
Abstract
Our cells are constantly exposed to insults from endogenous and exogenous agents that can introduce damage into our DNA and generate genomic instability. Many of these lesions cause structural damage to DNA and can alter or eliminate fundamental cellular processes, such as DNA replication or transcription. DNA lesions commonly include base and sugar modifications, single- and double-strand breaks, DNA-protein cross-links, and base-free sites. To counteract the harmful effects of DNA damage, cells have developed a specialized DNA repair system, which can be subdivided into several distinct mechanisms based on the type of DNA lesion. These processes include base excision repair, mismatch repair, nucleotide excision repair, and double-strand break repair, which comprise both homologous recombination and non-homologous end-joining. Although a complex set of cellular responses are elicited following DNA damage, this chapter provides an introduction to the specific molecular mechanisms of recognition, removal, and repair of DNA damage.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Lynch Syndrome
- Mismatch Repair Mismatch Repair
- Nucleotide Excision Repair
- Base Excision Repair
- Mismatch Repair Mismatch Repair Protein
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
2.1 Overview
It is estimated that each of the ~ 1013 cells within the human body incurs tens of thousands of DNA-damaging events per day [1]. DNA exclusively serves as the repository for the genetic information in each living cell and its integrity and stability are of much greater consequence than other cellular components, such as RNA and proteins. DNA damage can interfere with essential cellular processes, such as transcription or replication, and can compromise the viability of the cell. Specific DNA lesions can also induce mutations that cause cancer or other diseases as well as contribute to the aging process [2]. Thus, cells have evolved a network of DNA repair mechanisms to remove different types of DNA damage. Regardless of the type of lesion and the mechanism required for its repair, cells initiate a highly coordinated cascade of events—collectively known as the DNA damage response (DDR)—that senses the DNA damage, signals its presence, and mediates its repair. For example, the DDR may transiently arrest the cell cycle to allow for efficient DNA damage repair prior to replication or mitosis [3, 4] or signal cells to activate apoptosis under circumstances of persistent or irreparable DNA damage [5]. The importance of DDR is underscored by the prevalence of neurological and cancer susceptibility disorders, such as Ataxia-telangiectasia , Fanconi anemia, and Xeroderma pigmentosum, that are caused by DNA repair deficiencies [6]. In this chapter the major types of DNA damage and the respective molecular pathways that function in their repair will be introduced (see Fig. 2.1).

DNA damage and repair mechanisms. The diagram illustrates common DNA damaging agents, examples of DNA lesions caused by these agents, and the relevant DNA repair mechanism responsible for their removal. (Figure adapted from [83])
2.2 Types of DNA Damage
As a prelude to the repair of damaged DNA, we must first take into consideration the collection of damage products. DNA, like any other molecule, is subject to chemical reactions. DNA damage may result from either intrinsic or extrinsic agents. In general, the vast majority of DNA modifications are endogenous in origin (for review, see [7]). The simplest form of endogenous DNA damage is spontaneous hydrolysis [8]. The N-glycosidic bond between the DNA base and the deoxyribose is particularly prone to acid-catalyzed hydrolysis. Abasic or AP sites (apurinic/apyrimidinic sites), which are the products of hydrolytic nucleobase loss, are estimated to occur at a rate of approximately 10,000 per cell per day [8, 9]. In fact, abasic sites are also created by cellular design during the course of BER (see Sect. 2.3.1). Furthermore, abasic sites are chemically liable and can undergo b-elimination that results in DNA strand scission [10]. Another common reaction involving hydrolysis is the deamination of DNA bases carrying exocyclic amino groups [8, 11]. The most frequent of these lesions is the formation of uracil from cytosine occurring at an estimated 100–500 times per cell per day [12, 13]. Adenine and guanine, may also spontaneously deaminate to form hypoxanthine and xanthine, respectively, although at a much lower rate [14].
DNA is also susceptible to chemical modification by reactive molecules that are created during normal cellular metabolism. Among the most important of these molecules are reactive oxygen species (ROS), which include O2 −, H2O2, and •OH (for reviews, see [15, 16]). ROS generate over one hundred different oxidative DNA adducts, such as base modification, deoxyribose oxidation, single- or double-strand breakage, and DNA-protein cross-links [17]. Endogenous reactive nitrogen species, primarily nitric oxide (NO•) and its by-products, can also produce similar oxidative adducts [18]. The most extensively studied oxidative DNA lesion is the 8-oxoguanine, which is routinely used as an analytical measure of oxidative DNA damage in biological systems [19]. An additional type of DNA damage related to endogenous reactive molecules is alkylation. The putative candidates of such agents include the endogenous methyl donor, S-adenosylmethionine (SAM), nitrosated amines, and methyl radicals generated by lipid peroxidation [7, 20]. The primary sites of alkylation are the O- and N-atoms of nucleobases.
