
Summary
“It seems likely that most if not all the genetic information in any organism is carried by nucleic acid – usually by DNA […].” Plant organellar genomes have a spelling problem. If the genome were a book, many words with “U”s (uridines) would be spelled with “C”s (cytidines) instead, and in certain plant species, the reverse would also be seen, with Cs replaced by Us. However, plants change these “mistakes” at the RNA level, correcting U to C and C to U at non-random positions, via a phenomenon called RNA editing. We hope Francis Crick would have forgiven us for messing up the above quote from his 1962 Nobel Laureate acceptance speech. You can return the sentence to its original meaning easily by following the rules of plant organellar RNA editing. However, even when spelled right, the statement still has a hole in it, maybe one that Francis Crick anticipated and thus started the sentence with, “It seems likely….” Because here’s the rub: Organellar genetic information cannot be read the easy way, by identifying open reading frames based on start and stop codons and predicting the protein sequences based on codons. Instead, it is far better to read the RNA itself or, better yet in experimental terms, look at the cDNA.
In this review, we will attempt to summarize the state of knowledge regarding RNA editing in plant organelles. We will mostly focus on the mechanistic aspects of RNA editing, with considerable space devoted to our understanding of editing site recognition. Following that, and at the center of this review, we will examine the latest developments in our understanding of the editing machinery. In the end, we will dare to take a quick look at some of the reasons behind the seemingly futile process of plant organellar RNA editing.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
I. The Essentials of Organellar RNA Editing: C to U and U to C
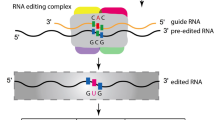
RNA editing was initially discovered in the transcriptome of trypanosome mitochondria (Benne et al. 1986), which undergo insertional/deletional RNA editing: scores of uracil residues are added to or removed from mitochondrial messages. In the 1980s and 1990s, various examples of RNA editing were described in organisms from diverse taxa (Gott and Emeson 2000; Knoop 2010). In all cases, the primary RNA sequences were found to be altered by base modifications, nucleotide insertions, nucleotide deletions, or (rarely) nucleotide replacements. The diverse editing processes discovered to date arose independently from each other and employ widely different mechanisms (Smith et al. 1997; Gott and Emeson 2000; Knoop 2010). In plant organelles, RNA editing is restricted to nucleotide conversions. In mRNAs, only changes from C to U or (less frequently) from U to C have been observed so far, while tRNAs additionally show conversions from A to I (inosine). Plant organellar RNA editing was first discovered in 1989 in wheat and evening primrose mitochondria (Covello and Gray 1989; Gualberto et al. 1989; Hiesel et al. 1989), followed 2 years later by the discovery of editing in maize chloroplasts (Hoch et al. 1991). Since then, researchers have shown that organellar RNA editing in mitochondria and chloroplasts shares many features, including the position of cis-regulatory sequences, the types of nucleotide conversions, the frequency of particular codons affected and, more recently, the types of trans-factors required for RNA editing (see Sect. IV). Also, RNA editing in mitochondria and chloroplasts shows a strict phylogenetic co-occurrence in embryophyte evolution (see Sect. II).
Editing sites do not seem to be strewn randomly across organellar genomes; in fact, most RNA-editing events restore conserved codon identities that had been lost on the DNA level (Walbot 1991; Gray 1996; Hanson et al. 1996; Maier et al. 1996; Knoop 2004). Several of the codons restored by RNA editing have been mutagenized and shown to be essential for protein function (Bock et al. 1994; Zito et al. 1997; Schmitz-Linneweber et al. 2005b), and many editing events regenerate/remove stop or start codons and can therefore be regarded as essential (e.g. Hoch et al. 1991; Wintz and Hanson 1991). In recent years, however, it has become clear that there are also a number of editing events, especially in mitochondria, that do not seem to be required for the encoded proteins to remain functional. For example, many mitochondrial null mutants for factors that are essential for the editing of individual sites or clusters of sites are indistinguishable from their wild-type siblings, at least under standard growth parameters (Bentolila et al. 2010; Takenaka 2010; Takenaka et al. 2010). The same holds true for several recently discovered null mutants of editing factors for chloroplast sites (Hammani et al. 2009). It will be necessary to analyze these mutants more thoroughly in the future, and possibly identify conditions under which the “unedited” versions of these proteins fail to entirely replace the “edited” ones.
