanoma, ovarian, and nasopharyngeal cancer cells), NE induces a significant increase in the synthesis and release of other pro-angiogenic factors, including IL-6, IL-8, MMP-2, and MMP-9 [21–24]. Interestingly, nicotine has been shown to increase xenografted human colon tumor growth in nude mice. This increased tumor growth is associated with elevated plasma E levels and tumor angiogenesis. However, blocking the β-adrenoceptors with specific antagonists significantly abrogated the nicotine-induced tumor growth through the down-regulation of tumor angiogenesis [25]. In addition, NE has also been shown to stimulate VEGF mRNA synthesis in endothelial cells through the cAMP-PKA pathway and to promote proliferation of these cells by activating ERK [26].
Molecular Mechanisms of NE- and E-induced Tumor Angiogenesis
By acting through β2-adrenoceptors, NE has been shown to promote angiogenesis in the orthotopically grown ovarian cancers HEY-8 and SKOV3ipI [19]. The underlying molecular mechanisms of this phenomenon were determined to be increased VEGF synthesis and the overexpression of matrix metalloproteinases, such as MMP-2 and MMP-9 [19]. Further investigations demonstrated that this VEGF-induced overexpression of matrix metalloproteinases is mediated through the cAMP-PKA signaling pathway following stimulation of β2-adrenoceptors by NE, indicating a novel signaling pathway, such as NE-β2-adrenoceptors-cAMP-PKA-VEGF [19]. NE treatment has also shown similar results in the human nasopharyngeal cell line HONE 1 [23]. In several human multiple myeloma cell lines (NCI-H-929, MM-M1, and FLAM-76) NE treatment has also shown similar results by acting through β1 and β2 adrenoceptors present in these cells [27]. However, a recent study has demonstrated that in vitro treatment of human prostate (PC3), breast (MDA-MB-231), and liver (HCC SK-Hep1) cancer cells with NE or isoproterenol stimulated the expression of HIF-1α and synthesis of VEGF in a dose-dependent manner [28]. This increased VEGF synthesis was decreased when the tumor cells were transfected with HIF-1α siRNA [28]. This observation was further strengthened when HIF-1α was up- or down-regulated in these tumor cells following pretreatment with the adenylate cyclase activator forskolin or the protein kinase A (PKA) inhibitor H-89, respectively [28]. Finally, pretreatment of tumor cells with a β-adrenergic blocker propranolol completely abolished the expression of VEGF and HIF-1α protein amount in these cells [28]. Therefore, in brief, NE induces VEGF expression in cancer cells through NE-β-adrenoceptor-PKA-HIF-1α-VEGF signaling pathway (Fig. 7.1).

NE stimulates VEGF synthesis in the tumor cells. NE by activating β-adrenergic receptors activates cAMP-PKA axis and stimulates VEGF synthesis by up-regulating the transcription factor Hypoxia Inducible Factor-1α (HIF-1α) in tumor cells
This catecholamine neurotransmitter also stimulates the synthesis and release of another pro-angiogenic factor, IL-6, in the human ovarian tumor cell lines SKOV3ip1, HEY-A8 and EG in vitro [24]. By acting through β-adrenoceptors in these tumor cells, NE significantly increased both IL-6 mRNA synthesis and promoter activity [24]. Additional results have demonstrated an abrogation of this NE-mediated effect on IL-6 synthesis following treatment with β-adrenoceptor antagonists, confirming the NE-mediated regulation of IL-6 gene transcription through the activation of β-adrenoceptors [24]. NE-mediated β-adrenoceptor activation was also shown to increase Src kinase phosphorylation, which subsequently increased IL-6 mRNA synthesis through the up-regulation of the IL-6 promoter activity [24]. This suggestion of a NE-β-adrenoceptor-Src kinase-IL-6 pathway for increased tumor angiogenesis was further strengthened by immunohistochemical analysis of human ovarian cancer tissues, in which a significant correlation between the overexpression of Src kinase and the degree of tumor neovascularization was observed [24]. Src activation was also instrumental in increasing the synthesis of other pro-angiogenic molecules, such as VEGF and IL-8 [24]. However, another alternate signaling pathway was recently identified in which NE and E stimulated MMPs in human ovarian tumor cell lines independent of the β1, β2-adrenoceptors-PKA pathway. This pathway involves STAT-3, a transcription factor known to initiate several signaling pathways in cancer cells [29].