Endogenous genomic damage can also arise due to unavoidable errors resulting from physiological DNA processing reactions. For example, DNA mismatches as well as insertions and deletions are occasionally introduced (at a rate of 10−4 to 10−6) as a result of misincorporation of bases by replicative DNA polymerases [21]. At the same time, erroneous incorporation of chemically altered nucleotide precursors, such as 8-oxo-dGTP and dUTP [22], also represents a significant source of replication-related DNA damage. In addition, abortive topoisomerase activity yields an irregular type of lesion wherein DNA strand breaks feature covalent linkage of the enzyme to the DNA termini [23, 24]. Likewise, the DNA repair processes themselves may also be error prone and introduce supplemental DNA damage [25].
Besides the numerous endogenous sources of DNA damage, cellular DNA is also under constant attack from exogenous or environmental DNA-damaging agents. These include physical stresses, such as ultraviolet light (UV) from the sun, which primarily causes two types of DNA lesions, namely cyclobutane pyrimidine dimers and 6–4 pyrimidone photoproducts, both of which consist of an atypical covalent bond between adjacent pyrimidine bases [26]. Another external, physical source of DNA damage is ionizing radiation, which can originate from both natural (e.g., cosmic and gamma radiation) and artificial sources (e.g., medical treatments, such as X-rays and radiotherapy). Ionizing radiation induces a variety of DNA lesions, the most harmful of these being double-strand breaks. DNA can also incur damage indirectly from ionizing radiation through the production of ROS [27].
In addition to the physical insults, the cell must also contend with several chemical sources of DNA damage (for reviews, see [28, 29]). For example, a variety of chemical agents (i.e., clinical drugs) have been developed over the years to target DNA as a means to treat cancer or other diseases. These include alkylating agents, such as methyl methanesulfonate and temozolomide, which induce alkylation of the DNA bases as well as bifunctional alkylating agents, such as nitrogen mustards, platinum compounds, and the natural product mitomycin C, that cause DNA damage in the form of intrastrand and interstrand cross-links [30]. Chemotherapeutic drugs, such as topoisomerse I or II inhibitors (e.g., camptothecin or etoposide, respectively), generate single-strand or double-strand breaks by trapping topoisomerase–DNA covalent complexes, respectively [31]. Other well-studied environmentally occurring DNA-damaging chemicals include N-nitrosoamines, heterocyclic amines, and polycyclic aromatic hydrocarbons (e.g., benzo[a]pyrene), which are commonly found in the diet, with the latter also being produced in air emissions, such as cigarette smoke and vehicle exhaust. In general, these types of compounds covalently bond to various sites on the DNA bases to form the so-called bulky DNA adducts. Similar adducts are generated between DNA and aflatoxins, which are naturally occurring toxins produced by fungi in the genus Aspergillus that grow in several types of food crops [32].
2.3 DNA Repair Mechanisms
To compensate for the many types of DNA damage that occur, cells have developed multiple repair mechanisms wherein each corrects a different subset of lesions. At a minimum, most would agree that mammalian cells utilize five major DNA repair mechanisms: base excision repair (BER), mismatch repair (MMR), nucleotide excision repair (NER), and double-strand break repair, which includes both homologous recombination (HR) and non-homologous end joining (NHEJ) (for comprehensive review, see [33]). For reference, Table 2.1 outlines specific genes that are associated with each DNA repair mechanism.
2.3.1 Base Excision Repair (BER)
BER, as the name implies, is the predominant mechanism responsible for the repair of damaged DNA bases that, in contrast to NER (see Sect. 2.3.3), do not significantly distort the overall structure of the DNA helix (for detailed review of BER, see [34]). BER is described as a highly coordinated pathway of consecutive enzymatic reactions. However, several distinct BER sub-pathways occur, which are contingent on the type of damage encountered at the onset as well as throughout the BER process. BER is typically initiated by the series of lesion-specific DNA glycosylases that remove the damaged base by cleaving the N-glycosidic bond linking the base to its corresponding deoxyribose, leading to the production of an AP or abasic site. At least twelve DNA glycosylases have been identified to date, each acting upon a single or small number of partially overlapping base lesions [35]. Despite their structural diversity, all DNA glycosylases utilize a base-flipping mechanism in which the target base is ‘flipped’ to an extra helical position for excision from DNA [36]. The resultant AP site is both an intermediate product of BER and a highly prevalent DNA lesion produced by spontaneous base loss. In either case, AP sites are generally repaired by apurinc/apyrmidinic endonuclease 1 (APE1), the second enzyme in the canonical BER pathway. APE1 hydrolyzes the phosphodiester backbone immediately 5′ to the AP site, creating a single-strand break flanked by 3′-OH and 5′-deoxyribose phosphate (5′-dRP) termini [37]. Alternatively, some DNA glycosylases have an associated AP lyase activity and are also capable of cleaving AP sites via a b-elimination reaction to produce 3′-phospho-α, β-unsaturated aldehyde and 5′-phosphate at the margins of the break. A subset of these bifunctional enzymes, such as the oxidized base-specific DNA glycosylase/lyases NEIL1 and NEIL2, catalyze successive b- and d-elimination converting the 3′-phospho-α, β-unsaturated aldehyde to a 3′-phosphate.
