
Abstract
Solitary fibrous tumors (SFT) are rare mesenchymal tumors that can affect central nervous system at any level. Histologically, they are characterized by round to spindle-shaped fibroblastic cells set in a collagenous matrix with variable amounts of hyalinized collagen bundles and strongly positive stain for CD34 on immunohistochemistry. Clinical presentation is related location, size of the tumor and affected structures in the CNS. Headache is the chief complaint in the majority of patients with intracranial tumors. When SFT involve the spinal cord, progressive limbs weakness, paresthesia, and radicular pain are the most consistent findings. On magnetic resonance imaging (MRI), these tumors are frequently isointense with normal brain parenchyma on T1-weighted images, hyperintense on T2-weighted-images, and show intense homogeneous enhancement after intravenous administration of gadolinium. Complete surgical resection is probably the only curative option and is regarded as the treatment of choice for all patients suited to it. Incomplete surgical resection is regarded as a bad prognosis factor for which radiation therapy may be an option.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Cerebellar Ataxia
- Solitary Fibrous Tumor
- Complete Surgical Resection
- Normal Brain Parenchyma
- Incomplete Surgical Resection
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Solitary fibrous tumors (SFT) are rare spindle-cell neoplasms of mesenchymal origin. The first detailed histological description of SFT was made by Wagner (1870), and they were first recognized as a distinct soft tissue neoplasm by Klemperer and Rabin (1931) in the visceral pleura, the most common site of occurrence. However, the identity of SFT continued to be debated, with the tumor subsequently categorized under mesothelial neoplasms based on tissue culture submesothelial characteristics, until the general consensus of SFT as a distinct mesenchymal neoplasm finally emerged (Park and Araujo 2009).
These tumors have been described in many extra pleural sites including pericardium, peritoneum, lung, liver, upper respiratory tract, mediastinum, nasal cavities, thyroid and parathyroid glands, orbits, and the central nervous system (CNS). SFT in the CNS are usually dura-based, meningioma-like masses, that may be intracranial or spinal. Primary meningeal SFT was first described by Carneiro et al. (1996), and the first description of SFT in the spinal cord was made by Alston et al. (1997). Since then, approximately 100 cases of SFT have been reported both in the cranial and the spinal compartments of the CNS (Yilmaz et al. 2009). SFTs were included in the 2000 World Health Organization classification of CNS tumors in the category of mesenchymal neoplasm of the meninges (Kleihues and Cavanee 2000).
The cellular origin of SFT is still a matter of debate. SFTs were initially thought to originate from CD34-positive dura-based fibroblasts or dendritic cells (Carneiro et al. 1996; Cummings et al. 2001). However, the recent description of SFT arising in deep cortical structures argues against this hypothesis, and a possible origin from the mesenchyma of the cerebral vasculature has been proposed (Kim et al. 2004).
Pathology
SFT appears in gross pathologic examination as a firm and elastic well-circumscribed tumor, often with attached fragments of dura mater, and smooth, glistening pseudo-capsules. On cutting, their surface displays a variety of colors. Histologically, the tumors are composed of round to spindle-shaped fibroblastic cells set in a collagenous matrix with variable amounts of hyalinized collagen bundles. Cellularity ranges from hypercellular to lax areas. The vasculature shows a hemangiopericytoma-like growth pattern and vessels with thickened hyalinized walls. The mitotic rate is usually <4/10 high power field (HPF) (Insabato et al. 2009). Ultrastructural analysis has demonstrated features of pericytic, fibroblastic, and myofibroblastic differentiation with vascular prominence with intercapillary stroma containing various mesenchymal cells, including pericytes in close proximity to capillaries (Ide et al. 2005). Necrosis, marked nuclear hyperchromasia, and abundant mitosis are rarely observed. The features commonly proposed to be associated with aggressive SFT are increased cellularity, pleomorphism, increased mitotic activity (mitotic index >4/10 HPF), necrosis, hemorrhage, and atypical location (Mekni et al. 2009).