DA as an Endogenous Inhibitor of Tumor Angiogenesis
In addition to acting as a precursor molecule in the biosynthetic pathway of NE and E, DA also acts as an important neurotransmitter in both the brain and the peripheral organs [30]. In the brain, DA regulates several major functions, including cognition, motor activities, and the reward effect in the form of pleasure [30, 31]. In peripheral systems, DA regulates cardiac and renal functions. In addition, recent evidence has indicated that DA influences other diverse functions, such as blood pressure, insulin synthesis in beta cells of the pancreas, and the functions of immune effector cells [32–34]. Recently, another role of this neurotransmitter in the peripheral system has been demonstrated: DA functions as an endogenous inhibitor of angiogenesis by acting through its D2 class of receptors present in the endothelial cells and EPCs [35–41].
A significant increase in B-16 melanoma growth has been found in D2 DA receptor (−/−) mice [37]. Another study has revealed significantly decreased growth of mammary carcinoma in hyperdopaminergic Wistar rats (APO-SUS), and increased growth of the same tumors was observed in hypodopaminergic rats (APO-UNSUS) [36]. This increased or decreased tumor growth in hypo- or hyperdopamineregic rats is closely associated with increased or decreased angiogenesis in tumors [36]. Furthermore, in human gastric cancer patients, a significant reduction of DA in malignant stomach tissues compared to the surrounding normal tissues has been observed, and exogenous administration of DA or a D2 DA receptor agonist significantly inhibited stomach tumor growth [38]. The mechanism of this phenomenon has been attributed to the inhibition of angiogenesis in the tumor tissues [38].
Molecular Mechanisms of DA-induced Inhibition of Tumor Angiogenesis
VEGF is the predominant cytokine that regulates angiogenesis by mediating proliferation, migration, and tube formation in endothelial cells from pre-existing vessels [3, 10]. VEGF also plays a pivotal role in the migration and subsequent mobilization of EPCs from the bone marrow into the neovessels of tumors by acting through VEGFR-2 receptors present on these cells [41]. In vivo studies have demonstrated that DA treatment significantly inhibits tumor angiogenesis [35–41]. Tumor endothelial cells isolated from human breast (MCF-7) and colon (HT29) tumor-bearing mice displayed suppression of VEGFR-2 phosphorylation with subsequent inhibition of its downstream signaling cascades (e.g., MAPK and focal adhesion kinase (FAK)), which regulate the proliferation and migration of endothelial cells; this regulation is essential for tumor neovessel formation (Fig. 7.2) [40]. Recent studies also reveal important contributions of bone marrow-derived EPCs in tumor angiogenesis [4, 5, 42]. Indeed, additional reports indicate that in the bone marrow niche, DA is synthesized in stromal cells [43] and is depleted in tumor-bearing mice [41], thus indicating a role of DA in the regulation of the mobilization of these precursor cells from bone marrow into circulation [41]. Importantly, the administration of exogenous DA, which inhibited tumor angiogenesis, also inhibited the mobilization of these cells from bone marrow [41]. This inhibitory effect of DA is abrogated when the animals are treated with a D2 DA receptor antagonist [41]. The inhibitory effect on the mobilization of EPCs from the bone marrow and hence on tumor angiogenesis is due to the D2 DA receptor-mediated down-regulation of MMP-9 synthesis by these bone marrow progenitor cells through the inhibition of the ERK1/ERK2 pathway (Fig. 7.3) [41]. These observations were further supported in D2 DA receptor (−/−) mice: increased numbers of circulating EPCs were evident in tumor-bearing mice compared to wild type controls, and the D2 DA receptor antagonist treatment failed to elicit any effect in the animals [41].

Dopamine inhibits tumor endothelial cell proliferation and migration. Dopamine by activating its D2 receptors inhibits VEGFR-2 phosphorylation and downstream signaling molecules like mitogen-activated protein kinase (MAPK) and Focal adhesion kinase (FAK)

Dopamine inhibits mobilization of bone marrow-derived endothelial progenitor cells for tumor neovessel formation. Activation of D2 dopamine receptors in endothelial progenitor cells (EPCs), dopamine inhibits migration of these cells from bone marrow to the circulation and subsequently to neovessels of the tumors by inhibiting synthesis of matrix metalloproteinase-9 (MMP-9) in these progenitor cells
These studies have clearly demonstrated that DA acts as an endogenous inhibitor of angiogenesis, and hence of tumor growth, by targeting the VEGF-induced proliferation and migration of endothelial cells and EPCs through several newly uncovered mechanisms [35–41].