Regardless of mechanism, incision of the phosphodiester bond results in a BER intermediate strand break harboring 3′- and 5′-blocking lesions. To allow completion of the repair process, these blocked termini must be restored to conventional 3′-OH and 5′-phosphate ends, which are essential for DNA polymerase and subsequent DNA ligase reactions Different DNA end-processing enzymes carry out the removal of these abnormal ends depending on whether cleavage occurred 3′ or 5′ to the AP site. APE1, for example in addition to its major AP endonuclease activity also has intrinsic 3′-phosphodiesterase activity, permitting restoration of 3′-OH from 3′-phospho-α, β-unsaturated aldehyde. The 3′-phophate product that is generated by specific bifunctional DNA glycosylases is converted to a 3′-OH by the 3′-phosphatase activity of PNKP (polynucleotide kinase 3′-phosphatase). Conversely, removal of the 5′-dRP occurs following template-guided gap filling by DNA polymerase b via its associated dRP lyase activity.
Besides the scheduled DNA single-strand breaks that arise as BER intermediates, numerous involuntary DNA single-strand breaks can also occur both through direct and indirect mechanisms (see Sect. 2.2). Such single-strand interruptions are processed and repaired by many of the same enzymes that are responsible for the later stages of BER. The termini of most, if not all, single-strand breaks contain 3′- and/or 5′-blocking lesions. For example, the most common blocking lesions at ROS-induced DNA strand breaks are 3′-phosphoglycolate and 3′-phosphoglycolaldehyde, which are generally processed by the 3′-phosphodesterase activity of APE1, or 3′ phosphate, which is removed by PNKP. Tyrosyl-DNA phosphodiesterase 1 (Tdp1) is an end-processing enzyme that repairs several 3′-blocking termini including 3′-phosphoglycolate; however, its preferred substrate is the 3′-phosphotyrosyl bond, which stems from dead-end topoisomerase I reactions [38]. Likewise, aprataxin (APTX) is another specific end-processing enzyme, which specifically repairs abortive 5′-adenylate intermediates of DNA ligase activity [39]. Thus, DNA end-processing is perhaps the most diverse, yet often redundant, enzymatic step of BER, largely due to the broad range of termini that can be generated [40].
The next steps in the BER process involve repair of the DNA strand break through DNA synthesis and ligation. The synthesis/ligation step is divided into two sub-pathways, short-patch and long patch BER, based on whether a single or several nucleotides are incorporated at the DNA strand break site, respectively [41]. The paradigm for short-patch BER encompasses single nucleotide gap filling and removal of the 5′-dRP by DNA polymerase b and successive ligation of the DNA ends by either DNA ligase I or the complex of DNA ligase III and XRCC1. Short-patch BER represents approximately 80–90 % of all BER. Long-patch BER is normally only initiated as a result of 5′-blocking lesions that are refractory to DNA polymerase b lyase activity. Long-patch BER demands several proteins associated with DNA replication, including DNA polymerase d or e, PCNA (proliferating cell nuclear antigen), RFC (replication factor-C), FEN1 (flap endonuclease-1), and DNA ligase I. Specifically, DNA polymerase b, d, or e accompanied by PCNA elongate the 3′-OH into the repair gap and displace the 5′-lesion as part of a DNA fragment or ‘flap’ oligonucleotide. The flap structure is then removed by FEN1 and DNA ligase I sequentially seals the nick that has been relocated downstream of the original nucleotide damage site.
In addition to the factors mentioned above, there are secondary proteins that are known to play a facilitative role in BER. Most notably among these are X-ray repair cross-complementing protein 1 (XRCC1) and poly (ADP-ribose) polymerase 1 (PARP1). XRCC1 has no known enzymatic activity, but rather functions as a molecular scaffold that orchestrates the assembly of several enzymatic components involved in the BER process. For instance, XRCC1 has been shown to interact with several BER proteins, including multiple DNA glycosylases, DNA polymerase b, APE1, ligase III, PNKP, Tdp1, and APTX [42]. Although no catalytic function has been ascribed to XRCC1, direct binding to nicked and gapped DNA has been demonstrated via its N-terminal domain [43]. Additionally, PARP-1 also physically interacts with XRCC1. PARP1 is an abundant nuclear protein that acts as a molecular sensor of DNA strand breaks. Upon binding to its DNA target, PARP-1 catalyzes the poly (ADP-ribosyl)ation (PAR) of itself, in addition to several other protein substrates. Once formed, this PAR modification allows for recruitment of repair proteins, such as XRCC1. At the same time, the dense negative charge of PAR results in the release of PARP-1 from DNA, which permits access of repair proteins to the DNA damage site [44]. Overall, BER is a multistep process that requires the sequential activity of several proteins and consists of numerous entry points based on the type of damage encountered.