II. Phylogenetic Distribution of RNA Editing Sites in Land Plants
With the exception of the marchantiid liverworts, species from all other major embryophyte taxa have been found to display organellar RNA editing (Malek et al. 1996; Freyer et al. 1997; Steinhauser et al. 1999; Duff and Moore 2005). This includes all of the angiosperms and gymnosperms investigated to date, including Arabidopsis (for example in Arabidopsis: Giegé and Brennicke 1999; Tillich et al. 2005), the fern Adiantum capillus-veneris (Wolf et al. 2004), the lycophyte Isoetes engelmannii, the mosses Physcomitrella patens (Miyata and Sugita 2004; Rüdinger et al. 2009) and Takakia lepidozioides (Sugita et al. 2006), and the hornwort Anthoceros formosae (Yoshinaga et al. 1996, 1997; Kugita et al. 2003; Duff and Moore 2005). With regard to the marchantiid liverworts, the sister group of jungermanniid liverworts were found to have editing of mitochondrial messages. This suggests that Marchantia polymorpha and the Marchantiales underwent a secondary loss of RNA editing (Groth-Malonek et al. 2005, 2007), and that organellar RNA editing can be considered a common trait of embryophytes (Fig. 13.1). Editing frequencies differ however dramatically between different taxa. For example, the chloroplast of A. formosae was found to have 509 C-to-U and 433 U-to-C editing sites (Kugita et al. 2003), whereas spermatophytes exhibit only about 30 C-to-U editing events and no U-to-C editing (Maier et al. 1996; Tsudzuki et al. 2001). The record holder on the high side is the lycopodiophyte, Selaginella, which has 2,139 editing sites in its mitochondrial genome (Hecht et al. 2011b). At the lower end, Physcomitrella has so far been found to have only two and 11 editing events in its chloroplast and mitochondrial transcriptomes, respectively (Miyata et al. 2002; Miyata and Sugita 2004; Rüdinger et al. 2009).

Phylogeny of RNA editing in chlorophytes. Based on current experimental data, it is most parsimonious to assume that organellar RNA editing was gained in the ancestor of all land plants (filled triangle). This is suggested by the finding that the few members of the chlorophytes analysed to date do not show organellar RNA editing. Because of pervasive RNA editing in members of its sister groups, the Haplomitriopsida and Jungermanniopsida, it is most likely that RNA editing was lost in the lineage leading to the liverwort Marchantia polymorpha (open triangle) (Adapted from (Knoop 2010).
Of the thousands of known organellar editing sites, very few are conserved between embryophytes (Tillich et al. 2006). This is true within a narrower range of taxa (Freyer et al. 1997; Schmitz-Linneweber et al. 2002; Fiebig et al. 2004), and even between species of the same genus (Sasaki et al. 2003), suggesting that RNA-editing sites undergo rapid evolution. The few sites analyzed across a large set (>100) of species from diverse branches of angiosperm evolution all seem to be ancient, and were likely present in the ancestor of all present-day angiosperms. However, these sites have been far from stable. For example, an editing site in the chloroplast matK gene was independently lost at least 36 times in angiosperm evolution (Tillich et al. 2009a). Similarly, a site in the chloroplast psbE gene was also lost multiple times (Hayes and Hanson 2008). Given that many basal tracheophyte chloroplast transcriptomes boast large numbers of editing sites, we can assume that the ancestor of spermatophytes had a complex, large editotype that became reduced to the 30-something sites presently found in the extant angiosperm and gymnosperm species. In mitochondria, however, this reduction never took place in angiosperms. It has been speculated that variations in genomic evolution between chloroplasts and mitochondria could explain these differences in RNA editing frequencies (see Sect. V).
III. cis-Requirements for Plant Organellar RNA Editing
The C-to-U editing of the human apoB mRNA depends on an 11-nucleotide (nt)-long sequence element called the “mooring sequence,” which is located right next to (upstream of) the editing site (Smith et al. 1997). This sequence is recognized by the Apobec1/ACF editing machinery and ensures that the correct C is converted to U. Based on this model, early efforts to identify cis-elements for RNA editing in organelles started from the hypothesis that sequences surrounding the target nucleotide would participate in its recognition by trans-factors. The work on chloroplast cis-elements initially progressed much faster than the corresponding work on mitochondria because (unlike mitochondria) chloroplasts were amenable to genetic engineering, which allowed for direct testing of cis-sequences. More recently, in vitro editing systems have been developed for both chloroplasts and mitochondria, facilitating the analysis of sequence requirements in both compartments.