Discussion
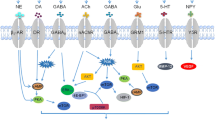
Together, current findings show that catecholamine neurotransmitters act as regulators of tumor angiogenesis and, hence, regulate the growth of malignant tumors [13]. The available evidence suggests that DA acts through its D2 class of receptors to inhibit tumor angiogenesis by targeting the proliferation and migration of tumor endothelial cells as well as the mobilization of EPCs [35–41]. In contrast, NE and E act through β-adrenoceptors to stimulate angiogenesis by stimulating the synthesis of pro-angiogenic cytokines (e.g., VEGF, IL-8, and IL-6) and MMPs (e.g., MMP-2 and MMP-9) in tumor cells through different signaling pathways [13]. Briefly, in the D2 DA receptor-mediated down-regulation of angiogenesis, the target cells are endothelial cells and EPCs, whereas in the NE-mediated up-regulation of tumor angiogenesis, the target cells are principally tumor cells [13]. Therefore, based on these opposing effects of stimulation and inhibition of tumor angiogenesis by the catecholamine neurotransmitters (DA, NE, and E), it is suggested that catecholamines function as an angiogenic switch in the tumor microenvironment (Fig. 7.4). The expression profile of D2 DA receptors or β-adrenoceptors in any organ may be altered in response to the onset of malignancy in that organ. These alterations may tilt the microenvironment of the tumor in favor of angiogenesis, thereby transforming an avascular, dormant tumor mass into a vascular, rapidly growing tumor. Epidemiological evidence has also indicated that the use of NE antagonists reduces the risk of cancer incidence [44]. Therefore, it will be prudent to undertake further detailed investigations to dissect the specific roles of catecholamines in relation to their function as an angiogenic switch in tumor growth. These studies will enable clinicians to develop DA or NE/E receptor agonists or antagonists as anti-angiogenic drugs.

Model of an angiogenic switch in tumor: Diagram of catecholamine-mediated operation of an angiogenic switch in tumor microenvironment. Dopamine inhibits angiogenesis, whereas norepinephrine and epinephrine up-regulate angiogenesis in tumor tissues
References
Folkman J, Shing Y (1992) Angiogenesis. J Biol Chem 267:10931–10934
Carmeliet P, Jain RK (2000) Angiogenesis in cancer and other diseases. Nature 407:249–257
Dvorak HF (2005) Angiogenesis: update 2005. J Thromb Haemost 3:1835–1842
Asahara T, Murohara T, Sullivan A et al (1997) Isolation of putative progenitor endothelial cells for angiogenesis. Science 275:964–967
Kopp HG, Ramos CA, Rafii S (2006) Contribution of endothelial progenitors and proangiogenic hematopoietic cells to vascularization of tumor and ischemic tissue. Curr Opin Hematol 13:175–181
Moserle L, Amadori A, Indraccolo S (2009) The angiogenic switch: implications in the regulation of tumor dormancy. Curr Mol Med 9:935–941
Naumov GN, Akslen LA, Folkman J (2006) Role of angiogenesis in human tumor dormancy: animal models of the angiogenic switch. Cell Cycle 5:1779–1787
Baeriswyl V, Christofori G (2009) The angiogenic switch in carcinogenesis. Semin Cancer Biol 19:329–337
Cai J, Han S, Qing R et al (2011) In persuit of new anti-angiogenic therapies for cancer treatment. Front Biosci 16:803–814
Ferrara N (2009) Vascular endothelial growth factor. Arterioscler Thromb Vasc Biol 29:789–791
Laverty R (1978) Catecholamines: role in health and disease. Drugs 16:418–440
Antoni MH, Lutgendorf SK, Cole SW et al (2006) The influence of bio-behavioral factors on tumor biology: pathways and mechanisms. Nat Rev Cancer 6:240–248
Chakroborty D, Sarkar C, Basu B et al (2009) Catecholamines regulate tumor angiogenesis. Cancer Res 69:3727–3730
Tilan J, Kitlinska J (2010) Sympathetic neurotransmitters and tumor angiogenesis-link between stress and cancer progression. J Oncol 2010:539706. doi:10.1155/2010/539706
Hasegawa H, Saiki I (2002) Psychosocial stress augments tumor development through beta-adrenergic activation in mice. Jpn J Cancer Res 93:729–735
Thaker PH, Sood AK (2008) Neuroendocrine influence on cancer biology. Semin Cancer Biol 18:164–170
Thaker PH, Lutgendorf SK, Sood AK (2007) The neuroendocrine impact of chronic stress on cancer. Cell Cycle 6:430–433
Armaiz-Pena GN, Lutgendorf SK, Cole SW, Sood AK (2009) Neuroendocrine modulation of cancer progression. Brain Behav Immun 23:10–15
Thaker PH, Han LY, Kamat AA et al (2006) Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med 12:939–944
Lutgendorf SK, Cole S, Costanzo E et al (2003) Stress-related mediators stimulate vascular endothelial growth factor secretion by two ovarian cancer cell lines. Clin Cancer Res 9:4514–4521