2.3.2 Mismatch Repair (MMR)
The MMR system plays an essential role in post-replication repair of misincorporated bases that have escaped the proofreading activity of replication polymerases. In addition to mismatched bases, MMR proteins also correct insertion/deletion loops (IDLs) that result from polymerase slippage during replication of repetitive DNA sequences. The significance of this pathway is corroborated by the fact that MMR deficient cells are said to display a mutator phenotype, which is characterized by invariably microsatellite instability and an elevated mutation frequency. More importantly, germline mutations in MMR genes are predisposed to a variety of cancers, including hereditary non-polyposis colon cancer, also known as Lynch syndrome [45]. The MMR pathway can be divided into three principle steps: a recognition step where mispaired bases are recognized, an excision step where the error-containing strand is degraded resulting in a gap, and a repair synthesis step, where the gap is filled by the DNA resynthesis (for detailed reviews of MMR, see [46–48]).
The MMR process is highly conserved from E.coli to humans. The canonical human MMR pathway is carried out by two major protein complexes, which are so-called MutS and MutL, based on their homology to the E.coli MMR proteins [49]. While MutS is responsible for mismatch recognition, MutL couples the recognition of the mispaired bases by the MutS complexes to downstream MMR events, which lead to the removal of the strand containing the error. In mammalians, the initial mismatch recognition step is fulfilled by two MutS activities that function as heterodimers. The MSH2-MSH6 heterodimer, also known as MutSa, preferentially recognizes base-base mismatches and small IDLs of one or two nucleotides, while MutSb, the heterodimer of MSH2 and MSH3 recognizes larger IDLs. Formation of the MutS-DNA complex is followed by ATP-dependent recruitment of MutL homolog (MLH) complexes. Three MutL activities have been identified and, like MutS, also function as heterodimeric complexes. MutLα, a heterodimer of MLH1 and PMS2, which contains the primary MutL activity (~ 90 %) in humans and supports the repair initiated by both MutSα and MutSβ. The two additional MutL heterodimers consist of MLH1/PMS2 (MutLb) and MLH1/MLH3 (MutLg), which may play minor roles in MMR.
Assembly of the ATP-dependent MutS-MutL-DNA heteroduplex ternary complex is necessary to activate exonuclease mediated degradation of the error-containing strand [50]. In humans, this degradation is performed by exonuclease 1 (Exo1) through its 5′ to 3′ exonucleolytic activity [51]. The entry point for Exo1, which may be thousands of nucleotides from the mismatch, is generated via single-strand scission by the PCNA/replication factor C (RFC)-dependent endonuclease activity of MutLα [52]. The extensive gap left by Exo1 is then resynthesized by DNA polymerase d, which is accompanied by at least two other proteins, PCNA and replication protein A (RPA). Lastly, MMR is completed by DNA ligase I sealing of the remaining nick.
2.3.3 Nucleotide Excision Repair (NER)
NER is a highly versatile repair pathway that can recognize and remove a wide variety of bulky, helix-distorting lesions from DNA. The most significant of these lesions are pyrimidine dimers, such as cyclobutane pyrimidine dimers (CPD) and 6–4 photoproducts, which are produced by the UV component of sunlight. Another noteworthy substrate of NER is cisplatin -DNA intrastrand crosslinks. NER is mediated by the sequential assembly of repair proteins at the site of the DNA lesion. While mechanistically similar to BER, the NER pathway is more complex, requiring some thirty different proteins to carry out a multi-step ‘cut-and-patch’-like mechanism. These steps involve DNA damage recognition, local opening of the DNA helix around the lesion, excision of a short single-strand segment of DNA spanning the lesion, and sequential repair synthesis and strand ligation (for detailed reviews of NER, see [53–55]). The biological importance of NER is supported by the fact that defects in NER cause several human genetic disorders, including xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy, which are all characterized by extreme sun sensitivity. In addition, these diseases demonstrate overlapping symptoms associated with cancer, developmental delay, immunological defects, neurodegeneration, and premature aging [56, 57].
The NER system consists of two related subpathways, termed global genome NER (GG-NER) and transcription-coupled NER (TC-NER). As the names imply, GG-NER eliminates DNA lesions throughout the genome, while TC-NER is preferentially responsible for repairing lesions located on the coding strand of actively transcribed genes. Both pathways are mechanistically the same, apart from the initial damage recognition step. In GG-NER, the principle damage recognition factor is the XPC/HR23B/CEN2 (XP complementation group C/Rad23 homolog B/Centrin-2) protein complex [58]. HR23B and CEN2 are accessory proteins that increase both the affinity and specificity of XPC binding to helix-distorting DNA damage. In addition, the DNA binding affinity of XPC generally correlates with the degree of helical distortion [59]. For example, XPC has low affinity to lesions that are caused by only minor distortions, such as UV-induced CPDs. Thus, an auxiliary damage-recognizing complex called the UV-damaged DNA binding complex (UV-DDB), which consists of two subunits, DDB1 and XPE (DDB2), initially detects these types of lesions. The binding of UV-DDB to damaged DNA induces an increase in helix distortion (i.e., DNA bending), which subsequently facilitates the recruitment of the XPC complex to the damage site [60]. In contrast, damage recognition in TC-NER is initiated when an elongating RNA polymerase II (RNAPII) is arrested upon encountering a site of DNA damage [61]. Subsequently, two TC-NER-specific proteins, Cockayne syndrome A (CSA) and B (CSB), are thought to displace the stalled RNAPII to allow NER proteins access to the lesion [62].