A. Chloroplast cis-Elements for RNA Editing
Early after the detection of RNA editing in chloroplasts, within-species sequence comparisons of the identified sites led to the detection of nucleotide biases at certain positions. Notably, position -1 seemed to be critical for editing, since 29 of 31 tobacco editing sites were found to have pyrimidines at this position (Maier et al. 1992a, b; Hirose et al. 1999), and point mutations at this site yielded pronounced reductions in editing efficiencies in vivo (Bock et al. 1996). The minimal sequence requirements for editing-site recognition and processing were tested by introducing mini-RNAs into the plastid genome of tobacco, which showed that the recognition of most editing sites relied on short (mostly <20 nt) upstream sequences (Chaudhuri et al. 1995; Bock et al. 1996; Chaudhuri and Maliga 1996; Reed et al. 2001a). However, researchers were unable to detect a core consensus sequence, providing an early indication that site recognition involved specific factors for individual sites. In addition, researchers failed to find common secondary structures in the vicinity of editing sites, indicating that this is not the manner in which the to-be-edited Cs are recognized. However, some inter-site homologies were found among small subsets of sites, always within 15 nt upstream of the editing site (Chateigner-Boutin and Hanson 2002, 2003; Tillich et al. 2005, 2006). Recent advances in our understanding of editing factors allow us to speculate that the members of these clusters are recognized by pentatricopeptide repeat (PPR) proteins, which were recently identified as acting on small sets of editing sites (see Sect. IV.A).
Over the past decade, the laborious plastid transformation techniques for editing site analyses used in the 1990s has been complemented by in vitro editing systems that have become available for four species: tobacco (Hirose and Sugiura 2001), pea (Miyamoto et al. 2002; Nakajima and Mulligan 2005), maize (Hayes et al. 2006), and Arabidopsis (Hegeman et al. 2005). These systems have allowed the thorough mutagenesis of cis-elements, which has confirmed that the core cis-elements are located in the 20-nt region upstream of most editing sites. Only rarely do nucleotides 3′ of the editing site contribute substantially to editing efficiency (Hayes and Hanson 2007). Further mutational analyses demonstrated that the nucleotides immediately preceding the editing site (−1 to−4) and the editing site itself are not essential for binding of the trans-factor(s), although their specific recognition is required for the nucleotide conversion itself (Miyamoto et al. 2002, 2004). Thus, the 5′-proximal bases of editing sites do not act merely as spacers, but rather must be bound in sequence-specific interactions in order for catalysis to occur.
However, although the in vivo and in vitro data have shown that the most important sequences for site recognition lie predominantly in the immediate 5′ vicinity of the nucleotide to be edited (Fig. 13.2), this is not the entire story. Several studies have suggested that there are also more complex cis-elements involved. For example, the tobacco ndhF mRNA shows a bipartite recognition site in which essential elements are 19 nt apart (Sasaki et al. 2006). For several editing sites, increasing the length of the 5′ region has been shown to increase editing efficiency, although these distal sequence elements are not essential (Hayes et al. 2006). An upstream-sequence effect has also been reported for the rpoBeU158SL editing site (Hayes et al. 2006; editing sites are identified in this review by their position in the respective reading frame following a recent nomenclature proposal by Rüdinger and colleagues Rüdinger et al. 2009). Also, in the case of the ndhBeU156PL and ndhBeU196HY plastid editing sites, 42 nt of both 5′ and 3′ adjacent sequences were insufficient to direct editing in vivo (Bock et al. 1996). Compared to native editing efficiencies (which usually reach 100%), the experimental editing efficiencies are generally poor (often below 10% of wild-type levels) for both in vitro and in vivo experiments (Bock et al. 1996; Reed et al. 2001b; Sasaki et al. 2006). The poor performance of these artificial systems may be due to overexpression problems and other technical limitations, but it could also reflect the lack of necessary distal sequence elements. Future work will be required to determine the specific role of additional modifiers located at a greater distance from the processed site. Such modifying sequences may not necessarily be involved directly in site recognition; indirect effects via regulation of translation (Karcher and Bock 1998) or modification of the processing status of other sequence elements in the same message (Schmitz-Linneweber et al. 2001) could also impact the processing of an editing site (Fig. 13.2).

