
Abstract
Protein tyrosine kinases are enzymes that are capable of adding a phosphate group to specific tyrosines on target proteins. A receptor tyrosine kinase (RTK) is a tyrosine kinase located at the cellular membrane and is activated by binding of a ligand via its extracellular domain. Protein phosphorylation by kinases is an important mechanism for communicating signals within a cell and regulating cellular activity; furthermore, this mechanism functions as an “on” or “off” switch in many cellular functions. Ninety unique tyrosine kinase genes, including 58 RTKs, were identified in the human genome; the products of these genes regulate cellular proliferation, survival, differentiation, function, and motility. Tyrosine kinases play a critical role in the development and progression of many types of cancer, in addition to their roles as key regulators of normal cellular processes. Recent studies have revealed that RTKs such as epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), c-Met, Tie, Axl, discoidin domain receptor 1 (DDR1), and erythropoietin-producing human hepatocellular carcinoma (Eph) play a major role in glioma invasion. Herein, we summarize recent advances in understanding the role of RTKs in glioma pathobiology, especially the invasive phenotype, and present the perspective that RTKs are a potential target of glioma therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Glioma
- Glioblastoma
- Invasion
- Tyrosine kinase receptor
- EGFR
- PDGFR
- c-Met
- Tie
- Axl
- DDR1
- Eph
- TrkA
- Cross-talk
- Tyrosine kinase inhibitor
- Clinical trial
8.1 Introduction
Glioblastoma (GBM) is an extremely aggressive, highly vascularized, infiltrative tumor with a median survival duration of 12–15 months after initial diagnosis (Nagane 2011). GBMs invade the surrounding brain tissue, making complete surgical excision highly improbable. Despite current therapeutic strategies, these tumors almost universally recur because of the invading GBM cells left after excision and are associated with a poor survival rate (Nakada et al. 2007).
Currently, numerous studies are attempting to decipher the molecular mechanism of invasion and to better understand the molecular mechanisms responsible for invasion processes (Chuang et al. 2004; Salhia et al. 2005; Nakada et al. 2007; Onishi et al. 2011). Among the many molecules that were reported as invasion promoters, members of the protein tyrosine kinase (PTK) family play a major role in modulating invasion (Nakada et al. 2007). The human protein kinase genome contains 518 protein kinase genes, including PTK genes that encode transmembrane receptor tyrosine kinases (RTKs) and soluble cytoplasmic tyrosine kinases that are also known as non-RTKs (Manning et al. 2002). More than 58 mammalian RTKs and 37 non-RTKs have been identified. RTKs contain an intracellular catalytic PTK domain and regulatory sequences, a transmembrane domain, and an extracellular ligand-binding domain. RTKs modulate a wide range of cellular events, including proliferation, migration, metabolism, differentiation, and apoptosis, under physiological as well as pathological conditions (Schlessinger 2000). The phosphorylation of tyrosine residues in RTKs is essential for maintaining cellular homeostasis and modulating gene expression in various intercellular and intracellular signaling pathways. Because the complex signaling network triggered by RTKs eventually leads to either activation or repression of various gene subsets, RTKs regulate intercellular communication and control cell proliferation, mitogenesis, survival, differentiation, motility, and metabolism (Schlessinger 2000). The individual cellular consequences of RTK activation are complex and depend on the cell type and the activated signal transduction pathway.
Several lines of experimental evidence have revealed that aberrant RTK activation frequently occurs during glioma initiation and progression and that these tumorigenic cascades may cooperate through multiple signaling cross-talks in the malignant transformation of cells, treatment resistance, and disease relapse (Table 8.1). Because GBMs actively synthesize a substantial variety of RTKs that contribute to invasion, a systematic approach to inhibiting RTKs is being undertaken as a treatment adjunct.
In this chapter, the most recent advancements in the structural and functional characterization of invasion signal transduction elements of the RTK signaling network and the molecular mechanisms involved in glioma invasion are described. We provide an overview of this field, highlighting areas with the strongest research evidence for the translational potential of the use of tyrosine kinase inhibitors (TKIs).
8.2 EGFR/EGF
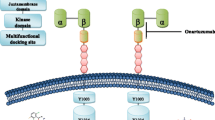
Epidermal growth factor receptor (EGFR) belongs to a large family of cell surface receptors with intrinsic protein tyrosine kinase activity. The EGFR family comprises four members designated EGFR (also known as ErbB1/HER1), ErbB2 (HER2/Neu), ErbB3 (HER3), and ErbB4 (HER4) (Jorissen et al. 2003). Six ligands are known to activate EGFR: EGF, transforming growth factor alpha (TGF-α), amphiregulin, betacellulin, heparin-binding EGF-like growth factor (HB-EGF), and epiregulin (Bogdan and Klambt 2001; Normanno et al. 2006). These ligands are secreted by glioma cells as well as by tumor microenvironmental cells such as microglias and reactive astrocytes (Hoelzinger et al. 2007). Upon binding of extracellular ligands, EGFR undergoes dimerization, resulting in trans-autophosphorylation of its cytoplasmic domain. EGFR can pair with another EGFR to form an active homodimer or with another member of the EGFR family to create a heterodimer (Yarden and Sliwkowski 2001). For example, EGFR can easily form heterodimers with ErbB2, which has a reduced internalization capacity compared with that of EGFR and thus prolongs EGFR signaling (Jones et al. 2006) (Fig. 8.1).

Cell signaling pathways induced by receptors tyrosine kinase (RTK). Homo-/heterodimerization of RTKs are caused by their ligands in autocrine or paracrine fashion. The dimerized receptors can initiate signal transduction cascades involved in cell survival, proliferation, motility and angiogenesis and so on, e.g.: phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR; RAS/RAF/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK); Janus kinase (JAK)/signal transducer and activator of transcription (STAT); and phospholipase Cγ (PLC)γ/protein kinase C (PKC). Examples of cross-talk between RTKs’ signaling and proteins associated with cell invasion (e.g. urokinase-type plasminogen activator (uPA), matrix metalloproteinase (MMP)s, focal adhesion kinase (FAK) and phosphatase and tensin homolog deleted from chromosome 10 (PTEN)) are demonstrated. Red arrows and blue bars indicate activation and suppression, respectively (ECM, extracellular matrix)
EGFR and its ligands are often over-expressed in human carcinomas. In GBM, EGFR gene amplification is the most frequent RTK alteration (approximately 40 %) (Libermann et al. 1985). EGFR overexpression and/or gene alteration is frequently observed in primary (de novo) GBM, which develops rapidly, has a short clinical history, and does not show evidence of less malignant precursor lesions (Ekstrand et al. 1992; Ohgaki et al. 2004; Wong et al. 1987). Genomic analysis by The Cancer Genome Atlas (TCGA) network revealed that EGFR aberration is related to the classical subtype of GBM (Network 2008). Amplification of the EGFR gene is also associated with structural alterations and the most common of these is called EGFR variant III (EGFRvIII). EGFRvIII is a mutant with an in-frame deletion of exons 2–7 from the extracellular region and can transmit constitutive growth signaling in a ligand-independent manner (Ekstrand et al. 1992; Yamazaki et al. 1988). EGFRvIII expression in glioma cells stimulates expression of TGF-α and HB-EGF, suggesting that EGFRvIII plays a role in generating an autocrine loop with wild-type EGFR expression (Ramnarain et al. 2006). Genetic alterations that affect EGFR signaling result in the activation of several downstream pathways such as the phosphatidylinositol 3-kinase (PI3K)/Akt and Ras/Raf/MEK (MAPK kinase)/MAPK (mitogen-activated protein kinase) pathways, which mediate cell proliferation, survival, and mobility (Kita et al. 2007; Ohgaki and Kleihues 2009). The PI3K/Akt pathway is negatively regulated by the tumor suppressor phosphatase and tensin homolog deleted from chromosome 10 (PTEN), which is mutated in 20–40 % of GBMs and is a hallmark of this disease (Ohgaki et al. 2004). PTEN dephosphorylates focal adhesion kinase (FAK), which is a key molecule for cell interaction with the extracellular matrix (ECM) (Gu et al. 1999; Kita et al. 2001; Tamura et al. 1998). Dephosphorylated FAK interferes with EGFR vIII-mediated glioma cell invasion, indicating that the EGFR-PTEN-FAK interaction plays an important role in glioma invasion (Cai et al. 2005) (Fig. 8.1).
Several reports have showed that GBM patients with EGFR overexpression or mutation have shorter survival, suggesting that EGFR alterations are associated with highly aggressive GBMs (Barker et al. 2001; Feldkamp et al. 1999; Shinojima et al. 2003). One report indicated that coexpression of EGFRvIII and PTEN was a positive indicator of responsiveness to EGFR inhibitors (e.g., gefitinib and erlotinib) in patients with GBM (Mellinghoff et al. 2005).
8.3 PDGFR/PDGF
Following EGFR/EGF signaling, aberrant platelet-derived growth factor receptor (PDGFR)/PDGF signaling is one of the hallmarks of GBM biology. Overexpression of PDGFR subtypes α and β and PDGF ligands A–D has been observed in glial tumors of all grades and is possibly associated with malignant progression (Fleming et al. 1992; Guha et al. 1995; Hermanson et al. 1992; Lokker et al. 2002; Nister et al. 1988; Ozawa et al. 2010). An experimental study revealed that glioma-like tumors can be induced after overproduction of PDGFB in the mouse brain (Uhrbom et al. 1998). Histochemical studies revealed that PDGFRα and PDGFA are expressed in glioma cells, whereas PDGFRβ and PDGFB have been found in the surrounding endothelial cells (ECs) (Hermanson et al. 1992; Plate et al. 1992). The expression of this receptor in blood vessels suggests that paracrine activation is also possible with respect to tumor cell migration and colony formation (Hoelzinger et al. 2007; Shih and Holland 2006). Although the expression of PDGFC and D ligands has also been demonstrated in gliomas, the clinical and biological significance of their expression has not been determined (Lokker et al. 2002). Ligand-receptor co-expressions in tumor cells allow for both autocrine and paracrine forms of activation. Some reports suggest that PDGFR/PDGF signaling thorough this autocrine/paracrine pair results in increased GBM cell motility in vivo (Cattaneo et al. 2006; Natarajan et al. 2006). Furthermore, these autocrine and/or paracrine loops can stimulate downstream signal transduction pathways including Ras/MAPK, PI3K/Akt and Janus kinase (JAK)/signal transducer and activator of transcription (STAT) and have pivotal roles in proliferation, differentiation, survival, and invasion (Blume-Jensen and Hunter 2001; Khoshyomn et al. 1999; Valius and Kazlauskas 1993) (Fig. 8.1).
The association between PDGFR expression and the prognosis of glioma patients is controversial. PDGFRα expression in low-grade gliomas was reportedly associated with both poor (Varela et al. 2004) and favorable (Ribom et al. 2002) prognosis. Furthermore, PDGFA expression, but not PDGFRα expression, was useful in predicting tumor grade in oligodendrogliomas (Majumdar et al. 2009). PDGFRα expression was not associated with survival in a series of high-grade pediatric gliomas and GBMs (Liang et al. 2008; Martinho et al. 2009). However, the absence of PDGFA expression was significantly associated with poor prognosis in patients with glioma (Martinho et al. 2009). PDGFR and ligand overexpression tend to be associated with loss of the TP53 tumor suppressor, which is characteristic of secondary GBMs that develop from less malignant precursors (Ohgaki and Kleihues 2009). According to the TCGA consortium, PDGFRA amplification (11 %) and isocitrate dehydrogenase-1 (IDH1) mutation are hallmarks of a proneural subtype of GBM, suggesting an association between this subtype and secondary GBM (Network 2008; Verhaak et al. 2010). Another study demonstrated that PDGFRA amplification was observed in 21 % of gliomas, although PDGFRA-activating mutations were not found (Martinho et al. 2009).