Following damage recognition, both GG-NER and TC-NER proceed through the common ‘core’ NER reactions. Initially, either the XPC complex in GG-NER or, presumably, CSB and CSA in TC-NER recruit the multi-subunit (ten protein complex) and the multi-functional transcription factor TFIIH to the site of damage. Next, two TFIIH-associated, ATP-dependent helicases XPB and XPD orchestrate the asymmetric unwinding of the DNA helix to form a ~ 30 nucleotide bubble flanking the lesion. Initial unwinding permits access of XPA to the damaged region, which provides a second level of damage recognition in addition to ensuring that undamaged DNA is not subjected to excision repair. The binding of XPA is accompanied by the heterotrimeric, single stranded DNA binding protein RPA (replication protein A), which allows for complete extension and subsequent stabilization of the so-called pre-incision complex. In the subsequent step, two structure-specific endonucleases XPG and XPF/ERCC1 cleave the DNA at positions 3′ and 5′ to the damage, respectively, leading to excision of the lesion-containing oligonucleotide of about 30 nucleotides. Lastly, DNA polymerase d or e uses the undamaged strand as a template to resynthesize the resulting gap. The nick of the repaired strand is then sealed by DNA ligase, thus completing the NER process.
2.3.4 Double-Strand Break Repair
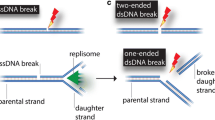
Double-strand breaks (DSBs) are amongst the most biologically hazardous types of DNA damage. For instance, a single unrepaired DSB is often sufficient to cause cell death. In addition, inaccurate repair can lead to deletions or chromosomal aberrations, events that associated with the development of cancer or other genomic instability syndromes. Thus, the repair of DSBs is both critical for cell survival and maintenance of genome integrity [63, 64]. The two main mechanisms by which mammalian cells repair DSBs are homologous recombination (HR) and non-homologous end-joining (NHEJ). These two repair systems differ in their requirement for a homologous template DNA and in the fidelity of DSB repair. HR-directed repair is largely an error-free mechanism as it utilizes the genetic information contained in the undamaged sister chromatid as a template (for review, see [65]). In contrast, NHEJ is normally error-prone and involves elimination of DSBs by direct ligation of the broken ends (for review, see [66]). NHEJ is reasoned to be the predominant pathway in mammalian cells operating in all phases of the cell cycle, while HR is restricted to the late-S and G2 phases. The basic mechanisms of these pathways and the factors involved are briefly outlined below.
2.3.4.1 Homologous Recombination (HR)
Much of our current knowledge concerning the mechanism of eukaryotic homology-directed repair is contributed to studies in bacteria and yeast, where HR is most efficient. HR can be conceptually divided into three phases: presynapsis, synapsis, and postsynapsis. During presynapsis, the DNA ends surrounding the DSB are processed through 5′ to 3′ end resection to generate molecules with 3′-single-stranded tails. The heterotrimeric MRN complex (Mre11-Rad50-Nbs1) together with CtIP (RBBP8) are responsible for the initiation of resection in which the 5′-ends on either side of the DSB are trimmed back to create short 3′-overhangs of single-strand DNA [67]. The second step in the 5′ to 3′ resection is presumably continued by the combined action of BLM helicase (Bloom syndrome, RecQ helicase-like) and Exo1 exonuclease[68]. Following end resection, single-stranded DNA tails are bound by RPA to remove disruptive secondary structures that would otherwise obstruct binding of Rad51 recombinase. RPA is subsequently replaced by Rad51 in conjunction with several mediator proteins, such as Rad52, BRCA2, and a group of proteins known as Rad51 paralogs (RAD51B, RAD51C, RAD51D, XRCC2, and XRCC3) [69]. The Rad51-coated single-stranded DNA tail, also referred to as the Rad51 nucleoprotein filament, then executes the DNA sequence homology search, which is the central reaction of HR. Once the homologous DNA has been identified, Rad51 mediates DNA strand invasion reaction, wherein the damaged DNA strand invades the template DNA duplex (i.e., sister chromatid). Next, DNA synthesis from the 3′-end of the invading strand is carried out by DNA polymerase h followed by successive ligation by DNA ligase I to yield a four-way junction intermediate structure known as a Holliday junction [70]. This recombination intermediate is resolved in one of three ways, by ‘dissolution’ mediated by the BLM-TopIIIa complex, by symmetrical cleavage by GEN1/Yen1 or Slx1/Slx4, or by asymmetric cleavage by the structure-specific endonuclease Mus81/Eme1 [71–73], resulting in the error-free correction of the DSB.