Models for cis-elements impacting plant organellar editing efficiencies. (a) For most editing sites (here shown as unedited “C”), a short (10–25 nt) sequence element immediately upstream of the editing sites (bold black line) is sufficient for RNA editing. At some sites, the editing efficiency is modulated by additional, more distal sequence elements that are mostly unknown (bold grey line). Possibly, such elements base-pair with the core cis-element and could thus modulate binding of trans-factors (b). Alternatively, additional cis-elements could recruit proteins (light grey) that help to position or activate the editing machinery (dark grey; c). Finally, modulating sequence elements could influence other processes during the RNA life cycle, for example ribosome processivity, which secondarily could impact RNA editing.
B. Mitochondrial cis-Elements for RNA Editing
A general consensus editing-site-recognition sequence for mitochondria has not yet been found in silico (Giegé and Brennicke 1999), nor have common secondary structures been detected in the vicinity of editing sites (Mulligan et al. 1999). Because mitochondria are not amenable to standard stable transformation techniques, most of the data on cis-elements for mitochondrial editing sites has come from in organello and in vitro editing systems. However, the first evidence for the location of such cis-elements was obtained through the evaluation of intra-mitochondrial recombination events that deleted the sequences 5′ or 3′ of an editing site. Such studies showed that editing occurred only when the immediate upstream sequences were retained (Lippok et al. 1994; Kubo and Kadowaki 1997).
The in organello systems, which were first developed for wheat (Farre and Araya 2001) and later for maize mitochondria (Staudinger and Kempken 2003), are based on the electroporation of artificial editing-site-containing genes into mitochondria. Deletional and point mutagenesis approaches allowed the delineation of mitochondrial cis-elements resembling those found in chloroplasts: For most of the tested sites, 16 nt upstream and 6 nt downstream of the editing site were found to be sufficient for editing, while more distant nucleotides did not seem to play a role (Farre et al. 2001; Choury et al. 2004). However, there were exceptions to this rule. For example, no editing was observed when the Sorghum atp6 gene was introduced into maize mitochondria, but partial editing was seen following the introduction of chimeric constructs consisting of the 5′-UTR and part of the 5′ coding region of maize atp6 fused with Sorghum atp6 sequence. In cases like these, where the coding regions of the two species are virtually identical, distant UTR sequences appear to serve as signals for editing (Staudinger et al. 2005). Such long-distance effects do not appear to be an artifact of the utilized heterologous approach because other heterologous experiments have yielded high editing frequencies, (for example Arabidopsis sequences in maize mitochondria; Bolle and Kempken 2006). When cis-elements in the vicinity of editing sites were analyzed with regard to the contribution of individual nucleotides, no consensus could be detected. However, the importance of individual nucleotides clearly differed between sites, providing an early indication that the various editing sites are served by individual trans-factors.
The results from in vitro experiments on mitochondrial cis-elements of the dicot plants, cauliflower and pea, paralleled and extended the above-described findings from in organello systems. The dominant influence of the 20 nt immediately upstream of editing sites was repeatedly found for different sites in vitro (Takenaka et al. 2004; Neuwirt et al. 2005), and optimal editing was found when the upstream sequences were extended to ∼40 and sometimes even 70 nucleotides (Neuwirt et al. 2005; van der Merwe et al. 2006). Competition experiments with mutated versus non-mutated templates, as well as the direct mutation of identified cis-elements, helped researchers delineate the cis-elements and the importance of individual bases within them. The findings from these studies largely supported the idea that individual sites have individual cis-elements (Takenaka et al. 2004). Furthermore, an interesting effect was observed when multiple cis-elements were concatenated: The tandemly repeated recognition elements dramatically increased RNA-editing efficiencies, suggesting that local enrichment of a site-recognition factor can enhance RNA editing (Verbitskiy et al. 2008).
In sum, the basic parameters for editing-site recognition have been conserved (at least among angiosperms) in chloroplasts and mitochondria. In both organelles, short individual upstream sequences seem to serve as site-recognition elements. As we will discuss below, members of the PPR protein family utilize such short sequence stretches in a highly specific manner, an interaction essential for editing to occur (see Sect. IV.A). For groups other than angiosperms, the situation is less well understood, mostly because we do not yet have access to comparable in vivo, in vitro or in organello systems for these organisms. However, preliminary comparisons of the editing-site sequence environment in ferns suggests the presence of similar short upstream cis-elements (Tillich et al. 2006).