8.4 ERBB2
ErbB2 (HER2/Neu) belongs to the EGFR receptor family that contains the other three members: EGFR, ErbB3, and ErbB4 (see EGFR/EGF section). Although the intracellular tyrosine kinase domain of the EGFR family is highly conserved, none of the EGFR family ligands bind ErbB2 because of its extracellular region structure. Thus, the role of ErbB2 depends on the patterns of dimerization within the family (Normanno et al. 2006). For example, among all possible ErbB2-containing heterodimeric receptor complexes, the most potent signaling module in terms of cell proliferation and in vitro transformation is the ErbB2/ErbB3 heterodimer, even though ErbB3 lacks kinase activity (Citri et al. 2003). These homo- or heterodimers have various potencies for the induction of signaling pathways such as the Ras/Raf/MEK/MAPK pathway for proliferation, the PI3K/Akt pathway for survival (Ben-Levy et al. 1994; Prigent and Gullick 1994), the phospholipase C (PLC)-γ pathway for cell migration and invasion (Khoshyomn et al. 1999), and the STAT pathway for cell cycle regulation (Gao et al. 2010) (Fig. 8.1).
Forced ErbB2 expression can transform cells into the invasive phenotype in association with expression of membrane-type1 (MT1) matrix metalloproteinase (MMP), which is a key enzyme in glioma invasion (Miyamori et al. 2000; Nakada et al. 2001; Sato et al. 1994). In addition, post-transcriptional shedding of cell surface ErbB2 has been reported to be processed by proteases that can degrade the ECM (e.g., MMP2, MMP9, MMP13, and uPA) (Gondi et al. 2009; Spencer et al. 2000; Yong et al. 2010), indicating a strong association between ErbB2 expression and glioma invasion.
Although ErbB2 protein expression is mainly seen in high-grade gliomas (Andersson et al. 2004; Engelhard et al. 1995), evidence for the prognostic value of ErbB2 expression levels in GBMs is presently sparse (Gulati et al. 2010; Haapasalo et al. 1996; Hiesiger et al. 1993; Mineo et al. 2007; Schwechheimer et al. 1994). On the other hand, both ErbB2 and ErbB4 expression levels have been shown to predict prognosis in childhood medulloblastoma and ependymoma (Gilbertson et al. 1997; 2002). Although few reports have described mutation of the ErbB2 gene (Stephens et al. 2004), a recent study of the TCGA consortium revealed that mutation was observed in 7 of 91 (8 %) GBMs (Network 2008; Verhaak et al. 2010).
Furthermore, ErbB2 overexpression has been well described in human breast cancers (20–30 %), which correlates with more aggressive tumors and a poorer prognosis (Hortobagyi 2005).
8.5 c-Met/HGF
Hepatocyte growth factor (HGF) was originally identified as a polypeptide growth factor for hepatocytes and is believed to play an important role in liver regeneration (Yamada et al. 1994). HGF functions as a mitogen for a variety of cell types and as a morphogen and motogen for some epithelial cells that express its receptor (Moriyama et al. 1995). The receptor protein for HGF, c-Met, is encoded by the c-met proto-oncogene, which has tyrosine kinase activity and was originally described as an activated oncogene in a human osteosarcoma cell line (Cooper et al. 1984). Recently, attention has been focused on the role of the HGF/c-Met system because of its multiple biological activities including motility, proliferation, survival, and morphogenesis.
Under normal conditions, HGF-induced c-Met activation is tightly regulated by paracrine ligand delivery, ligand activation at the target cell surface, and ligand activated receptor internalization and degradation (Cecchi et al. 2010). Despite its functions under normal conditions, HGF/c-Met signaling contributes to oncogenesis and tumor progression in several cancers and promotes aggressive cellular invasiveness that is strongly linked to tumor metastasis (Rosario and Birchmeier 2003; Zhang and Vande Woude 2003).
The HGF/c-Met pathway has been implicated in a wide variety of human malignancies. Overexpres-sion of HGF and/or c-Met is frequently observed and amplification of the c-met gene has been reported in several tumor types (Burgess et al. 2006). Several studies have described the expression of HGF and c-met as well as HGF activator mRNAs in glioma cell lines and tissues, particularly GBM (Koochekpour et al. 1997; Moriyama et al. 1995). On the other hand, the expression of HGF and c-Met was low or barely detectable in low-grade astrocytoma and c-Met immunoreactivity was correlated with the histological grade of the tumor.
A few studies suggested that HGF and c-Met are expressed in human gliomas and that expression levels correlated with tumor grade (Moriyama et al. 1998b). Likewise, HGF expression was significantly higher in high-grade tumors than in low-grade tumors based on HGF content detection in samples of different clinical grades (Abounader and Laterra 2005; Lamszus et al. 1999). c-Met receptor expression has also been detected in malignant brain tumors including all gliomas, medulloblastomas, ependymomas, and schwannomas (Koochekpour et al. 1997; Moriyama et al. 1998b). The results of an overexpression study provided sufficient evidence implicating the HGF/c-Met pathway in brain tumorigenesis and malignant progression; this study demonstrated that HGF/c-Met plays important and critical roles in brain tumor formation and growth.
The HGF/c-Met system in vascular ECs works as signal transduction molecules/pathways in gliomas that mediate neovascularization. Numerous in vivo and in vitro studies have indicated that HGF and c-Met are expressed and functional in neuromicrovascular and brain tumor vascular cells. The HGF/c-Met system is highly activated in cultured neural microvascular ECs. In gliomas, HGF stimulates the proliferation of neuromicrovascular ECs by paracrine and autocrine mechanisms. Other studies demonstrated that HGF-dependent interactions between glioma cells, and between glioma cells and the endothelium, can contribute to the heterogeneous proliferative and angiogenic phenotypes of malignant gliomas in vivo (Abounader and Laterra 2005). Moriyama et al. explored the effect of HGF on vascular endothelial growth factor (VEGF) expression in c-Met-positive human glioma cell lines and their results suggest that HGF can act as an indirect angiogenic factor through autocrine induction of VEGF expression and secretion in malignant gliomas in addition to its direct angiogenic activities (Moriyama et al. 1998a). Taken together, these in vitro and in vivo findings suggest the multifunctional and multilevel involvement of the HGF/c-Met pathway in brain tumor angiogenesis as well as brain tumor growth.
Activation of the HGF/c-Met axis promotes proliferation and survival through a variety of downstream effectors including Gab1, Grb2, and PI3K (Abounader and Laterra 2005; Ponzetto et al. 1994). Following activation of the HGF/c-Met pathway, the RAS-MAPK signaling pathway plays an essential role in morphogenesis. The activation of c-Met prevents apoptosis through activation of PI3K and subsequent Akt activation. Cross-talk between the PI3K/Akt and the RAS-MAPK pathways has been implicated in promoting cell survival, migration, and invasion (Zeng et al. 2002) (Fig. 8.2).

Major signaling pathways by the receptor tyrosine kinase c-Met. Growth factor receptor-bound protein 2 (GRB2), Grb2-associated adaptor protein 1 (GAB1), hepatocyte growth factor (HGF), phosphatidylinositol 3-kinase (PI3K), son of sevenless (SOS), rat sarcoma oncogene homolog (RAS), extracellular receptor kinase (ERK), mitogen-activated protein kinase (MAPK), signal transducer and activator of transcription (STAT), SRC, SRC homology protein tyrosine phosphatase 2 (SHP2), SRC homology domain c-terminal adaptor homolog (SHC), mammalian target of rapamycin (mTOR)
8.6 Tie/Ang
The Angiopoietin/Tie system acts as a vascular-specific ligand/receptor system and plays an essential role in tumor-associated angiogenesis. The angiopoietin family includes four ligands, namely, angiopoietin-1 (Ang-1), angiopoietin-2 (Ang-2), angiopoietin-3 (Ang-3), and angiopoietin-4 (Ang-4), and two corresponding RTKs (Tie1 and Tie2). Ang-1 acts as an agonist, whereas Ang-2 acts as an antagonist, of the Tie2 receptor (Maisonpierre et al. 1997). Tie2 activation promotes vessel assembly and maturation by mediating EC survival signals and regulating the recruitment of mural cells. The Tie/Ang-1 system may function during vessel maturation and stabilization and Ang-1 is a prominent regulator of vascular development (Lee et al. 2009; Machein et al. 2004). In contrast, Ang-2 is produced by ECs and acts as an autocrine antagonist of Ang-1-mediated Tie2 activation. Ang-2 is an angiogenic factor that antagonizes Ang-1 activity by competitively inhibiting the binding of Ang-1 to its cognate endothelial receptor, Tie2, causing vasculature destabilization. Ang-2 also acts in concert with VEGF to regulate vessel growth (Hu et al. 2006) (Fig. 8.3).

The major Ang/Tie signaling pathways in glioma angiogenesis and migration. Angiopoietin (Ang), growth factor receptor-bound protein 2 (GRB2), phosphatidylinositol 3-kinase (PI3K), rat sarcoma oncogene homolog (RAS), SH2 domain-containing phosphatase (SHP2), focal adhesion kinase (FAK), endothelial nitric oxide synthase (eNOS), docking protein R (Dok-R), extracellular receptor kinase (ERK), p21-activated kinase (PAK)
The role of Ang-1 in tumor-associated angiogenesis remains controversial because some reports imply that Ang-1 induction impairs angiogenesis and inhibits tumor growth whereas other authors suggest that Ang-1 overexpression promotes tumor growth in some animal models (Hawighorst et al. 2002; Hayes et al. 2000; Machein et al. 2004). However, Machein et al. reported that Ang-1 can promote tumor angiogenesis in a rat glioma model (Machein et al. 2004).
Ang-2 immunoreactivity is higher in malignant gliomas than in low-grade gliomas, which documents a strong correlation between the expression of Ang-2 and increasing glioma tumor grade (Koga et al. 2001). Increasing data suggest that Ang-2 expression is negatively correlated with vessel maturation in malignant gliomas and that VEGF expression is positively correlated with vessel maturation in low-grade gliomas.
Conflicting results have been reported in the literature regarding the role of the Tie2/Ang system in tumor angiogenesis. The tyrosine kinase receptor Tie2 was initially reported as a specific vascular receptor present in both normal and tumoral ECs, including ECs in astrocytomas, and its levels correlate positively with increasing malignancy (Liu et al. 2010; Stratmann et al. 1998). Tie2 is also expressed in glioma cells and brain tumor stem cells present in malignant gliomas and its expression and activation increases with increasing astrocytoma malignancy grade (Zadeh et al. 2004b).
Studies on in vivo human glioma biopsies have showed that Ang-2, MMP-2, MT1-MMP, and laminin 5γ2 are co-overexpressed in the invasive areas but not in the central regions of the glioma tissues. In vitro data also demonstrated that Ang-2 promoted the expression and activation of MMP-2, MT1-MMP, and laminin 5γ2, which may be essential for malignant glioma invasiveness (Brinckerhoff and Matrisian 2002; Lohi 2001). In addition, inhibition of Tie2 activation significantly decreased GBM xenograft growth by disrupting tumor vascularity (Zadeh et al. 2004b).
Accumulating evidence suggests that Tie2 activation regulates angiogenesis in a highly context- and tissue-dependent manner and closely collaborates with VEGF and possibly with other angiogenesis regulators (Gale et al. 2002; Zadeh et al. 2004b). Activation of the Tie/Ang functional axis promotes glioma proliferation and migration through various downstream effectors, including FAK, VEGF-A, PI3K, αγβ1, and extracellular signal-regulated kinase 1/2 (ERK1/2) (Brinckerhoff and Matrisian 2002; Lee et al. 2009; Zadeh et al. 2004 b). Following activation of the Tie/Ang pathway, the FAK signaling pathway plays an essential role in malignant cell migration. The activation of Tie-2 induces VEGF-A to bind its EC-specific receptors, VEGFR1 and VEGFR2, which induces internalization of vascular endothelial cadherin (Zadeh et al. 2004a).