2.3.4.2 Non-Homologous End-Joining (NHEJ)
The molecular mechanism of NHEJ is mediated by a relatively small number of essential factors that are sequentially recruited to DSB sites. The initial step in the NHEJ process entails recognition and binding of the Ku70/Ku80 heterodimer (Ku) to the exposed DNA termini of the DSB. Structurally, Ku adopts a preformed ring-shaped structure that completely encircles the DNA duplex [74]. Upon binding to DNA, the Ku-DNA complex recruits the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) to generate the so-called DNA-PK holoenzyme, which exhibits protein kinase activity. The recruitment of DNA-PKcs induces an inward translocation of Ku along the DNA, allowing DNA-PKcs to contact DNA termini [75]. More importantly, the binding of the DNA-PKcs molecules on opposing DSB ends promotes synapsis or tethering of the two DNA molecules. Synapsis of DNA-PKcs also results in autophosphorylation of DNA-PKcs, which allows the DNA termini to become accessible [76]. Like most DNA repair processes, depending on the type and complexity of the DSB break, DNA ends may require modification prior to ligation. For example, DNA termini containing single-stranded overhangs can be made ligatable through either DNA polymerase-mediated fill-in or nucleolytic resection. The resynthesis of missing nucleotides during NHEJ has been associated with two members of the X family DNA polymerases, Pol m and Pol l [77]. Alternatively, the NHEJ-specific nuclease Artemis, whose activities include a DNA-PK independent 5′ to 3′ exonuclease activity as well as a DNA-PK dependent endonuclease activity, which is acquired through phosphorylation by DNA-PK, can excise single-stranded overhangs [78]. Other candidates that may also participate in DNA end ‘cleaning’ process include several of the lesion-specific BER enzymes, such as APE1, Tdp1, and PNKP [79] (see above), as well as the two functional exonucleases Exo1 and WRN, which is mutated in Werner syndrome patients [80, 81]. Consequently, the same enzymes that participate in the end-processing step of NHEJ are considered to be responsible for the overhang mispairing and the gain or loss of nucleotides associated with NHEJ-mediated repair. After appropriate (or sometimes inappropriate) processing of the DNA termini, ligation of the DNA ends is carried out by DNA ligase IV in conjunction with its binding partner XRCC4. An additional factor, XLF (XRCC4-like factor), interacts with the XRCC4-DNA ligase IV complex to promote DNA ligation [82].
2.4 Conclusion
The biological significance of DNA repair mechanisms is underscored by the fact that their deregulation can contribute to the initiation and progression of cancer. On the other hand, DNA repair can confer resistance to front line cancer treatments (i.e. chemotherapy and radiation), which rely on the generation of DNA damage to kill cancer cells. Thus, the sensitivity of cancer cells to DNA damaging agents is most likely related to intrinsic deficiencies in DNA repair mechanisms. The capacity of cancer cells (or cancer stem cells) to recognize DNA damage and initiate DNA repair is a key mechanism for therapeutic resistance or recurrence. The following chapters will discuss the DNA repair mechanisms that ensure protection of cancer stem cells.
References
Lindahl T, Barnes DE (2000) Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol 65:127–133
Hoeijmakers JH (2009) DNA damage, aging, and cancer. N Engl J Med 361(15):1475–1485
Stracker TH, Usui T, Petrini JH (2009) Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair (Amst) 8(9):1047–1054
Zhou BB, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408(6811):433–439
Rich T, Allen RL, Wyllie AH (2000) Defying death after DNA damage. Nature 407(6805):777–783
McKinnon PJ (2009) DNA repair deficiency and neurological disease. Nat Rev Neurosci 10(2):100–112
De Bont R, van Larebeke N (2004) Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 19(3):169–185
Lindahl T (1993) Instability and decay of the primary structure of DNA. Nature 362(6422):709–715
Lindahl T, Nyberg B (1972) Rate of depurination of native deoxyribonucleic acid. Biochemistry 11(19):3610–3618
Sugiyama H, Fujiwara T, Ura A et al (1994) Chemistry of thermal degradation of abasic sites in DNA. Mechanistic investigation on thermal DNA strand cleavage of alkylated DNA. Chem Res Toxicol 7(5):673–683
Yonekura S, Nakamura N, Yonei S, Zhang-Akiyama QM (2009) Generation, biological consequences and repair mechanisms of cytosine deamination in DNA. J Radiat Res (Tokyo) 50(1):19–26
Frederico LA, Kunkel TA, Shaw BR (1990) A sensitive genetic assay for the detection of cytosine deamination: determination of rate constants and the activation energy. Biochemistry 29(10):2532–2537
Krokan HE, Drablos F, Slupphaug G (2002) Uracil in DNA–occurrence, consequences and repair. Oncogene 21(58):8935–8948
Kow YW (2002) Repair of deaminated bases in DNA. Free Radic Biol Med 33(7):886–893
Apel K, Hirt H (2004) Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol 55:373–399
Marnett LJ (2000) Oxyradicals and DNA damage. Carcinogenesis 21(3):361–370.