The picture is far less clear regarding the infrequent effect of more distant (usually upstream) sequences. It is conceivable that the overall structure of the RNA including long-distant tertiary interactions, is important for editing-site recognition (Fig. 13.2). Such long-range interactions could potentially help make the editing site available for PPR proteins, perhaps with the involvement of additional protein factors. Alternatively, undesirable secondary structures could impair the access of PPR proteins to their target sites, and might therefore require helicases to open the sites and allow the PPR proteins to interact. In the future, it is likely that a better understanding of the nature and components of the plant organellar editing machinery (‘editosome’) will help provide insights into these and other possibilities (see also discussion in Takenaka et al. 2008).
C. Tools for Analyzing RNA-Editing Sites In Silico
The mitochondrial transcriptomes of embryophytes usually have 100 or more RNA-editing sites. Therefore, prediction tools are essential for a comprehensive analysis of RNA editing in any organellar genome that has not yet been experimentally investigated. Originally, simple algorithms were developed based on sets of known editing sites versus non-edited sites. However, the predictive value of such algorithms was low (Cummings and Myers 2004). Later, a larger set of parameters was used to describe a likely editing site in an improved tool called REGAL (RNA Editing site prediction by Genetic Algorithm Learning; Thompson and Gopal 2006). This tool scored characteristics known to show biases between edited and unedited Cs in mitochondrial genomes, namely: the base at the −1 position of the editing site; the base at the +1 position of the editing site; the increase in hydrophobicity between the pre- and post-editing-encoded amino acids; the position of the edited C within the codon; the kind of codon that is edited; and the kind of amino acids that are consequently exchanged by the editing event. Using these parameters, the algorithm was trained based on the known Arabidopsis mitochondrial editing sites, which could be used to correctly predict more than 80% of the editing sites in related genomes (e.g. Brassica). An advantage of REGAL-like algorithms over the phylogeny-based analyses (see below) is that they can predict RNA-editing sites in intergenic regions and for species-specific ORFs, whereas homology-based searches are not applicable to such regions. A first tool to use such homology-based, phylogenetic information as a basis for editing-site prediction was PREP (predictive RNA editors for plants). The tool was initially tailored for the analysis of mitochondrial genomes (Mower 2005), but more recent versions have been made suitable for the analysis of chloroplast genomes or user-defined alignments (Mower 2009). The principle behind phylogenetic editing-site prediction is that RNA editing leads to an increase in protein conservation across species because codons for non-conserved amino acids are corrected to those for conserved amino acids (reviewed in Bock 2000; Wakasugi et al. 2001). Thus, editing sites are expected at positions where a C-to-U conversion would increase the conservation of a protein with respect to its homologs in other plants. This principle is also used by PREPACT (plant RNA editing prediction and analysis computer tool; Lenz et al. 2010), which extends the previous programs by predicting both C-to-U and U-to-C editing events. In addition, the output generated by PREPACT highlights different types of editing events (including partial editing) and offers a broader set of user-modified parameters for the graphical output.
The most elaborate RNA editing site prediction software developed to date uses phylogenetic information in conjunction with biochemical information on RNA-editing sites. This algorithm, called CURE (for cytidine-to-uridine recognizing editor) was initially designed for the analysis of mitochondrial genomes (Du and Li 2008), but was later adapted for seed plant chloroplast genomes (Du et al. 2009). So far, CURE has outperformed PREP (Du et al. 2009) but not REGAL (Thompson and Gopal 2006). CURE still has problems making accurate predictions for non-seed plant genomes (Lenz et al. 2010), but the quality of prediction should increase as the number of available training sets (i.e. experimentally determined editing sites) continues to climb.