Furthermore, Tie receptor-independent signaling and non-vascular Ang effects can produce other intracellular signaling outcomes. ECs can adhere to immobilized Ang via the αγβ1 integrin and FAK signaling pathway to induce glioma cell invasion by stimulating MMP-2 expression (Hu et al. 2006). The activation of the Tie-2/Ang-4 functional axis promotes the in vivo growth of human GBM cells by promoting tumor angiogenesis and directly activating ERK1/2 in GBM cells (Brinckerhoff and Matrisian 2002).
Although the preceding studies focused on elucidating the role of Ang-2 in cancer biology, its exact role in glioma angiogenesis remains elusive. A recent report demonstrated that Ang-2 significantly enhances vascular growth and induces aberrant pathological changes in malignant astrocytomas; this report also found that Ang-2 is not consistently elevated throughout all growth stages of malignant astrocytomas (Zadeh et al. 2010).
Although Ang-1 and Ang-2 play known roles in tumor angiogenesis, it is unknown how Ang-4 affects GBM angiogenesis progression and the mechanism underlying its effects. Brunckhorst et al. found a novel mechanism of Ang-4 on glioma progression that directly activates the ERK1/2 kinase pathway (Brunckhorst et al. 2010). Taken together, these data indicate that the Tie-2/Ang-4 functional axis can be considered an attractive therapeutic target for GBM.
8.7 Axl/Gas6
The receptor tyrosine kinase Axl, a member of the Tyro3, Axl, and Mer family of receptor tyrosine kinases (TAMRs), is characterized by an extracellular domain consisting of two immunoglobulin-like domains in juxtaposition to two fibronectin type III domains (Janssen et al. 1991). TAMRs, in particular Axl, have transforming properties: overexpression of a truncated version of Axl in premalignant cells is sufficient to induce tumors in mice (Zhang et al. 1996). Growth arrest–specific gene 6 (Gas6), which is the natural ligand of Axl, was discovered because its expression is upregulated in fibroblasts under growth-arrest conditions (Manfioletti et al. 1993). Axl/Gas6 signaling has been shown to regulate survival, proliferation, and migration in a variety of cells in vitro including tumor-derived cell lines of epithelial, mesenchymal, and hematopoietic origin (Hafizi and Dahlback 2006).
Axl and Gas6 are overexpressed in human gliomas of malignancy grades WHO II to IV. In contrast, Axl staining was not detectable in non-neoplastic brain tissue and Gas6 was strongly expressed in neurons (Hutterer et al. 2008). The receptor tyrosine kinase Axl is a mediator of glioma growth and invasion. Axl is predominantly expressed in pseudopalisading glioma cells, which are characterized by an accumulation of tumor cells around necrotic areas. Furthermore, an accumulation of Axl-positive tumor cells was observed adjacent to microvascular neoformations, which is a characteristic feature of invading glioma tumor cells spreading along perivascular regions. Inhibition of Axl signaling by overexpression of a dominant-negative receptor mutant suppressed experimental gliomagenesis and resulted in long-term survival of mice after intracerebral glioma cell implantation when compared with Axl wild-type transfected tumor cells. Inhibition of Axl signaling interfered with cell proliferation (30 % inhibition versus Axl wild-type), glioma cell migration (90 % inhibition versus Axl wild-type), and invasion (79 % inhibition versus Axl wild-type) However, an analysis of tumor vessel density and diameter failed to reveal an attenuated tumor vasculature as an explanation for the reduced tumorigenesis in Axl dominant-negative cells (Vajkoczy et al. 2006). GBM patients with high Axl expression and Axl/Gas6 coexpression showed a significantly shorter time to tumor progression and poorer overall survival, indicating that Axl and Gas6 expression predict poor prognosis in GBM patients (Hutterer et al. 2008).
8.8 DDR1/Collagen
Discoidin domain receptor 1 (DDR1) tyrosine kinases constitute a family of non-integrin collagen receptors that contain a discoidin homology region in the ectodomain (Alves et al. 1995). DDR1, which is mainly expressed in epithelial cells, is primarily activated by collagens I to IV and VIII and facilitates cell adhesion (Vogel et al. 1997). DDR1 has five isoforms generated by alternative splicing: DDR1a, b, c, d and e. DDRs have been implicated in the expression of pro-inflammatory mediators and matrix-degrading enzymes and play an important role in migration, proliferation, ECM remodeling, and wound repair (Vogel et al. 2006).
In GBM, collagen IV is present in virtually all tumor vessels, in some giant glioma cells, and in tumor cells around vascular proliferations. Collagens VII and VIII are absent from normal brain but may be expressed in glioma tissues supporting DDR1 signaling (Senner et al. 2008). Furthermore, DDR1 is overexpressed in glioma. Although overexpression of either DDR1a or DDR1b in cell-based glioma models caused increased cell attachment, glioma cells overexpressing DDR1a exhibit enhanced invasion and migration concomitant with increased levels of MMP-2. Inhibition of MMP activity suppressed DDR1a-stimulated cell-invasion and inhibition of DDR1 reduced DDR1a-mediated invasion and enhanced adhesion of DDR1a and DDR1b overexpressing cells. DDR1a plays a critical role in inducing tumor cell adhesion and invasion, and this invasive phenotype is caused by activation of MMP-2 (Ram et al. 2006; Yamanaka et al. 2006) (Fig. 8.4). DDR1 expression is more closely correlated with survival than histological grade in gliomas, suggesting that DDR1 expression might be a better predictive factor of patient survival than WHO grading (Yamanaka et al. 2006).

DDR1 signaling in glioma. In GBM, DDR1 is overexpressed and activated by collagen IV which is present in virtually all tumor vessels, as well as in some giant glioma cells and in tumor cells around vascular proliferations; also collagens VII and VIII may be expressed in glioma tissues supporting DDR1 signaling. Glioma cells overexpressing DDR1a display enhanced migration and invasion associated with the increase levels of matrix metalloproteinase 2 (MMP-2). (DDR1, discoidin domain receptor 1)
8.9 Eph/Ephrin
Erythropoietin-producing human hepatocellular carcinoma (Eph) receptors comprise the largest family of RTKs in mammals. Eph receptors have been divided into an EphA subclass (nine members) and an EphB subclass (six members) on the basis of extracellular domain sequence homology and ligand affinity (Gale et al. 1996). Ephrin ligands are also transmembrane proteins and have been divided into two subclasses: glycosylphosphatidyl-inositol (GPI)-linked ephrin-As (five members), which are anchored to the cell membrane, and transmembrane ephrin-Bs (three members). Ephrin-As preferentially bind to EphA receptors, while ephrin-Bs preferentially bind to EphB receptors, although promiscuity has been observed.
Eph/ephrins form an essential cell-cell communication system capable of bi-directional intracellular signaling between adjacent cells, where receptor signaling is designated “forward” and ephrin signaling is “reverse” (Heroult et al. 2006; Kullander and Klein 2002). Eph/ephrin members are plentiful and their relationships are complex. Generally, Eph/ephrin interactions are repulsive because cells containing a given Eph are repelled by cells containing the corresponding Ephrin. Through this mechanism, the Eph/ephrin system plays a role in numerous biological processes including cell adhesion and migration during development, especially in the central nervous system (Wimmer-Kleikamp and Lackmann 2005). Recently, a role for the Eph/ephrin system has also emerged in cancer, especially in the area of invasive behavior (Campbell and Robbins 2008).
Genes for Eph/ephrins are overexpressed or differentially expressed in numerous human cancers (Surawska et al. 2004). Mounting evidence documents a strong correlation between the expression and phosphorylation levels of many Eph/ephrin family members and increasing glioma tumor grade, suggesting that elevated Eph/ephrin expression levels may be diagnostic for GBM and reduced patient survival rates (Nakada et al. 2011). Based on the microarray data obtained from two distinct GBM cell phenotypes (invading cells and tumor core cells) collected from GBM specimens, pathway enrichment analysis indicated that EphB/ephrin-B is the most tightly linked system to the invading cell phenotype (Nakada et al. 2010). According to these results, it is likely that the Eph/ephrin system contributes to glioma invasion. In spite of the overexpression of Eph/ephrin members in glioma, no evidence of gene amplification or mutation has been reported.
Numerous Ephs and ephrins have been noted to influence or correlate with the malignant behavior of cancer cells. In glioma, EphA/ephrin-A was associated with proliferation (Wykosky et al. 2005) whereas EphB/ephrin-B was involved in invasion and neovascularization (Erber et al. 2006). EphB2, ephrin-B2, and ephrin-B3 mRNA levels were shown to significantly increase with histological grade in glioma and contribute invasive properties in GBM (Nakada et al. 2004, 2006, 2010). Additionally, a high ephrin-B2 level was shown to confer poor survival (Nakada et al. 2010). The co-expression of EphB2 and ephrin-B in GBM cells suggests the existence of an EphB/ephrin-B interaction through cell–cell contact in GBM. Data showing that glioma invasion is inhibited by blocking Eph/ephrin suggest that the Eph/ephrin system is a potential therapeutic target for invasive glioma.
Different Eph/ephrin molecules are conceivably linked to different intracellular signaling pathways in a cell-type-specific manner, which allows this system to perform a variety of functions. The key signaling molecules in the invasion-signaling pathway induced by EphB/ephrin B in GBM appear to be small GTPases such as Rac and Ras. Ephrin-B2 and ephrin-B3 can activate EphB2 through cell–cell contact, inducing invasion via EphB2 forward signaling. A previous study showed that EphB2 plays a functional role in promoting GBM cell invasion by eliciting signaling through R-Ras and affecting integrin activity (Nakada et al. 2005). In contrast, reverse signaling of the ephrin-B2 and ephrin-B3 ligands was demonstrated to be an important factor in the regulation of glioma cell invasion through the Rac1 GTPase (Fig. 8.5).

Putative model of Eph/ephrin function in glioma. Signaling induced by cell-cell contact via EphB/ephrin-B induces repulsion. Ligand independent signaling via autophosphorylation of Eph without ephrin stimulation can also promote the invasion without cell-cell communication
A greater depth of investigation has occurred with respect to involvement of the Eph/ephrin subsystem in invasion. Fundamental effect of Eph/ephrin necessitates direct cell-cell contact. However, it was recently revealed that ligand-independent signaling via autophosphorylation of Eph without ephrin stimulation can promote invasion without cell-cell communication (Miao et al. 2009) (Fig. 8.5). EphB2 was overexpressed in invading GBM cells compared with stationary cells in the tumor core, suggesting that EphB2 is autophosphorylated in invading glioma cells (Nakada et al. 2004, 2006). This indicates that invading glioma cells overexpressing Eph/ephrin far from the tumor can migrate by themselves without interaction of other cells.
8.10 TrkA
Neurotrophic tyrosine kinase receptor type 1 (TrkA) is the high affinity receptor for nerve growth factor (NGF), neurotrophin-3, and neurotrophin-4/5. Phosphorylated TrkA may play an important role in mitotic spindle assembly because it is colocalized with α-tubulin at the mitotic spindle from prophase to anaphase, whereas in interphase, phosphorylated TrkA is localized on the membrane and processes of glioma cells (Zhang et al. 2005).