Using immunohistochemistry, SFTs are strongly positive for CD34 and variably positive for CD99, Bcl2, and vimentin. Some tumors express progesterone receptor in the nucleus of the cells. SFTs usually are negative for desmin, cytokeratins, and S-100 (Insabato et al. 2009). Immunohistochemical staining for Ki-67 (MIB-1) antibody ranges from less than 2 to 25% (Mekni et al. 2009). In addition, the staining for p53 and cyclin-D1 may be important to assess the biological behavior of the tumors (Mosquera and Fletcher 2009; Mekni et al. 2009). Only a few cases of SFT with anaplastic histological features were reported with invasion of parenchyma and bone, local recurrence, and distant metastasis (Mekni et al. 2009).
The main differential diagnoses are schwannoma, fibrous meningioma and hemangiopericytoma (HPC). Schwannomas are easily distinguished because of their microscopic features (nuclear pseudopalisading, wavy nuclei) and their strong immunoreactivity to S-100. Cellular whorls, storiform cell arrangements, and psammomas bodies are usually present in meningiomas. The cells in SFTs tend to be plumper than in meningiomas. Immunohistochemistry is also used for showing the complete absence of S-100 or EMA expression in SFTs as opposed to meningiomas. CD34 may be focally positive in meningiomas. Ultrastructural features of meningiomas include cytoplasmic interdigitations and well-formed desmosomes, which are lacking in SFTs. There is considerable overlap in the histological features of SFT and HPC. In a typical case of SFT, the distinction is based on the presence of frequent areas of hyalinization and collagen deposition, diffuse immunoreactivity for CD34, sparse reticulin staining around clusters of cells rather than abundant and fine reticulin around individual cells, and absence of basement membrane-like material at ultrastructural level, a hallmark of HPCs (Fig. 13.1).

Spindle cell tumor consistent with solitary fibrous tumor. Panel (a): Highly cellular spindle cell proliferation with a dense, hyalinized collagenous stroma and dilated vascular spaces, some showing a staghorn-like appearance. Areas of cellular pleomorphism and increased cellularity were present, but mitoses were not identified (H&E 50×); Panel (b): Strong immunoreactivity for CD34 (50×); Panel (c): Strong immunoreactivity for vimentin (50×) (From Furlanetto et al. 2009)
Cytogenetics
Analyses of a limited number of SFT to date have not found any consistent characteristic cytogenetic abnormalities. A comparative genomic hybridization (CGH) analysis of three meningeal SFTs reported by Martin et al. (2002) showed one case with loss of chromosome 3 and two tumors with deletions of the region 3p21–p26. Other chromosomal losses included 4p15, 8q22–q24, 10, 11q14–q25, 17q11–q23, 20 and 21. Chromosomal gains were reported on 18p11–p13, 1p11–p36, and 20q11–q13. Cytogenetical abnormalities on chromosome 3 had not been previously described in SFTs from other primary sites.
Clinical and Imaging Features
SFTs can be present in any site of the CNS, involving the spinal cord in approximately one quarter of the patients (Tihan et al. 2003; Rodriguez et al. 2004; Caroli et al. 2004; Kim et al. 2004; Pizzolitto et al. 2004; Pakasa et al. 2005; Metellus et al. 2007; Furlanetto et al. 2009). When intracranial, they are more commonly found within the supratentorial space and the ventricular system, followed by infratentorial space, including posterior fossa structures, cerebellum and cerebellopontine angles. Tentorial SFT corresponds to a minority of cases (Hakan et al. 2009). Among the spinal SFT, the lesions were intradural in 73% and intramedullary in 27% of reported cases (Ciappetta et al. 2010).
The mean age at the time of diagnosis from 2 series of 18 patients each was, respectively, 47 and 56 years, with a sex ratio of approximately 1:1 (Tihan et al. 2003; Metellus et al. 2007). Clinical presentation is related to a few factors: location, size of the tumor and affected structures in the CNS. Headache is the chief complaint in the majority of patients with intracranial tumors. In some cases, SFT were found during evaluation for seizures. When SFT involve the spinal cord, progressive limbs weakness, paresthesia, and radicular pain were the most consistent findings. In a series, cranial nerve paresis and cerebellar ataxia were the most frequent symptoms for tumors at the infratentorial level; whereas, for supratentorial tumors, intracranial hypertension, epilepsy, and lateral hemianopsia were usually found (Metellus et al. 2007).