Lutgendorf SK, Lamkin DM, Jennings NB (2008) Biobehavioral influences on matrix metalloproteinase expression in ovarian carcinoma. Clin Cancer Res 14:6839–6846
Yang EV, Kim SJ, Donovan EL et al (2009) Norepinephrine upregulates VEGF, IL-8, and IL-6 expression in human melanoma tumor cell lines: implications for stress-related enhancement of tumor progression. Brain Behav Immun 23:267–275
Yang EV, Sood AK, Chen M et al (2006) Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res 66:10357–10364
Nilsson MB, Armaiz-Pena GN, Takahashi R et al (2007) Stress hormones regulate interleukin-6 expression by human ovarian carcinoma cells through a Src-dependent mechanism. J Biol Chem 282:29919–29926
Wong HP, Yu L, Lam EK et al (2007) Nicotine promotes colon tumor growth and angiogenesis through beta-adrenergic activation. Toxicol Sci 97:279–287
Seya Y, Fukuda T, Isobe K et al (2006) Effect of norepinephrine on RhoA, MAP kinase, proliferation and VEGF expression in human umbilical vein endothelial cells. Eur J Pharmacol 553:54–60
Yang EV, Donovan EL, Benson DM, Glaser R (2008) VEGF is differentially regulated in multiple myeloma-derived cell lines by norepinephrine. Brain Behav Immun 22:318–323
Park SY, Kang JH, Jeong KJ et al (2011) Norepinephrine induces VEGF expression and angiogenesis by a hypoxia-inducible factor-1α protein-dependent mechanism. Int J Cancer 128:2306–2316
Landen CN Jr, Lin YG, Armaiz Pena GN et al (2007) Neuroendocrine modulation of signal transducer and activator of transcription-3 in ovarian cancer. Cancer Res 67:10389–10396
Beaulieu JM, Gainetdinov RR (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63:182–217
Missale C, Nash SR, Robinson SW, Jaber M, Caron MG (1998) Dopamine receptors: from structure to function. Physiol Rev 78:189–225
Rubi B, Maechler P (2010) Minireview: new roles for peripheral dopamine on metabolic control and tumor growth: let’s seek the balance. Endocrinology 151:5570–5581
Basu S, Dasgupta PS (2000) Dopamine, a neurotransmitter, influences the immune system. J Neuroimmunol 102:113–124
Sarkar C, Basu B, Chakroborty D et al (2010) The immunoregulatory role of dopamine: an update. Brain Behav Immun 24:525–528
Basu S, Nagy JA, Pal S et al (2001) The neurotransmitter dopamine inhibits angiogenesis induced by vascular permeability factor/vascular endothelial growth factor. Nat Med 7:569–574
Teunis MA, Kavelaars A, Voest E et al (2002) Reduced tumor growth, experimental metastasis formation, and angiogenesis in rats with a hyperreactive dopaminergic system. FASEB J 16:1465–1467
Basu S, Sarkar C, Chakroborty D et al (2004) Ablation of peripheral dopaminergic nerves stimulates malignant tumor growth by inducing vascular permeability factor/vascular endothelial growth factor-mediated angiogenesis. Cancer Res 64:5551–5555
Chakroborty D, Sarkar C, Mitra RB et al (2004) Depleted dopamine in gastric cancer tissues: dopamine treatment retards growth of gastric cancer by inhibiting angiogenesis. Clin Cancer Res 10:4349–4356
Sarkar C, Chakroborty D, Mitra RB et al (2004) Dopamine in vivo inhibits VEGF-induced phosphorylation of VEGFR-2, MAPK, and focal adhesion kinase in endothelial cells. Am J Physiol Heart Circ Physiol 287:H1554–H1560
Sarkar C, Chakraborty D, Chowdhury UR et al (2008) Dopamine increases the efficacy of anticancer drugs in breast and colon cancer preclinical models. Clin Cancer Res 14:2502–2510
Chakroborty D, Chowdhury UR, Sarkar C et al (2008) Dopamine regulates endothelial progenitor cell mobilization from mouse bone marrow in tumor vascularization. J Clin Invest 118:1380–1389
Gao D, Nolan D, McDonnell K et al (2009) Bone marrow-derived endothelial progenitor cells contribute to the angiogenic switch in tumor growth and metastatic progression. Biochim Biophys Acta 1796:33–40
Marino F, Cosentino M, Bombelli R et al (1997) Measurement of catecholamines in mouse bone marrow by means of HPLC with electrochemical detection. Haematologica 82:392–394
Friedman GD, Udaltsova N, Habel LA (2011) Norepinephrine antagonists and cancer risk. Int J Cancer 128:737–738
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Basu, S., Dasgupta, P.S. (2013). Catecholamine Neurotransmitters: An Angiogenic Switch in the Tumor Microenvironment. In: Mousa, S., Davis, P. (eds) Angiogenesis Modulations in Health and Disease. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6467-5_7
Download citation
DOI: https://doi.org/10.1007/978-94-007-6467-5_7
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6466-8
Online ISBN: 978-94-007-6467-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)