TrkA is strongly expressed in the subpopulation of highly infiltrating glioma cells in vivo but not in the glioma cells that remain within the bulk of the tumor. Thus, TrkA expression is dependent on both the cell type and the location within the tumor (Edwards et al. 2011). TrkA activation typically leads to the activation of survival- and growth-mediating pathways through the cytoplasmic proteins SHC, PI3K, and PLC-γ1 (Escalante et al. 2000; Meakin et al. 1999). GBM cell growth can be enhanced by NGF acting via TrkA receptor phosphorylation (Singer et al. 1999). Switching between anti-mitogenic and mitogenic TrkA signaling is controlled by PLC-γ1 activity (Ye et al. 2000, 2002) (Fig. 8.6). Consequently, it was shown that TrkA expression in GBMs attenuates tumor progression in vivo (Rabin et al. 1998) by inducing differentiation of tumor cells from undifferentiated glioma to oligodendrocytes (Pflug et al. 2001).

TrkA signaling pathways in glioma. GBM cell growth can be enhanced by TrkA receptor (neurotrophic tyrosine kinase receptor type 1) phosphorylation activated by NGF (nerve growth factor). TrkA activation leads to the activation of survival and growth mediating pathways through cytoplasmic proteins SHC, PI3K and PLC-γ1. Switching between anti-mitogenic and mitogenic TrkA signaling is controlled by PLC-γ1 activity. Glioma initiating/stem cells require integrin β1–TrkA complex for connective tissue growth factor (CTGF) signaling; binding of the complex with CTGF causes NF-kB-mediated activation of the ZEB-1 promoter with subsequent induction of the ZEB-1 transcriptional repressor resulting in decreased expression of E-cadherin, accordingly enhancing glioma cell invasion and migration
Glioma initiating/stem cells require the cell surface protein receptors integrin β1 and TrkA, which constitute the integrin β1–TrkA complex, for connective tissue growth factor (CTGF) signaling. The integrin β1–TrkA complex can bind to CTGF and cause nuclear factor-kappa B (NF-kB)-mediated activation of the ZEB-1 promoter with subsequent induction of the ZEB-1 transcriptional repressor, resulting in decreased expression of E-cadherin and subsequent enhancement of glioma cell invasion and migration (Fig. 8.6). TrkA knockdown resulted in decreased CTGF-induced cell migration and the absence of tumor cell invasion into normal mice cortex. However, TrkA knockdown did not affect CTGF-induced proliferation or the clonogenicity of the glioma stem cells in vitro. Thus, CTGF, TrkA, and NF-kB may be potential therapeutic targets to alleviate tumor cell infiltration (Edwards et al. 2011).
8.11 Cross-Talk
As described above, many RTKs lead to the signaling of PI3K/Akt and Rac1 in invading glioma cells, suggesting that these molecules confer critical downstream signaling for invasion. A previous study showed coexpression of multiple activated RTKs in individual dissociated cells from a primary GBM (Stommel et al. 2007). Accordingly, multiple RTKs may be simultaneously or sequentially used by GBM cells to maintain invasion-signaling pathways via molecules such as PI3K/Akt and Rac1.
Given that individual cells express multiple RTKs, it is reasonable to speculate that these RTKs are interacting with each other. For example, the c-Met receptor is strongly phosphorylated as a function of EGFRvIII receptor levels, suggesting the presence of cross-talk between c-Met and EGFRvIII signaling, although the intermediary molecule has not yet been elucidated (Huang et al. 2007). The Axl RTK follows a similar phosphorylation response as a function of EGFRvIII levels (Huang et al. 2007). It was previously reported that EGFR and EphA2 are expressed in GBM and co-localize to the cell surface. EphA2 phosphorylation is dependent on EGFR activity and EphA2 downregulation inhibits EGFR phosphorylation, downstream signaling, and EGF-induced cell viability (Ramnarain et al. 2006). Previous studies reported that EphA4, whose expression is correlated with increasing glioma grade, forms a heteroreceptor complex with fibroblast growth factor receptor 1 (FGFR1) in glioma cells and that the EphA4-FGFR1 complex potentiated FGFR-mediated downstream signaling such as Akt/MAPK, Rac1, and Cdc42 pathways, resulting in the promotion of invasion (Fukai et al. 2008).
In such a multiple-input system with cross-talk among RTKs, a single-agent of anti-RTK inhibition might be incapable of sufficiently suppressing invasion signaling, resulting in insensitivity to invasion inhibition by any single agent and a lack of clinical efficacy. It is anticipated that combinations of drugs against different activated RTKs or single drugs with inhibitory activities against multiple activated RTKs will have more favorable outcomes.
8.12 Targeting Receptor Type Tyrosine Kinases
As mentioned in the previous paragraphs, TKIs that target multiple signaling pathways and critical growth factors essential for tumor progression have become a major focus of interest in various clinical studies. Although all studies remain experimental, several of these specific molecular agents that are most expectant or have been widely evaluated are reviewed below (Table 8.2).
8.12.1 Targeting EGFR
8.12.1.1 Gefitinib
Gefitinib is a low molecular weight, selective inhibitor of EGFR tyrosine kinase and was the first drug of this type. Although an early study suggested that gefitinib was active in patients with malignant gliomas (Franceschi et al. 2007; Mellinghoff et al. 2005), gefitinib therapy showed limited efficacy in multicenter phase II studies in patients with malignant glioma (Franceschi et al. 2007; Hegi et al. 2011; Rich et al. 2004), either for recurrent disease or as part of the initial treatment regimen. Because EGFR kinase domain mutations, which are associated with the greatest sensitivity to EGFR inhibitors (Pedersen et al. 2005), are uncommon in malignant gliomas (Lassman et al. 2005), it remains to be determined whether other molecular alterations involving EGFR signaling pathways occur selectively in treatment responders.
8.12.1.2 Erlotinib
Erlotinib is an orally active, reversible EGFR TKI. Because erlotinib is metabolized by the cytochrome P450 isoenzymes 3A4 (70 %) and CYP 1A2 (30 %), patients taking enzyme-inducing anti-epileptic drugs are not eligible for this treatment (Raizer et al. 2010). Similar to gefitinib, clinical studies examining the therapeutic efficacy of erlotinib have so far failed to demonstrate a major therapeutic break-through in the setting of GBM (Haas-Kogan et al. 2005; Mellinghoff et al. 2005; Raizer et al. 2010; Van Den Bent et al. 2009), including newly diagnosed GBM treated with temozolomide (TMZ) and radiotherapy (Peereboom et al. 2010). Although Raizer and colleagues reported that the development of a rash during the initial erlotinib administration cycle correlates with survival in patients with non-progressive GBM after radiotherapy, the significance of this finding remains unclear (Raizer et al. 2010).
8.12.1.3 125I-mAb 425
125I-mAb 425, an 125I-labeled anti–EGFR 425 murine monoclonal antibody (mAb), is an IgG2a isotype developed from mice immunized with A-431 epidermoid carcinoma cells. mAb 425 binds to the tumor and produces anticarcinogenic effects mediated by direct cell growth inhibition, complement-dependent cytotoxicity, and activation of the humoral response. Initial clinical studies have shown survival benefits of adjuvant 125I-425 mAb in GBM patients (Emrich et al. 2002; Quang and Brady 2004). In a phase II study of 192 patients with GBM treated with anti–EGFR 125I-mAb 425 radioimmunotherapy, the reported survival was 15.7 months and treatment was safe and well tolerated (Li et al. 2010).
8.12.1.4 Nimotuzumab
Nimotuzumab (h-R3), a humanized monoclonal antibody directed against the EGFR, consequently inhibits tyrosine kinase activation. Recently, several reports of pediatric diffuse intrinsic pontine glioma treated with nimotuzumab showed a relatively favorable prognosis (Lam et al. 2009; Mateos et al. 2011; Saurez et al. 2009). Thus, nimotuzumab is currently being evaluated in conjunction with radiotherapy in a phase III trial in children with diffuse pontine glioma and adult GBM.
8.12.1.5 Cetuximab
Cetuximab, IMC-C225, is a chimeric (mouse/human) monoclonal IgG1 antibody that binds to EGFR with high specificity and affinity. Encouragingly, cetuximab enhanced the cytotoxicity produced by radiation therapy in vivo in EGFR-amplified GBM (Belda-Iniesta et al. 2006; Eller et al. 2005). In 2006, however, a phase II trial of cetuximab in 55 recurrent high-grade glioma patients revealed no significant correlation between response, survival, and EGFR amplification (Neyns et al. 2009).
8.12.1.6 Lapatinib
Lapatinib is a dual TKI that interrupts the HER2/neu (ErbB2) growth receptor pathway. It is used in therapy for HER2-positive breast cancer. However, a phase I/II trial of lapatinib in patients with relapsed GBM failed to show significant activity of this agent independent of the presence of EGFRvIII mutation and PTEN immunohistochemical status (Thiessen et al. 2010).
8.12.2 Targeting PDGFR
8.12.2.1 Imatinib
Imatinib mesylate (Gleevec; formerly known as STI571) is a potent inhibitor of the PDGFRα, PDGFRβ, Bcr-Abl, c-Fms, and c-Kit tyrosine kinases. Its antitumor activities in chronic myelogenous leukemia and gastrointestinal stromal tumors result from the inhibitions of Bcr-Abl (Druker et al. 2001) and c-Kit (Demetri et al. 2002), respectively. An initial study showed that imatinib mesylate plus hydroxyurea was well tolerated and associated with durable antitumor activity in some patients with recurrent GBM (Reardon et al. 2005). However, more extensive phase II and III studies have shown only minimal evidence of single agent activity (Dresemann et al. 2010; Raymond et al. 2008; Wen et al. 2006).
8.12.2.2 Sunitinib
Sunitinib malate is an oral small-molecule inhibitor of VEGFRs, PDGFRs, c-Kit, Flt3, and RET kinases (Chow and Eckhardt 2007). Sunitinib has substantial clinical activity against hypoxia-inducible factor (HIF)/VEGF-dependent, PDGFR-dependent, and KIT-dependent cancers. Recently, a phase II study of sunitinib failed to demonstrate a relevant clinical benefit from single-agent sunitinib (37.5 mg/day) in patients with alkylator-refractory recurrent glioma. Furthermore, substantial treatment-related toxicity was observed in several patients (Neyns et al. 2011). Although sunitinib affected the glioma vasculature in a small subgroup of patients, no objective tumor responses were observed regarding apparent reduced cerebral blood-flow and blood-volume within the lesion compared with the normal brain or reduced gadolinium enhancement of the tumor. A possible explanation for the failure of sunitinib to have meaningful clinical activity is the lower potency of sunitinib to selectively inhibit VEGF/VEGFR signaling within the tumor vasculature compared to the VEGF-targeted mAb bevacizumab (Friedman et al. 2009) or the more potent VEGFR-specific small-molecule TKI cediranib (Batchelor et al. 2010a, b).
8.12.2.3 Dasatinib
Dasatinib is an aminotriazole analog with high specificity for several kinases including Bcr-Abl, Src, c-Kit, PDGFRβ, and EphA2, which was approved for use in patients with chronic myelogenous leukemia after imatinib treatment and Philadelphia chromosome-positive acute lymphoblastic leukemia. Dasatinib appears to be a more potent inhibitor of Bcr-Abl than imatinib (Tokarski et al. 2006). Recently, investigators have shown that Src is frequently phosphorylated in GBM cell lines such as T98G and U87 compared with normal tissue and is activated in human GBM tumors (Du et al. 2009). These findings support the possible role of dasatinib for malignant gliomas, and future clinical trials will further assess the clinical value of SRC inhibition with dasatinib incorporating TMZ and other cytotoxic agents.