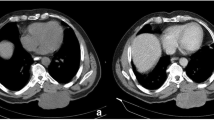
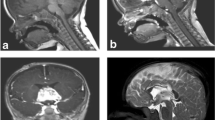
On computed tomography scan (CT), SFT appear usually as relatively well circumscribed, partially calcified heterogeneous masses, demonstrating variable degrees of enhancement upon intravenous contrast infusion. On magnetic resonance imaging (MRI), these tumors are frequently isointense with normal brain parenchyma on T1-weighted images, hyperintense on T2-weighted-images, and show intense homogeneous enhancement after intravenous administration of gadolinium. In a series described by Mekni et al. (2009), the tumors averaged 5 cm in largest diameter. On imaging studies alone, these lesions were indistinguishable from other SNC tumors, like meningiomas or gliomas (Fig. 13.2).

MRI sagital (left) and coronal (right) T1WI views with IV Gadolinium showing, at the suprasellar level, a tumor above the pituitary gland with strong heterogeneous contrast enhancement. The pathological specimen was consistent with solitary fibrous tumor (From Furlanetto et al. 2009)
Management
Despite lack of prospective data evaluating different treatment modalities for SFT, complete surgical resection is probably the only curative option and is regarded as the treatment of choice for all patients suited to it. In patients with incomplete resection, the role of postoperative radiation therapy remains uncertain. Management of patients who develop local or distant relapses has been challenging, since no clearly effective therapy exists. A new attempt of resection should be considered if technically feasible as it can lead to improvement in progression free survival.
Radiation therapy has been used in some cases, both as adjuvant therapy in patients at high risk for local recurrence or as primary therapy in unresectable tumors. Retrospective analyses of adjuvant radiation therapy have cautiously supported its utility, with studies showing at best a non-significant trend toward prolonged recurrence-free survival and overall survival (Park and Araujo 2009). Its use should not be recommended routinely in the adjuvant setting, especially in patients with completely resected tumors lacking adverse prognostic features. It should, therefore, be considered for those patients with aggressive tumors displaying adverse prognostic markers, until more data on its efficacy become available. Stereotaxic radiosurgery in recurrent unresectable SFTs appears to be a potentially promising therapeutic modality. The role of systemic therapy for advanced unresectable tumors remains unknown. Inhibition of angiogenesis has emerged as a potential promising therapy, but further studies are needed to determine its efficacy (Park and Araujo 2009).
Prognosis
The most important prognostic factor in SFT is complete surgical resection. Metellus et al. (2007) reported a 50% recurrence or progression rate in their series of 18 patients with a mean follow-up of 45 months. Incomplete surgical resection was significantly associated with recurrence, while near 90% of patients with gross total resection were free from disease at follow-up. The histological criteria for aggressive extrapleural SFT, known to have a worse prognosis and a higher index of recurrences, are hypercellularity, moderate-to-marked cellular atypias, necrosis, and more than 4 mitoses per 10 HPF (Mekni et al. 2009). No study, to date, has defined criteria for malignant SFT in the CNS, so the same criteria are usually used.