A different sort of tool, called RedIdb (Picardi et al. 2007, 2010), seeks to categorize editing sites in the organellar genomes of eukaryotic organisms. RedIdb tries to present each editing event in its biological context by giving the corresponding DNA, cDNA and protein sequences together with gene ontologies and InterPro domains. Links are also established to the RESOPS (RNA-editing sites of land plant organelles on protein three-dimensional (3D) structures) database, which maps the amino acids affected by RNA editing onto the available 3D protein structures (Yura et al. 2009). RedIdb can be used directly for simple analyses because sequence analysis tools (e.g., BLAST and CLUSTAL algorithms, Thompson et al. 1994; Altschul et al. 1997) are directly implemented in the database. Furthermore, RedIdb is linked with the EdiPy tool, a script designed to allow the evolutionary simulation of highly edited mitochondrial sequences that are not amenable to analysis using standard statistical analysis tools (e.g., bootstrap analysis). RedIdb has the advantage of manual curation of entries over more general databases, such as dbRES (He et al. 2007), which collects all of the editing sites (not just those of organelles) deposited in GenBank, or ChloroplastDB (Cui et al. 2006) and GOBASE (O’Brien et al. 2009), which are general organellar-genome databases that do not emphasize RNA editing.
In all, current in silico tools have greatly helped to access RNA editing in novel organellar genomes and to move on swiftly from sequence analysis to editing site prediction and experimental analysis. Hopefully, our gain in knowledge on editing trans-factors (see next section), will at one point allow to connect in silico site prediction with the automated prediction of target specificities of editing site recognition factors in any embryophyte genome.
IV. Trans-Factors for C-to-U RNA Editing in Plant Organelles
Although the hunt for plant organellar editing factors was initially long and frustrating, recent years have seen tremendous progress in the field, and researchers have finally determined how editing specificity is assured. In short, proteins from the pentatricopeptide repeat (PPR) family show highly specific recognition of cis-elements upstream of editing sites. Some auxiliary factors have also been identified, but the process of catalysis is still unclear and it is not yet known which factors contribute directly to base conversion.
A. Pentatricopeptide Repeat (PPR) Proteins Specify Editing Sites
The identification of the first editing factor for an organellar (in this case, plastid) site was not the outcome of an elaborate genetic or biochemical screen for editing factors, but instead came out of work on an unrelated problem. T. Shikanai’s group (Kyoto University, Japan) had a long-standing interest in the plastid NADH dehydrogenase (NDH) complex, a multi-subunit complex in the thylakoid membrane that has still not been functionally assigned with a high degree of certainty. Shikanai and colleagues identified mutants of the NDH complex by screening an ethane methyl sulfonate (EMS) induced mutant collection, looking for characteristic defects in chlorophyll fluorescence (Hashimoto et al. 2003).The isolated mutants included one harboring a lesion in the gene for a PPR protein called CRR4 (chloroplast respiratory reduction 4; Kotera et al. 2005). The loss of CRR4 abrogated editing of the start codon of the ndhD mRNA, which encodes a core subunit of the NDH complex. Given that almost half of the editing sites in the plastid genome reside in ndh genes, it is not surprising that the screen also uncovered several additional editing mutants showing defects in specific ndh sites; all of them were found to result from lesions in PPR genes, namely those encoding CRR21, CRR22 and CRR28 (Okuda et al. 2007, 2009b; for a complete list see Table 13.1). Other studies searching for mutants defective in chloroplast development also identified PPR proteins as being involved in editing, again with each protein serving a specific site (CLB19, Chateigner-Boutin et al. 2008; LPA66, Cai et al. 2009; AtECB2, Yu et al. 2009; Vac1, Tseng et al. 2010). Not surprisingly, the apparent importance of PPR proteins in RNA editing spurred reverse-genetic studies; these led to the identification of seven additional PPR proteins that functioned in the editing of specific chloroplast sites: (OTP80; OTP81; OTP85; OTP86; OTP82; OTP84; RARE1; Hammani et al. 2009; Okuda et al. 2009a; Robbins et al. 2009).