8.12.3 Multi-Kinase Inhibitors
8.12.3.1 Cediranib
Cediranib (AZD2171) is a potent oral inhibitor of all VEGF receptor tyrosine kinases and PDGF receptors (Batchelor et al. 2007). Cediranib caused a structural and functional normalization of tumor vasculature in all 16 patients with recurrent GBM and resulted in a significant reduction of tumor-associated vasogenic edema as measured by MRI techniques. This effect was paralleled by a potent steroid-sparing effect in most patients (Batchelor et al. 2007). In a subsequent phase II study, partial responses were seen in 57 % of patients based upon tumor measurements (Batchelor et al. 2010 b), and the median progression-free survival (PFS) and the median overall survival (OS) were 32.4 weeks and 16.7 weeks, respectively (Table 8.2), with manageable toxicity. Based upon these results, a phase III study was conducted in 325 patients with recurrent GBM (Batchelor 2010a). Although cediranib treatment regimens resulted in a statistically significant decrease in steroid use and a reduced contrast enhancing area on neuroimaging, there was no statistically significant improvement in PFS, the primary endpoint of the trial.
8.12.3.2 Sorafenib
Sorafenib is a multi-target oral TKI with inhibitory effects on the VEGF receptor, PDGF receptor, and the Ras/Raf signaling pathway (Wilhelm et al. 2004). Despite potentially complementary direct and indirect mechanisms of anti-tumor activity, Reardon et al. demonstrated that sorafenib combined with daily TMZ has minimal activity as a salvage regimen for recurrent GBM patients in their single-arm phase II study (Reardon et al. 2011). Meanwhile, Hainsworth et al. also failed to demonstrate the efficacy of treatment when compared with the results expected with standard therapy in newly diagnosed GBM (Hainsworth et al. 2010). A plausible explanation for this low effectiveness is the low to moderate ability of sorafenib to penetrate into the brain. However, the blood brain barrier (BBB) is compromised in patients with GBM and it is unlikely that sorafenib would have lower penetration than bevacizumab, a large protein. In general, the addition of small molecule TKIs to chemotherapy has not consistently improved treatment results, even when both components of therapy have individual efficacy.
8.12.3.3 Vandetanib
Vandetanib (ZD6474) is a potent oral TKI for various RTKs, in particular VEGFR2 and EGFR. A phase I study of 35 children with newly diagnosed diffuse intrinsic pontine glioma treated with vandetanib reported 1- and 2-year OS outcomes of 37.5 % ± 10.5 % and 21.4 % ± 11 %, respectively. Three patients remained alive with PFS for more than 2 years. The recommended phase II dose of vandetanib in children is 145 mg/m2 per day (Broniscer et al. 2010). Several phase II studies, including those in recurrent and newly diagnosed malignant glioma, are currently underway.
8.12.3.4 Cabozantinib (XL-184)
Recently, interim results have been reported from a phase II study of Cabozantinib (XL184) treatment in previously treated progressive GBM. Cabozantinib is an oral inhibitor of multiple RTKs that includes VEGFR2 as the main target followed by c-Met, RET, c-kit, Flt3, Tie-2, and Axl (Wen 2010; Yakes et al. 2011). In this study, the median PFS in antiangiogenic-naive cohorts was 16 weeks (Table 8.2). In total, 61 % of patients on corticosteroids at baseline had a reduction in corticosteroid dose of at least 50 %. The investigators concluded that XL184 demonstrates encouraging clinical activity in patients with progressive GBM. Cabozantinib, currently in phase III clinical trials, is a promising agent for inhibiting tumor angiogenesis and metastasis in glioma, especially in cases of dysregulated Met and VEGFR signaling.
8.13 Prospective
Future studies are required to more precisely establish the molecular mechanisms and specific downstream signaling elements that contribute to the cooperative or synergistic interactions of RTK signaling pathways in invading glioma cells. Further studies on the manipulation of the RTK systems involved in invasion will aid in engineering GBM therapies and in elucidating the complexity and additional functional implications of RTK systems. Moreover, it is of great therapeutic interest to define invasion-associated RTKs that could be targeted for blocking invasion. These works should help to develop novel potential pharmacological agents to modulate invasion processes and thereby counteract the activation of invasion-signaling pathways and promotion of invasion. The data obtained from GBM patients treated with TKIs should confirm the therapeutic benefit of TKIs and the safety of selectively targeting RTKs, alone or in combination with the current conventional therapies. The effort to combat GBM, along with emerging data regarding the underlying molecular invasion circuitry and the development of specific TKIs, will result in the development of an array of new treatment approaches.
Abbreviations
- Ang:
-
Angiopoietin
- BBB:
-
Blood brain barrier
- CTGF:
-
Connective tissue growth factor
- DDR1:
-
Discoidin domain receptor 1
- EC:
-
Endothelial cell
- ECM:
-
Extracellular matrix
- EGFR:
-
Epidermal growth factor receptor
- Eph:
-
Erythropoietin-producing human hepatocellular carcinoma
- ERK:
-
Extracellular signal-regulated kinase
- FAK:
-
Focal adhesion kinase
- FGFR:
-
Fibroblast growth factor receptor
- Gas6:
-
Growth arrest–specific gene 6
- GBM:
-
Glioblastoma multiforme
- GPI:
-
Glycosylphosphatidyl-inositol
- HB-EGF:
-
Heparin-binding EGF-like growth factor
- HGF:
-
Hepatocyte growth factor
- HIF:
-
Hypoxia inducible factor
- IDH1:
-
Isocitrate dehydrogenase-1
- JAK:
-
Janus kinase
- MAPK:
-
Mitogen-activated protein kinase
- MEK:
-
MAPK kinase
- MMP:
-
Matrix metalloproteinase
- mAb:
-
Monoclonal antibody
- MT1-MMP:
-
Membrane-type1-MMP
- NGF:
-
Nerve growth factor
- NF-kB:
-
Nuclear factor-kappa B
- OS:
-
Overall survival
- PDGFR:
-
Platelet derived growth factor receptor
- PFS:
-
Progression-free survival
- PI3K:
-
Phosphatidylinositol 3-kinase
- PKC:
-
Protein kinase C
- PLC:
-
Phospholipase C
- PTEN:
-
Phosphatase and tensin homolog deleted from chromosome 10
- PTK:
-
Protein tyrosine kinase
- RTK:
-
Receptor tyrosine kinase
- STAT:
-
Signal transducer and activator of transcription
- TAMR:
-
A member of the Tyro3, Axl, and Mer family of receptor tyrosine kinase
- TCGA:
-
The Cancer Genome Atlas
- TGF-α:
-
Transforming growth factor alpha
- TKI:
-
Tyrosine kinase inhibitor
- TMZ:
-
Temozolomide
- TrkA:
-
Neurotrophic tyrosine kinase receptor type 1
- uPA:
-
Urokinase-type plasminogen activator
- VEGF:
-
Vascular endothelial growth factor
References
Abounader R, Laterra J (2005) Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncol 7:436–451
Alves F, Vogel W, Mossie K, Millauer B, Hofler H, Ullrich A (1995) Distinct structural characteristics of discoidin I subfamily receptor tyrosine kinases and complementary expression in human cancer. Oncogene 10:609–618
Andersson U, Guo D, Malmer B, Bergenheim AT, Brannstrom T, Hedman H, Henriksson R (2004) Epidermal growth factor receptor family (EGFR, ErbB2-4) in gliomas and meningiomas. Acta Neuropathol 108:135–142
Barker FG 2nd, Simmons ML, Chang SM, Prados MD, Larson DA, Sneed PK, Wara WM, Berger MS, Chen P, Israel MA, Aldape KD (2001) EGFR overexpression and radiation response in glioblastoma multiforme. Int J Radiat Oncol Biol Phys 51:410–418
Batchelor TT, Sorensen AG, Di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP, Chen PJ, Zhu M, Ancukiewicz M, Mrugala MM, Plotkin S, Drappatz J, Louis DN, Ivy P, Scadden DT, Benner T, Loeffler JS, Wen PY, Jain RK (2007) AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 11:83–95
Batchelor T, Mulholland J, Neyns B et al (2010a) A phase III randomized study comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, with lomustine alone in recurrent glioblastoma patients. Ann Oncol 21:4
Batchelor TT, Duda DG, Di Tomaso E, Ancukiewicz M, Plotkin SR, Gerstner E, Eichler AF, Drappatz J, Hochberg FH, Benner T, Louis DN, Cohen KS, Chea H, Exarhopoulos A, Loeffler JS, Moses MA, Ivy P, Sorensen AG, Wen PY, Jain RK (2010b) Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol 28:2817–2823
Belda-Iniesta C, Carpeno Jde C, Saenz EC, Gutierrez M, Perona R, Baron MG (2006) Long term responses with cetuximab therapy in glioblastoma multiforme. Cancer Biol Ther 5:912–914
Ben-Levy R, Paterson HF, Marshall CJ, Yarden Y (1994) A single autophosphorylation site confers oncogenicity to the Neu/ErbB-2 receptor and enables coupling to the MAP kinase pathway. EMBO J 13:3302–3311
Blume-Jensen P, Hunter T (2001) Oncogenic kinase signalling. Nature 411:355–365
Bogdan S, Klambt C (2001) Epidermal growth factor receptor signaling. Curr Biol 11:R292–295
Brinckerhoff CE, Matrisian LM (2002) Matrix metalloproteinases: a tail of a frog that became a prince. Nat Rev Mol Cell Biol 3:207–214
Broniscer A, Baker JN, Tagen M, Onar-Thomas A, Gilbertson RJ, Davidoff AM, Pai Panandiker AS, Leung W, Chin TK, Stewart CF, Kocak M, Rowland C, Merchant TE, Kaste SC, Gajjar A (2010) Phase I study of vandetanib during and after radiotherapy in children with diffuse intrinsic pontine glioma. J Clin Oncol 28:4762–4768
Brunckhorst MK, Wang H, Lu R, Yu Q (2010) Angiopoietin-4 promotes glioblastoma progression by enhancing tumor cell viability and angiogenesis. Cancer Res 70:7283–7293
Burgess T, Coxon A, Meyer S, Sun J, Rex K, Tsuruda T, Chen Q, Ho SY, Li L, Kaufman S, Mcdorman K, Cattley RC, Elliott G, Zhang K, Feng X, Jia XC, Green L, Radinsky R, Kendall R (2006) Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-Met-dependent human tumors. Cancer Res 66:1721–1729
Cai XM, Tao BB, Wang LY, Liang YL, Jin JW, Yang Y, Hu YL, Zha XL (2005) Protein phosphatase activity of PTEN inhibited the invasion of glioma cells with epidermal growth factor receptor mutation type III expression. Int J Cancer 117:905–912
Campbell TN, Robbins SM (2008) The Eph receptor/ephrin system: an emerging player in the invasion game. Curr Issues Mol Biol 10:61–66
Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068
Cattaneo MG, Gentilini D, Vicentini LM (2006) Deregulated human glioma cell motility: inhibitory effect of somatostatin. Mol Cell Endocrinol 256:34–39
Cecchi F, Rabe DC, Bottaro DP (2010) Targeting the HGF/Met signalling pathway in cancer. Eur J Cancer 46:1260–1270
Chow LQ, Eckhardt SG (2007) Sunitinib: from rational design to clinical efficacy. J Clin Oncol 25:884–896
Chuang YY, Tran NL, Rusk N, Nakada M, Berens ME, Symons M (2004) Role of synaptojanin 2 in glioma cell migration and invasion. Cancer Res 64:8271–8275