References
Alston SR, Francel PC, Jane JA Jr (1997) Solitary fibrous tumor of the spinal cord. Am J Surg Pathol 21:477–483
Carneiro SS, Scheithauer BW, Nascimento AG, Hirose T, Davis DH (1996) Solitary fibrous tumor of the meninges: a lesion distinct from fibrous meningioma. A clinicopathologic and immunohistochemical study. Am J Clin Pathol 106:217–224
Caroli E, Salvati M, Orlando ER, Lenzi J, Santoro A, Giangaspero F (2004) Solitary fibrous tumors of the meninges: report of four cases and literature review. Neurosurg Rev 27:246–251
Ciappetta P, D’Urso PI, Cimmino A, Ingravallo G, Rossi R, Colamaria A, D’Urso OF (2010) Intramedullary solitary fibrous tumor of dorsal spinal cord. Neuropathology 30:273–278
Cummings TJ, Burchette JL, McLendon RE (2001) CD34 and dural fibroblasts: the relationship to solitary fibrous tumor and meningioma. Acta Neuropathol 102:349–354
Furlanetto TW, Pinheiro CF, Oppitz PP, de Alencastro LC, Asa SL (2009) Solitary fibrous tumor of the sella mimicking pituitary adenoma: an uncommon tumor in a rare location-a case report. Endocr Pathol 20:56–61
Hakan T, Turk CC, Aker FV (2009) Tentorial solitary fibrous tumour: case report and review of the literature. Neurol Neurochir Pol 43:77–82
Ide F, Obara K, Mishima K, Saito I, Kusama K (2005) Ultrastructural spectrum of solitary fibrous tumor: a unique perivascular tumor with alternative lines of differentiation. Virchows Arch 446:646–652
Insabato L, Siano M, Somma A, Gentile R, Santangelo M, Pettinato G (2009) Extrapleural solitary fibrous tumor: a clinicopathologic study of 19 cases. Int J Surg Pathol 17:250–254
Kim KA, Gonzalez I, McComb JG, Giannotta SL (2004) Unusual presentations of cerebral solitary fibrous tumors: report of four cases. Neurosurgery 54:1004–1009
Kleihues P, Cavanee WK (2000) World health organization classification of tumours: pathology and genetics: tumours of the nervous system world health organization
Klemperer P, Rabin CB (1931) Primary neoplasms of the pleura. A report of five cases. Arch Pathol 11:385–412
Martin AJ, Summersgill BM, Fisher C, Shipley JM, Dean AF (2002) Chromosomal imbalances in meningeal solitary fibrous tumors. Cancer Genet Cytogenet 135:160–164
Mekni A, Kourda J, Hammouda KB, Tangour M, Kchir N, Zitouna M, Haouet S (2009) Solitary fibrous tumour of the central nervous system: pathological study of eight cases and review of the literature. Pathology 41:649–654
Metellus P, Bouvier C, Guyotat J, Fuentes S, Jouvet A, Vasiljevic A, Giorgi R, Dufour H, Grisoli F, Figarella-Branger D (2007) Solitary fibrous tumors of the central nervous system: clinicopathological and therapeutic considerations of 18 cases. Neurosurgery 60:715–722
Mosquera JM, Fletcher CD (2009) Expanding the spectrum of malignant progression in solitary fibrous tumors: a study of 8 cases with a discrete anaplastic component – is this dedifferentiated SFT? Am J Surg Pathol 33:1314–1321
Pakasa NM, Pasquier B, Chambonniere ML, Morrison AL, Khaddage A, Perret AG, Dumollard JM, Barral FG, Péoc’h M (2005) Atypical presentations of solitary fibrous tumors of the central nervous system: an analysis of unusual clinicopathological and outcome patterns in three new cases with a review of the literature. Virchows Arch 447:81–86
Park MS, Araujo DM (2009) New insights into the hemangiopericytoma/solitary fibrous tumor spectrum of tumors. Curr Opin Oncol 21:327–331
Pizzolitto S, Falconieri G, Demaglio G (2004) Solitary fibrous tumor of the spinal cord: a clinicopathologic study of two cases. Ann Diagn Pathol 8:268–275
Rodriguez F, Scheithauer BW, Ockner DM, Giannini C (2004) Solitary fibrous tumor of the cerebellopontine angle with salivary gland heterotopia: a unique presentation. Am J Surg Pathol 28:139–142
Tihan T, Viglione M, Rosenblum MK, Olivi A, Burger PC (2003) Solitary fibrous tumors in the central nervous system. A clinicopathologic review of 18 cases and comparison to meningeal hemangiopericytomas. Arch Pathol Lab Med 127:432–439
Wagner E (1870) Das tuberkelahnliche lymphadenom (der cytogene oder reticulirte tuberkel). Arch Heilk (Leipig) 11:497
Yilmaz C, Kabatas S, Ozen OI, Gulsen S, Caner H, Altinors N (2009) Solitary fibrous tumor. J Clin Neurosci 16:1578–1581
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Geib, G., Furlanetto, T.W. (2013). Solitary Fibrous Tumors. In: Hayat, M. (eds) Tumors of the Central Nervous System, Volume 10. Tumors of the Central Nervous System, vol 10. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-5681-6_13
Download citation
DOI: https://doi.org/10.1007/978-94-007-5681-6_13
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-5680-9
Online ISBN: 978-94-007-5681-6
eBook Packages: MedicineMedicine (R0)