Cadet J, Berger M, Douki T, Ravanat JL (1997) Oxidative damage to DNA: formation, measurement, and biological significance. Rev Physiol Biochem Pharmacol 131:1–87
Burney S, Caulfield JL, Niles JC, Wishnok JS, Tannenbaum SR (1999) The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat Res 424(1–2):37–49
Ravanat J-L (2005) Measuring oxidized DNA lesions as biomarkers of oxidative stress: an analytical challenge FABAD. J Pharm Sci 30(2):100–113
Major GN, Collier JD (1998) Repair of DNA lesion O6-methylguanine in hepatocellular carcinogenesis. J Hepatobiliary Pancreat Surg 5(4):355–366
McCulloch SD, Kunkel TA (2008) The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res 18(1):148–161
Shimizu M, Gruz P, Kamiya H et al (2003) Erroneous incorporation of oxidized DNA precursors by Y-family DNA polymerases. EMBO Rep 4(3):269–273
McClendon AK, Osheroff N (2007) DNA topoisomerase II, genotoxicity, and cancer. Mutat Res 623(1–2):83–97
Pourquier P, Pommier Y (2001) Topoisomerase I-mediated DNA damage. Adv Cancer Res 80:189–216
Bridges BA (2005) Error-prone DNA repair and translesion synthesis: focus on the replication fork. DNA Repair (Amst) 4(5):618–619, 634
Ravanat JL, Douki T, Cadet J (2001) Direct and indirect effects of UV radiation on DNA and its components. J Photochem Photobiol B 63(1–3):88–102
Ward JF (1988) DNA damage produced by ionizing radiation in mammalian cells: identities, mechanisms of formation, and reparability. Prog Nucleic Acid Res Mol Biol 35:95–125
Wogan GN, Hecht SS, Felton JS, Conney AH, Loeb LA (2004) Environmental and chemical carcinogenesis. Semin Cancer Biol 14(6):473–486
Irigaray P, Belpomme D (2010) Basic properties and molecular mechanisms of exogenous chemical carcinogens. Carcinogenesis 31(2):135–148
Noll DM, Mason TM, Miller PS (2006) Formation and repair of interstrand cross-links in DNA. Chem Rev 106(2):277–301
Sinha BK (1995) Topoisomerase inhibitors. A review of their therapeutic potential in cancer. Drugs 49(1):11–19
Bedard LL, Massey TE (2006) Aflatoxin B1-induced DNA damage and its repair. Cancer Lett 241(2):174–183
Altieri F, Grillo C, Maceroni M, Chichiarelli S (2008) DNA damage and repair: from molecular mechanisms to health implications. Antioxid Redox Signal 10(5):891–937
Zharkov DO (2008) Base excision DNA repair. Cell Mol Life Sci 65(10):1544–1565
Jacobs AL, Schar P (2012) DNA glycosylases: in DNA repair and beyond. Chromosoma 121(1):1–20
Hitomi K, Iwai S, Tainer JA (2007) The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA Repair (Amst) 6(4):410–428
Abbotts R, Madhusudan S (2010) Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Cancer Treat Rev 36(5):425–435
Interthal H, Chen HJ, Champoux JJ (2005) Human Tdp1 cleaves a broad spectrum of substrates, including phosphoamide linkages. J Biol Chem 280(43):36518–36528
Ahel I, Rass U, El-Khamisy SF et al (2006) The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 443(7112):713–716
Caldecott KW (2008) Single-strand break repair and genetic disease. Nat Rev Genet 9(8):619–631
Fortini P, Dogliotti E (2007) Base damage and single-strand break repair: mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair (Amst) 6(4):398–409
Caldecott KW (2003) XRCC1 and DNA strand break repair. DNA Repair (Amst) 2(9):955–969
Marintchev A, Mullen MA, Maciejewski MW, Pan B, Gryk MR, Mullen GP (1999) Solution structure of the single-strand break repair protein XRCC1 N-terminal domain. Nat Struct Biol 6(9):884–893
Malanga M, Althaus FR (2005) The role of poly(ADP-ribose) in the DNA damage signaling network. Biochem Cell Biol 83(3):354–364
Peltomaki P (2001) Deficient DNA mismatch repair: a common etiologic factor for colon cancer. Hum Mol Genet 10(7):735–740
Li GM (2008) Mechanisms and functions of DNA mismatch repair. Cell Res 18(1):85–98
Fukui K (2010) DNA mismatch repair in eukaryotes and bacteria. J Nucleic Acids 2010:1–6
Larrea AA, Lujan SA, Kunkel TA (2010) SnapShot: DNA mismatch repair. Cell 141(4):730 e1
Modrich P (2006) Mechanisms in eukaryotic mismatch repair. J Biol Chem 281(41):30305–30309
Galio L, Bouquet C, Brooks P (1999) ATP hydrolysis-dependent formation of a dynamic ternary nucleoprotein complex with MutS and MutL. Nucleic Acids Res 27(11):2325–2331
Tran PT, Erdeniz N, Symington LS, Liskay RM (2004) EXO1-A multi-tasking eukaryotic nuclease. DNA Repair (Amst) 3(12):1549–1559
Kadyrov FA, Holmes SF, Arana ME et al (2007) Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease. J Biol Chem 282(51):37181–37190