Citri A, Skaria KB, Yarden Y (2003) The deaf and the dumb: the biology of ErbB-2 and ErbB-3. Exp Cell Res 284:54–65
Cooper CS, Park M, Blair DG, Tainsky MA, Huebner K, Croce CM, Vande Woude GF (1984) Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 311:29–33
Demetri GD, Von Mehren M, Blanke CD, Van Den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H (2002) Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 347:472–480
Dresemann G, Weller M, Rosenthal MA, Wedding U, Wagner W, Engel E, Heinrich B, Mayer-Steinacker R, Karup-Hansen A, Fluge O, Nowak A, Mehdorn M, Schleyer E, Krex D, Olver IN, Steinbach JP, Hosius C, Sieder C, Sorenson G, Parker R, Nikolova Z (2010) Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide. J Neurooncol 96:393–402
Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL (2001) Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 344:1031–1037
Du J, Bernasconi P, Clauser KR, Mani DR, Finn SP, Beroukhim R, Burns M, Julian B, Peng XP, Hieronymus H, Maglathlin RL, Lewis TA, Liau LM, Nghiemphu P, Mellinghoff IK, Louis DN, Loda M, Carr SA, Kung AL, Golub TR (2009) Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat Biotechnol 27:77–83
Edwards LA, Woolard K, Son MJ, Li A, Lee J, Ene C, Mantey SA, Maric D, Song H, Belova G, Jensen RT, Zhang W, Fine HA (2011) Effect of brain- and tumor-derived connective tissue growth factor on glioma invasion. J Natl Cancer Inst 103:1162–1178
Ekstrand AJ, Sugawa N, James CD, Collins VP (1992) Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci U S A 89:4309–4313
Eller JL, Longo SL, Kyle MM, Bassano D, Hicklin DJ, Canute GW (2005) Anti-epidermal growth factor receptor monoclonal antibody cetuximab augments radiation effects in glioblastoma multiforme in vitro and in vivo. Neurosurgery 56:155–162, discussion 162
Emrich JG, Brady LW, Quang TS, Class R, Miyamoto C, Black P, Rodeck U (2002) Radioiodinated (I-125) monoclonal antibody 425 in the treatment of high grade glioma patients: ten-year synopsis of a novel treatment. Am J Clin Oncol 25:541–546
Engelhard HH, Wolters M, Criswell PS (1995) Analysis of c-erbB2 protein content of human glioma cells and tumor tissue. J Neurooncol 23:31–40
Erber R, Eichelsbacher U, Powajbo V, Korn T, Djonov V, Lin J, Hammes HP, Grobholz R, Ullrich A, Vajkoczy P (2006) EphB4 controls blood vascular morphogenesis during postnatal angiogenesis. EMBO J 25:628–641
Escalante M, Courtney J, Chin WG, Teng KK, Kim JI, Fajardo JE, Mayer BJ, Hempstead BL, Birge RB (2000) Phosphorylation of c-Crk II on the negative regulatory Tyr222 mediates nerve growth factor-induced cell spreading and morphogenesis. J Biol Chem 275:24787–24797
Feldkamp MM, Lala P, Lau N, Roncari L, Guha A (1999) Expression of activated epidermal growth factor receptors, Ras-guanosine triphosphate, and mitogen-activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery 45:1442–1453
Fleming TP, Saxena A, Clark WC, Robertson JT, Oldfield EH, Aaronson SA, Ali IU (1992) Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res 52:4550–4553
Franceschi E, Cavallo G, Lonardi S, Magrini E, Tosoni A, Grosso D, Scopece L, Blatt V, Urbini B, Pession A, Tallini G, Crino L, Brandes AA (2007) Gefitinib in patients with progressive high-grade gliomas: a multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). Br J Cancer 96:1047–1051
Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R, Vredenburgh J, Huang J, Zheng M, Cloughesy T (2009) Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 27:4733–4740
Fukai J, Yokote H, Yamanaka R, Arao T, Nishio K, Itakura T (2008) EphA4 promotes cell proliferation and migration through a novel EphA4-FGFR1 signaling pathway in the human glioma U251 cell line. Mol Cancer Ther 7:2768–2778
Gale NW, Holland SJ, Valenzuela DM, Flenniken A, Pan L, Ryan TE, Henkemeyer M, Strebhardt K, Hirai H, Wilkinson DG, Pawson T, Davis S, Yancopoulos GD (1996) Eph receptors and ligands comprise two major specificity subclasses and are reciprocally compartmentalized during embryogenesis. Neuron 17:9–19
Gale NW, Thurston G, Hackett SF, Renard R, Wang Q, Mcclain J, Martin C, Witte C, Witte MH, Jackson D, Suri C, Campochiaro PA, Wiegand SJ, Yancopoulos GD (2002) Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev Cell 3:411–423
Gao L, Li F, Dong B, Zhang J, Rao Y, Cong Y, Mao B, Chen X (2010) Inhibition of STAT3 and ErbB2 suppresses tumor growth, enhances radiosensitivity, and induces mitochondria-dependent apoptosis in glioma cells. Int J Radiat Oncol Biol Phys 77:1223–1231
Gilbertson RJ, Perry RH, Kelly PJ, Pearson AD, Lunec J (1997) Prognostic significance of HER2 and HER4 coexpression in childhood medulloblastoma. Cancer Res 57:3272–3280
Gilbertson RJ, Bentley L, Hernan R, Junttila TT, Frank AJ, Haapasalo H, Connelly M, Wetmore C, Curran T, Elenius K, Ellison DW (2002) ERBB receptor signaling promotes ependymoma cell proliferation and represents a potential novel therapeutic target for this disease. Clin Cancer Res 8:3054–3064
Gondi CS, Dinh DH, Klopfenstein JD, Gujrati M, Rao JS (2009) MMP-2 downregulation mediates differential regulation of cell death via ErbB-2 in glioma xenografts. Int J Oncol 35:257–263
Gu J, Tamura M, Pankov R, Danen EH, Takino T, Matsumoto K, Yamada KM (1999) Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J Cell Biol 146:389–403
Guha A, Dashner K, Black PM, Wagner JA, Stiles CD (1995) Expression of PDGF and PDGF receptors in human astrocytoma operation specimens supports the existence of an autocrine loop. Int J Cancer 60:168–173
Gulati S, Ytterhus B, Granli US, Gulati M, Lydersen S, Torp SH (2010) Overexpression of c-erbB2 is a negative prognostic factor in anaplastic astrocytomas. Diagn Pathol 5:18
Haapasalo H, Hyytinen E, Sallinen P, Helin H, Kallioniemi OP, Isola J (1996) c-erbB-2 in astrocytomas: infrequent overexpression by immunohistochemistry and absence of gene amplification by fluorescence in situ hybridization. Br J Cancer 73:620–623
Haas-Kogan DA, Prados MD, Tihan T, Eberhard DA, Jelluma N, Arvold ND, Baumber R, Lamborn KR, Kapadia A, Malec M, Berger MS, Stokoe D (2005) Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst 97:880–887
Hafizi S, Dahlback B (2006) Signalling and functional diversity within the Axl subfamily of receptor tyrosine kinases. Cytokine Growth Factor Rev 17:295–304
Hainsworth JD, Ervin T, Friedman E, Priego V, Murphy PB, Clark BL, Lamar RE (2010) Concurrent radiotherapy and temozolomide followed by temozolomide and sorafenib in the first-line treatment of patients with glioblastoma multiforme. Cancer 116:3663–3669
Hawighorst T, Skobe M, Streit M, Hong YK, Velasco P, Brown LF, Riccardi L, Lange-Asschenfeldt B, Detmar M (2002) Activation of the tie2 receptor by angiopoietin-1 enhances tumor vessel maturation and impairs squamous cell carcinoma growth. Am J Pathol 160:1381–1392
Hayes AJ, Huang WQ, Yu J, Maisonpierre PC, Liu A, Kern FG, Lippman ME, Mcleskey SW, Li LY (2000) Expression and function of angiopoietin-1 in breast cancer. Br J Cancer 83:1154–1160
Hegi ME, Diserens AC, Bady P, Kamoshima Y, Kouwenhoven MC, Delorenzi M, Lambiv WL, Hamou MF, Matter MS, Koch A, Heppner FL, Yonekawa Y, Merlo A, Frei K, Mariani L, Hofer S (2011) Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib–a phase II trial. Mol Cancer Ther 10:1102–1112
Hermanson M, Funa K, Hartman M, Claesson-Welsh L, Heldin CH, Westermark B, Nister M (1992) Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res 52:3213–3219
Heroult M, Schaffner F, Augustin HG (2006) Eph receptor and ephrin ligand-mediated interactions during angiogenesis and tumor progression. Exp Cell Res 312:642–650
Hiesiger EM, Hayes RL, Pierz DM, Budzilovich GN (1993) Prognostic relevance of epidermal growth factor receptor (EGF-R) and c-neu/erbB2 expression in glioblastomas (GBMs). J Neurooncol 16:93–104
Hoelzinger DB, Demuth T, Berens ME (2007) Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J Natl Cancer Inst 99:1583–1593
Hortobagyi GN (2005) Trastuzumab in the treatment of breast cancer. N Engl J Med 353:1734–1736
Hu B, Jarzynka MJ, Guo P, Imanishi Y, Schlaepfer DD, Cheng SY (2006) Angiopoietin 2 induces glioma cell invasion by stimulating matrix metalloprotease 2 expression through the alphavbeta1 integrin and focal adhesion kinase signaling pathway. Cancer Res 66:775–783
Huang PH, Cavenee WK, Furnari FB, White FM (2007) Uncovering therapeutic targets for glioblastoma: a systems biology approach. Cell Cycle 6:2750–2754
Hutterer M, Knyazev P, Abate A, Reschke M, Maier H, Stefanova N, Knyazeva T, Barbieri V, Reindl M, Muigg A, Kostron H, Stockhammer G, Ullrich A (2008) Axl and growth arrest-specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin Cancer Res 14:130–138
Janssen JW, Schulz AS, Steenvoorden AC, Schmidberger M, Strehl S, Ambros PF, Bartram CR (1991) A novel putative tyrosine kinase receptor with oncogenic potential. Oncogene 6:2113–2120
Jones RB, Gordus A, Krall JA, Macbeath G (2006) A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature 439:168–174
Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW (2003) Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res 284:31–53
Khoshyomn S, Penar PL, Rossi J, Wells A, Abramson DL, Bhushan A (1999) Inhibition of phospholipase C-gamma1 activation blocks glioma cell motility and invasion of fetal rat brain aggregates. Neurosurgery 44:568–577, discussion 577–568
Kita D, Takino T, Nakada M, Takahashi T, Yamashita J, Sato H (2001) Expression of dominant-negative form of Ets-1 suppresses fibronectin-stimulated cell adhesion and migration through down-regulation of integrin alpha5 expression in U251 glioma cell line. Cancer Res 61:7985–7991
Kita D, Yonekawa Y, Weller M, Ohgaki H (2007) PIK3CA alterations in primary (de novo) and secondary glioblastomas. Acta Neuropathol 113:295–302
Koga K, Todaka T, Morioka M, Hamada J, Kai Y, Yano S, Okamura A, Takakura N, Suda T, Ushio Y (2001) Expression of angiopoietin-2 in human glioma cells and its role for angiogenesis. Cancer Res 61:6248–6254
Koochekpour S, Jeffers M, Rulong S, Taylor G, Klineberg E, Hudson EA, Resau JH, Vande Woude GF (1997) Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res 57:5391–5398
Kullander K, Klein R (2002) Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol 3:475–486
Lam C, Bouffet E, Bartels U (2009) Nimotuzumab in pediatric glioma. Future Oncol 5:1349–1361
Lamszus K, Laterra J, Westphal M, Rosen EM (1999) Scatter factor/hepatocyte growth factor (SF/HGF) content and function in human gliomas. Int J Dev Neurosci 17:517–530
Lassman AB, Rossi MR, Raizer JJ, Abrey LE, Lieberman FS, Grefe CN, Lamborn K, Pao W, Shih AH, Kuhn JG, Wilson R, Nowak NJ, Cowell JK, Deangelis LM, Wen P, Gilbert MR, Chang S, Yung WA, Prados M, Holland EC (2005) Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01–03 and 00–01. Clin Cancer Res 11:7841–7850
Lee HS, Han J, Bai HJ, Kim KW (2009) Brain angiogenesis in developmental and pathological processes: regulation, molecular and cellular communication at the neurovascular interface. FEBS J 276:4622–4635
Li L, Quang TS, Gracely EJ, Kim JH, Emrich JG, Yaeger TE, Jenrette JM, Cohen SC, Black P, Brady LW (2010) A phase II study of anti-epidermal growth factor receptor radioimmunotherapy in the treatment of glioblastoma multiforme. J Neurosurg 113:192–198
Liang ML, Ma J, Ho M, Solomon L, Bouffet E, Rutka JT, Hawkins C (2008) Tyrosine kinase expression in pediatric high grade astrocytoma. J Neurooncol 87:247–253
Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, Whittle N, Waterfield MD, Ullrich A, Schlessinger J (1985) Amplification and overexpression of the EGF receptor gene in primary human glioblastomas. J Cell Sci Suppl 3:161–172
Liu D, Martin V, Fueyo J, Lee OH, Xu J, Cortes-Santiago N, Alonso MM, Aldape K, Colman H, Gomez-Manzano C (2010) Tie2/TEK modulates the interaction of glioma and brain tumor stem cells with endothelial cells and promotes an invasive phenotype. Oncotarget 1:700–709
Lohi J (2001) Laminin-5 in the progression of carcinomas. Int J Cancer Journal international du cancer 94:763–767
Lokker NA, Sullivan CM, Hollenbach SJ, Israel MA, Giese NA (2002) Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res 62:3729–3735
Machein MR, Knedla A, Knoth R, Wagner S, Neuschl E, Plate KH (2004) Angiopoietin-1 promotes tumor angiogenesis in a rat glioma model. Am J Pathol 165:1557–1570
Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, Mcclain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD (1997) Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277:55–60
Majumdar K, Radotra BD, Vasishta RK, Pathak A (2009) Platelet-derived growth factor expression correlates with tumor grade and proliferative activity in human oligodendrogliomas. Surg Neurol 72:54–60
Manfioletti G, Brancolini C, Avanzi G, Schneider C (1993) The protein encoded by a growth arrest-specific gene (gas6) is a new member of the vitamin K-dependent proteins related to protein S, a negative coregulator in the blood coagulation cascade. Mol Cell Biol 13:4976–4985
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298:1912–1934
Martinho O, Longatto-Filho A, Lambros MB, Martins A, Pinheiro C, Silva A, Pardal F, Amorim J, Mackay A, Milanezi F, Tamber N, Fenwick K, Ashworth A, Reis-Filho JS, Lopes JM, Reis RM (2009) Expression, mutation and copy number analysis of platelet-derived growth factor receptor A (PDGFRA) and its ligand PDGFA in gliomas. Br J Cancer 101:973–982
Mateos ME, Lopez-Laso E, Izquierdo L, Perez-Navero JL, Garcia S, Garzas C (2011) Response to nimotuzumab in a child with a progressive diffuse intrinsic pontine glioma. Pediatr Int 53:261–263
Meakin SO, Macdonald JI, Gryz EA, Kubu CJ, Verdi JM (1999) The signaling adapter FRS-2 competes with Shc for binding to the nerve growth factor receptor TrkA. A model for discriminating proliferation and differentiation. J Biol Chem 274:9861–9870
Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF, Sawyers CL, Mischel PS (2005) Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 353:2012–2024
Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, Sloan AE, Cohen ML, Wang B (2009) EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell 16:9–20
Mineo JF, Bordron A, Baroncini M, Maurage CA, Ramirez C, Siminski RM, Berthou C, Dam Hieu P (2007) Low HER2-expressing glioblastomas are more often secondary to anaplastic transformation of low-grade glioma. J Neurooncol 85:281–287
Miyamori H, Hasegawa K, Kim KR, Sato H (2000) Expression of metastasis-associated mts1 gene is co-induced with membrane type-1 matrix metalloproteinase (MT1-MMP) during oncogenic transformation and tubular formation of Madin Darby canine kidney (MDCK) epithelial cells. Clin Exp Metastasis 18:51–56
Moriyama T, Kataoka H, Tsubouchi H, Koono M (1995) Concomitant expression of hepatocyte growth factor (HGF), HGF activator and c-met genes in human glioma cells in vitro. FEBS Lett 372:78–82
Moriyama T, Kataoka H, Hamasuna R, Yokogami K, Uehara H, Kawano H, Goya T, Tsubouchi H, Koono M, Wakisaka S (1998a) Up-regulation of vascular endothelial growth factor induced by hepatocyte growth factor/scatter factor stimulation in human glioma cells. Biochem Biophys Res Commun 249:73–77
Moriyama T, Kataoka H, Kawano H, Yokogami K, Nakano S, Goya T, Uchino H, Koono M, Wakisaka S (1998b) Comparative analysis of expression of hepatocyte growth factor and its receptor, c-met, in gliomas, meningiomas and schwannomas in humans. Cancer Lett 124:149–155
Nagane M (2011) Neuro-oncology: continuing multidisciplinary progress. Lancet Neurol 10:18–20
Nakada M, Kita D, Futami K, Yamashita J, Fujimoto N, Sato H, Okada Y (2001) Roles of membrane type 1 matrix metalloproteinase and tissue inhibitor of metalloproteinases 2 in invasion and dissemination of human malignant glioma. J Neurosurg 94:464–473
Nakada M, Niska JA, Miyamori H, Mcdonough WS, Wu J, Sato H, Berens ME (2004) The phosphorylation of EphB2 receptor regulates migration and invasion of human glioma cells. Cancer Res 64:3179–3185
Nakada M, Niska JA, Tran NL, Mcdonough WS, Berens ME (2005) EphB2/R-Ras signaling regulates glioma cell adhesion, growth, and invasion. Am J Pathol 167:565–576
Nakada M, Drake KL, Nakada S, Niska JA, Berens ME (2006) Ephrin-B3 ligand promotes glioma invasion through activation of Rac1. Cancer Res 66:8492–8500
Nakada M, Nakada S, Demuth T, Tran NL, Hoelzinger DB, Berens ME (2007) Molecular targets of glioma invasion. Cell Mol Life Sci 64:458–478
Nakada M, Anderson EM, Demuth T, Nakada S, Reavie LB, Drake KL, Hoelzinger DB, Berens ME (2010) The phosphorylation of ephrin-B2 ligand promotes glioma cell migration and invasion. Int J Cancer 126:1155–1165
Nakada M, Hayashi Y, Hamada J (2011) Role of Eph/ephrin tyrosine kinase in malignant glioma. Neuro Oncol 13:1163–1170
Natarajan M, Stewart JE, Golemis EA, Pugacheva EN, Alexandropoulos K, Cox BD, Wang W, Grammer JR, Gladson CL (2006) HEF1 is a necessary and specific downstream effector of FAK that promotes the migration of glioblastoma cells. Oncogene 25:1721–1732
Neyns B, Sadones J, Joosens E, Bouttens F, Verbeke L, Baurain JF, D’hondt L, Strauven T, Chaskis C, In’t Veld P, Michotte A, De Greve J (2009) Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann Oncol 20:1596–1603
Neyns B, Sadones J, Chaskis C, Dujardin M, Everaert H, Lv S, Duerinck J, Tynninen O, Nupponen N, Michotte A, De Greve J (2011) Phase II study of sunitinib malate in patients with recurrent high-grade glioma. J Neurooncol 103:491–501
Nister M, Libermann TA, Betsholtz C, Pettersson M, Claesson-Welsh L, Heldin CH, Schlessinger J, Westermark B (1988) Expression of messenger RNAs for platelet-derived growth factor and transforming growth factor-alpha and their receptors in human malignant glioma cell lines. Cancer Res 48:3910–3918
Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS (2006) Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366:2–16
Ohgaki H, Kleihues P (2009) Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci 100:2235–2241
Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, Burkhard C, Schuler D, Probst-Hensch NM, Maiorka PC, Baeza N, Pisani P, Yonekawa Y, Yasargil MG, Lutolf UM, Kleihues P (2004) Genetic pathways to glioblastoma: a population-based study. Cancer Res 64:6892–6899
Onishi M, Ichikawa T, Kurozumi K, Date I (2011) Angiogenesis and invasion in glioma. Brain Tumor Pathol 28:13–24
Ozawa T, Brennan CW, Wang L, Squatrito M, Sasayama T, Nakada M, Huse JT, Pedraza A, Utsuki S, Yasui Y, Tandon A, Fomchenko EI, Oka H, Levine RL, Fujii K, Ladanyi M, Holland EC (2010) PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev 24:2205–2218
Pedersen MW, Pedersen N, Ottesen LH, Poulsen HS (2005) Differential response to gefitinib of cells expressing normal EGFR and the mutant EGFRvIII. Br J Cancer 93:915–923
Peereboom DM, Shepard DR, Ahluwalia MS, Brewer CJ, Agarwal N, Stevens GH, Suh JH, Toms SA, Vogelbaum MA, Weil RJ, Elson P, Barnett GH (2010) Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J Neurooncol 98:93–99
Pflug BR, Colangelo AM, Tornatore C, Mocchetti I (2001) TrkA induces differentiation but not apoptosis in C6-2B glioma cells. J Neurosci Res 64:636–645
Plate KH, Breier G, Farrell CL, Risau W (1992) Platelet-derived growth factor receptor-beta is induced during tumor development and upregulated during tumor progression in endothelial cells in human gliomas. Lab Invest 67:529–534
Pollack IF, Stewart CF, Kocak M, Poussaint TY, Broniscer A, Banerjee A, Douglas JG, Kun LE, Boyett JM, Geyer JR (2011) A phase II study of gefitinib and irradiation in children with newly diagnosed brainstem gliomas: a report from the Pediatric Brain Tumor Consortium. Neuro Oncol 13:290–297
Ponzetto C, Bardelli A, Zhen Z, Maina F, Dalla Zonca P, Giordano S, Graziani A, Panayotou G, Comoglio PM (1994) A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 77:261–271
Prigent SA, Gullick WJ (1994) Identification of c-erbB-3 binding sites for phosphatidylinositol 3′-kinase and SHC using an EGF receptor/c-erbB-3 chimera. EMBO J 13:2831–2841
Quang TS, Brady LW (2004) Radioimmunotherapy as a novel treatment regimen: 125I-labeled monoclonal antibody 425 in the treatment of high-grade brain gliomas. Int J Radiat Oncol Biol Phys 58:972–975
Rabin SJ, Tornatore C, Baker-Cairns B, Spiga G, Mocchetti I (1998) TrkA receptors delay C6-2B glioma cell growth in rat striatum. Brain Res Mol Brain Res 56:273–276
Raizer JJ, Abrey LE, Lassman AB, Chang SM, Lamborn KR, Kuhn JG, Yung WK, Gilbert MR, Aldape KA, Wen PY, Fine HA, Mehta M, Deangelis LM, Lieberman F, Cloughesy TF, Robins HI, Dancey J, Prados MD (2010) A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol 12:95–103
Ram R, Lorente G, Nikolich K, Urfer R, Foehr E, Nagavarapu U (2006) Discoidin domain receptor-1a (DDR1a) promotes glioma cell invasion and adhesion in association with matrix metalloproteinase-2. J Neurooncol 76:239–248
Ramnarain DB, Park S, Lee DY, Hatanpaa KJ, Scoggin SO, Otu H, Libermann TA, Raisanen JM, Ashfaq R, Wong ET, Wu J, Elliott R, Habib AA (2006) Differential gene expression analysis reveals generation of an autocrine loop by a mutant epidermal growth factor receptor in glioma cells. Cancer Res 66:867–874