Shuck SC, Short EA, Turchi JJ (2008) Eukaryotic nucleotide excision repair: from understanding mechanisms to influencing biology. Cell Res 18(1):64–72
Costa RM, Chigancas V, Galhardo Rda S, Carvalho H, Menck CF (2003) The eukaryotic nucleotide excision repair pathway. Biochimie 85(11):1083–1099
Nouspikel T (2008) Nucleotide excision repair and neurological diseases. DNA Repair (Amst) 7(7):1155–1167
Cleaver JE, Lam ET, Revet I (2009) Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet 10(11):756–768
Vermeulen W, de Boer J, Citterio E et al (1997) Mammalian nucleotide excision repair and syndromes. Biochem Soc Trans 25(1):309–315
Sugasawa K, Ng JM, Masutani C et al (1998) Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell 2(2):223–232
Sugasawa K (2008) XPC: its product and biological roles. Adv Exp Med Biol 637:47–56
Sugasawa K (2010) Regulation of damage recognition in mammalian global genomic nucleotide excision repair. Mutat Res 685(1–2):29–37
Fousteri M, Mullenders LH (2008) Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res 18(1):73–84
Hanawalt PC, Spivak G (2008) Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol 9(12):958–970
van Gent DC, Hoeijmakers JH, Kanaar R (2001) Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet 2(3):196–206
Khanna KK, Jackson SP (2001) DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet 27(3):247–254
Li X, Heyer WD (2008) Homologous recombination in DNA repair and DNA damage tolerance. Cell Res 18(1):99–113
Lieber MR (2010) The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79:181–211
Sartori AA, Lukas C, Coates J et al (2007) Human CtIP promotes DNA end resection. Nature 450(7169):509–514
Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC (2008) Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci USA 105(44):16906–16911
Forget AL, Kowalczykowski SC (2010) Single-molecule imaging brings Rad51 nucleoprotein filaments into focus. Trends Cell Biol 20(5):269–276
McIlwraith MJ, Vaisman A, Liu Y, Fanning E, Woodgate R, West SC (2005) Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol Cell 20(5):783–792
Ip SC, Rass U, Blanco MG, Flynn HR, Skehel JM, West SC (2008) Identification of Holliday junction resolvases from humans and yeast. Nature 456(7220):357–361
Mimitou EP, Symington LS (2009) Nucleases and helicases take center stage in homologous recombination. Trends Biochem Sci 34(5):264–272
Seki M, Nakagawa T, Seki T et al (2006) Bloom helicase and DNA topoisomerase III alpha are involved in the dissolution of sister chromatids. Mol Cell Biol 26(16):6299–6307
Walker JR, Corpina RA, Goldberg J (2001) Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 412(6847):607–614
Yoo S, Dynan WS (1999) Geometry of a complex formed by double strand break repair proteins at a single DNA end: recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res 27(24):4679–4686
DeFazio LG, Stansel RM, Griffith JD, Chu G (2002) Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J 21(12):3192–3200
Lieber MR, Lu H, Gu J, Schwarz K (2008) Flexibility in the order of action and in the enzymology of the nuclease, polymerases, and ligase of vertebrate non-homologous DNA end joining: relevance to cancer, aging, and the immune system. Cell Res 18(1):125–133
Jeggo P, O’Neill P (2002) The Greek goddess, Artemis, reveals the secrets of her cleavage. DNA Repair (Amst) 1(9):771–777
Chappell C, Hanakahi LA, Karimi-Busheri F, Weinfeld M, West SC (2002) Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J 21(11):2827–2832
Perry JJ, Yannone SM, Holden LG et al (2006) WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat Struct Mol Biol 13(5):414–422
Bahmed K, Seth A, Nitiss KC, Nitiss JL (2011) End-processing during non-homologous end-joining: a role for exonuclease 1. Nucleic Acids Res 39(3):970–978
Ahnesorg P, Smith P, Jackson SP (2006) XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 124(2):301–313
Boland CR, Luciani MG, Gasche C, Goel A (2005) Infection, inflammation, and gastrointestinal cancer. Gut 54(9):1321–1331
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Dexheimer, T. (2013). DNA Repair Pathways and Mechanisms. In: Mathews, L., Cabarcas, S., Hurt, E. (eds) DNA Repair of Cancer Stem Cells. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-4590-2_2
Download citation
DOI: https://doi.org/10.1007/978-94-007-4590-2_2
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-4589-6
Online ISBN: 978-94-007-4590-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)