Raymond E, Brandes AA, Dittrich C, Fumoleau P, Coudert B, Clement PM, Frenay M, Rampling R, Stupp R, Kros JM, Heinrich MC, Gorlia T, Lacombe D, Van Den Bent MJ (2008) Phase II study of imatinib in patients with recurrent gliomas of various histologies: a European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. J Clin Oncol 26:4659–4665
Reardon DA, Egorin MJ, Quinn JA, Rich JN, Gururangan S, Vredenburgh JJ, Desjardins A, Sathornsumetee S, Provenzale JM, Herndon JE 2nd, Dowell JM, Badruddoja MA, Mclendon RE, Lagattuta TF, Kicielinski KP, Dresemann G, Sampson JH, Friedman AH, Salvado AJ, Friedman HS (2005) Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J Clin Oncol 23:9359–9368
Reardon DA, Vredenburgh JJ, Desjardins A, Peters K, Gururangan S, Sampson JH, Marcello J, Herndon JE 2nd, Mclendon RE, Janney D, Friedman AH, Bigner DD, Friedman HS (2011) Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. J Neurooncol 101:57–66
Ribom D, Andrae J, Frielingsdorf M, Hartman M, Nister M, Smits A (2002) Prognostic value of platelet derived growth factor alpha receptor expression in grade 2 astrocytomas and oligoastrocytomas. J Neurol Neurosurg Psychiatry 72:782–787
Rich JN, Rasheed BK, Yan H (2004) EGFR mutations and sensitivity to gefitinib. N Engl J Med 351:1260–1261, author reply 1260–1261
Rosario M, Birchmeier W (2003) How to make tubes: signaling by the Met receptor tyrosine kinase. Trends Cell Biol 13:328–335
Salhia B, Rutten F, Nakada M, Beaudry C, Berens M, Kwan A, Rutka JT (2005) Inhibition of Rho-kinase affects astrocytoma morphology, motility, and invasion through activation of Rac1. Cancer Res 65:8792–8800
Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E, Seiki M (1994) A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 370:61–65
Saurez G, Cabanas R, Zaldivar M, Garnier T, Iglesias B, Piedra P, Castillo MR, Longchong M, Iznaga N, Lage A (2009) Clinical experience with nimotuzumab in cuban pediatric patients with brain tumors, 2005 to 2007. MEDICC Rev 11:27–33
Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103:211–225
Schwechheimer K, Laufle RM, Schmahl W, Knodlseder M, Fischer H, Hofler H (1994) Expression of neu/c-erbB-2 in human brain tumors. Hum Pathol 25:772–780
Senner V, Ratzinger S, Mertsch S, Grassel S, Paulus W (2008) Collagen XVI expression is upregulated in glioblastomas and promotes tumor cell adhesion. FEBS Lett 582:3293–3300
Shih AH, Holland EC (2006) Platelet-derived growth factor (PDGF) and glial tumorigenesis. Cancer Lett 232:139–147
Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, Makino K, Saya H, Hirano H, Kuratsu J, Oka K, Ishimaru Y, Ushio Y (2003) Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res 63:6962–6970
Singer HS, Hansen B, Martinie D, Karp CL (1999) Mitogenesis in glioblastoma multiforme cell lines: a role for NGF and its TrkA receptors. J Neurooncol 45:1–8
Spencer KS, Graus-Porta D, Leng J, Hynes NE, Klemke RL (2000) ErbB2 is necessary for induction of carcinoma cell invasion by ErbB family receptor tyrosine kinases. J Cell Biol 148:385–397
Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, Stevens C, O’meara S, Smith R, Parker A, Barthorpe A, Blow M, Brackenbury L, Butler A, Clarke O, Cole J, Dicks E, Dike A, Drozd A, Edwards K, Forbes S, Foster R, Gray K, Greenman C, Halliday K, Hills K, Kosmidou V, Lugg R, Menzies A, Perry J, Petty R, Raine K, Ratford L, Shepherd R, Small A, Stephens Y, Tofts C, Varian J, West S, Widaa S, Yates A, Brasseur F, Cooper CS, Flanagan AM, Knowles M, Leung SY, Louis DN, Looijenga LH, Malkowicz B, Pierotti MA, Teh B, Chenevix-Trench G, Weber BL, Yuen ST, Harris G, Goldstraw P, Nicholson AG, Futreal PA, Wooster R, Stratton MR (2004) Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature 431:525–526
Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, Chin L, Depinho RA (2007) Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 318:287–290
Stratmann A, Risau W, Plate KH (1998) Cell type-specific expression of angiopoietin-1 and angiopoietin-2 suggests a role in glioblastoma angiogenesis. Am J Pathol 153:1459–1466
Surawska H, Ma PC, Salgia R (2004) The role of ephrins and Eph receptors in cancer. Cytokine Growth Factor Rev 15:419–433
Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM (1998) Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 280:1614–1617
Thiessen B, Stewart C, Tsao M, Kamel-Reid S, Schaiquevich P, Mason W, Easaw J, Belanger K, Forsyth P, Mcintosh L, Eisenhauer E (2010) A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother Pharmacol 65:353–361
Tokarski JS, Newitt JA, Chang CY, Cheng JD, Wittekind M, Kiefer SE, Kish K, Lee FY, Borzillerri R, Lombardo LJ, Xie D, Zhang Y, Klei HE (2006) The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res 66:5790–5797
Uhm JH, Ballman KV, Wu W, Giannini C, Krauss JC, Buckner JC, James CD, Scheithauer BW, Behrens RJ, Flynn PJ, Schaefer PL, Dakhill SR, Jaeckle KA (2011) Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int J Radiat Oncol Biol Phys 80:347–353
Uhrbom L, Hesselager G, Nister M, Westermark B (1998) Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res 58:5275–5279
Vajkoczy P, Knyazev P, Kunkel A, Capelle HH, Behrndt S, Von Tengg-Kobligk H, Kiessling F, Eichelsbacher U, Essig M, Read TA, Erber R, Ullrich A (2006) Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc Natl Acad Sci U S A 103:5799–5804
Valius M, Kazlauskas A (1993) Phospholipase C-gamma 1 and phosphatidylinositol 3 kinase are the downstream mediators of the PDGF receptor’s mitogenic signal. Cell 73:321–334
Van Den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, Clement PM, Frenay M, Campone M, Baurain JF, Armand JP, Taphoorn MJ, Tosoni A, Kletzl H, Klughammer B, Lacombe D, Gorlia T (2009) Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol 27:1268–1274
Varela M, Ranuncolo SM, Morand A, Lastiri J, De Kier Joffe EB, Puricelli LI, Pallotta MG (2004) EGF-R and PDGF-R, but not bcl-2, overexpression predict overall survival in patients with low-grade astrocytomas. J Surg Oncol 86:34–40
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110
Vogel W, Gish GD, Alves F, Pawson T (1997) The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell 1:13–23
Vogel WF, Abdulhussein R, Ford CE (2006) Sensing extracellular matrix: an update on discoidin domain receptor function. Cell Signal 18:1108–1116
Wen PY (2010) American Society of Clinical Oncology 2010: report of selected studies from the CNS tumors section. Expert Rev Anticancer Ther 10:1367–1369
Wen PY, Yung WK, Lamborn KR, Dahia PL, Wang Y, Peng B, Abrey LE, Raizer J, Cloughesy TF, Fink K, Gilbert M, Chang S, Junck L, Schiff D, Lieberman F, Fine HA, Mehta M, Robins HI, Deangelis LM, Groves MD, Puduvalli VK, Levin V, Conrad C, Maher EA, Aldape K, Hayes M, Letvak L, Egorin MJ, Capdeville R, Kaplan R, Murgo AJ, Stiles C, Prados MD (2006) Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99–08. Clin Cancer Res 12:4899–4907
Wilhelm SM, Carter C, Tang L, Wilkie D, Mcnabola A, Rong H, Chen C, Zhang X, Vincent P, Mchugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA (2004) BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 64:7099–7109
Wimmer-Kleikamp SH, Lackmann M (2005) Eph-modulated cell morphology, adhesion and motility in carcinogenesis. IUBMB Life 57:421–431
Wong AJ, Bigner SH, Bigner DD, Kinzler KW, Hamilton SR, Vogelstein B (1987) Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc Natl Acad Sci U S A 84:6899–6903
Wykosky J, Gibo DM, Stanton C, Debinski W (2005) EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res 3:541–551
Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, Orf J, You A, Laird AD, Engst S, Lee L, Lesch J, Chou YC, Joly A (2011) Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther 10:2298–2308
Yamada T, Tsubouchi H, Daikuhara Y, Prat M, Comoglio PM, Mcgeer PL, Mcgeer EG (1994) Immunohistochemistry with antibodies to hepatocyte growth factor and its receptor protein (c-MET) in human brain tissues. Brain Res 637:308–312
Yamanaka R, Arao T, Yajima N, Tsuchiya N, Homma J, Tanaka R, Sano M, Oide A, Sekijima M, Nishio K (2006) Identification of expressed genes characterizing long-term survival in malignant glioma patients. Oncogene 25:5994–6002
Yamazaki H, Fukui Y, Ueyama Y, Tamaoki N, Kawamoto T, Taniguchi S, Shibuya M (1988) Amplification of the structurally and functionally altered epidermal growth factor receptor gene (c-erbB) in human brain tumors. Mol Cell Biol 8:1816–1820
Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2:127–137
Ye K, Hurt KJ, Wu FY, Fang M, Luo HR, Hong JJ, Blackshaw S, Ferris CD, Snyder SH (2000) Pike. A nuclear gtpase that enhances PI3kinase activity and is regulated by protein 4.1 N. Cell 103:919–930
Ye K, Aghdasi B, Luo HR, Moriarity JL, Wu FY, Hong JJ, Hurt KJ, Bae SS, Suh PG, Snyder SH (2002) Phospholipase C gamma 1 is a physiological guanine nucleotide exchange factor for the nuclear GTPase PIKE. Nature 415:541–544
Yong HY, Kim IY, Kim JS, Moon A (2010) ErbB2-enhanced invasiveness of H-Ras MCF10A breast cells requires MMP-13 and uPA upregulation via p38 MAPK signaling. Int J Oncol 36:501–507
Zadeh G, Koushan K, Pillo L, Shannon P, Guha A (2004a) Role of Ang1 and its interaction with VEGF-A in astrocytomas. J Neuropathol Exp Neurol 63:978–989
Zadeh G, Qian B, Okhowat A, Sabha N, Kontos CD, Guha A (2004b) Targeting the Tie2/Tek receptor in astrocytomas. Am J Pathol 164:467–476
Zadeh G, Koushan K, Baoping Q, Shannon P, Guha A (2010) Role of angiopoietin-2 in regulating growth and vascularity of astrocytomas. J Oncol 2010:659231
Zeng Q, Chen S, You Z, Yang F, Carey TE, Saims D, Wang CY (2002) Hepatocyte growth factor inhibits anoikis in head and neck squamous cell carcinoma cells by activation of ERK and Akt signaling independent of NFkappa B. J Biol Chem 277:25203–25208
Zhang YW, Vande Woude GF (2003) HGF/SF-met signaling in the control of branching morphogenesis and invasion. J Cell Biochem 88:408–417
Zhang QK, Boast S, De Los SK, Begemann M, Goff SP (1996) Transforming activity of retroviral genomes encoding Gag-Axl fusion proteins. J Virol 70:8089–8097
Zhang Z, Yang Y, Gong A, Wang C, Liang Y, Chen Y (2005) Localization of NGF and TrkA at mitotic apparatus in human glioma cell line U251. Biochem Biophys Res Commun 337:68–74
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Nakada, M. et al. (2013). Receptor Tyrosine Kinases: Principles and Functions in Glioma Invasion. In: Barańska, J. (eds) Glioma Signaling. Advances in Experimental Medicine and Biology, vol 986. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-4719-7_8
Download citation
DOI: https://doi.org/10.1007/978-94-007-4719-7_8
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-4718-0
Online ISBN: 978-94-007-4719-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)