Within a few years after the PPR proteins were first identified as editing factors of chloroplast sites, other family members were identified as being required for mitochondrial sites (Table 13.1). Notably, none appears to dually target editing sites in both organelles. The first mitochondrial editing factor was identified as part of an elegant screen for ecotype-specific differences in editing efficiency (Zehrmann et al. 2008), in which differences found between Arabidopsis accessions Columbia and C24 were used to map the editing activity. The identified factor was named MEF1 (mitochondrial editing factor 1), and insertional mutagenesis was used to confirm that it is essential for multiple sites (Zehrmann et al. 2009). A similar screen for quantitative trait loci that affect RNA editing identified REME1, a PPR protein that was shown to support editing sites in the nad2 and tatC mRNAs but was not found to be essential for their editing (Bentolila et al. 2008, 2010). A different type of forward screen for editing defects in a population of EMS-induced Arabidopsis mutants made use of a multiplexed single-nucleotide-primer-extension assay (Takenaka and Brennicke 2009). This screen utilized multiple primers that annealed just downstream of editing sites and were then extended with two alternatively labeled dideoxy nucleotides corresponding to either the edited or the unedited nucleotide. The extension products were then detected and analyzed with standard Sanger sequencing technology. The screen was shown to be capable of identifying a single mutant out of a pool of 50 plant samples (Takenaka and Brennicke 2009). Multiple mutants were recovered using this screening technique, and some of the underlying genes have been identified, including those encoding MEF9 and MEF11 (Verbitskiy et al. 2009; Takenaka 2010). MEF11 also emerged in an unrelated screen for lovastatin-insensitive mutants and was therefore initially called LOI1 (Kobayashi et al. 2007).
Additional mitochondrial PPR proteins required for specific editing sites were found by a reverse genetic screen in the moss Physcomitrella patens (PpPPR_56, PpPPR_77, PpPPR_91, PpPPR_71, Ohtani et al. 2010; Tasaki et al. 2010). An unrelated screen that sought to identify T-DNA mutants in rice uncovered a PPR gene mutation that abrogated the editing of at least three sites (Kim et al. 2009). Based on its seed and seedling phenotype, the mutant was called OGR1 (opaque and growth retardation 1). A screen for mutants displaying slow and delayed growth led to the identification of SLOW GROWTH1, which is required for editing of the nad4 and nad9 sites (Sung et al. 2010). Finally, a number of PPR proteins that target mitochondrial editing sites were found using reverse genetics, including six identified by a screen of T-DNA insertions in Arabidopsis PPR genes (MEF-18, MEF-19, MEF-20, MEF-21, MEF-22, MEF8, Takenaka et al. 2010).
In sum, 33 PPR proteins have been shown to serve organellar RNA-editing sites, all in a highly specific manner. For 20 of the 33, only one target RNA-editing site has been described. Of the remaining 13 PPR proteins, 8 serve 2 sites, 4 serve 3 sites, and 1 (OGR1) is involved in editing 7 sites (Table 13.1).
While the specificity of PPR proteins for low numbers of editing sites is consistent with the findings of studies on non-editing PPR proteins that are also linked to few RNA processing events (Schmitz-Linneweber and Small 2008), several caveats should be kept in mind. First, most of the target editing sites for PPR proteins that have been described to date are based solely on genetic data. Most of the editing PPR proteins have not yet been directly shown to associate with their target sites. Thus, a genetically determined lesion in the editing of an individual site could be a secondary effect of a more general impairment in basic organellar function. For example, loss of overall plastid translation affects the processing of multiple RNA-editing sites (Karcher and Bock 1998; Halter et al. 2004). Therefore, it would be desirable to determine whether the identified editing-site-related factors directly associate with their editing sites, either in vitro or (even better) in vivo.
Only one editing factor has been examined for its association with RNA in vivo to date: LOl1. Two RNA targets of LOI1, cox3 and atp1, were identified by co-purification with overexpressed LOI1:FLAG proteins (Tang et al. 2010). However, only one of the two recovered RNAs, the cox3 mRNA, displayed an editing defect in LOI1 mutants. Furthermore, the LOI1:FLAG eluates failed to yield any of the six other editing-deficient RNAs that had been recovered from LOI1-deficient plants. This, together with the low frequency of cox3 cDNA clones found after reverse transcription of RNA bound to LOI1:FLAG, precluded a final conclusion on whether or not LOI1 directly binds the RNAs that fail to undergo editing in the LOI1 mutants (Tang et al. 2010). With regard to in vitro studies, two other PPR proteins with functions in RNA editing have been shown to bind directly to their cognate editing site in vitro (Okuda et al. 2006; Tasaki et al. 2010). A minimal CRR4-binding element was determined to lie within the region from −25 to +10 relative to the ndhD editing site (Okuda et al. 2006). Similarly, the moss PpPPR_71 editing site is contained in a sequence element spanning nucleotide −40 to +5 relative to the editing site ccmFCeU122SF (Tasaki et al. 2010). These in vitro studies on the RNA binding sites of the editing PPR proteins are in good agreement with the locations and sizes of the previously determined cis-sequences for editing (see Sect. III.B).
A second caveat concerns the completeness of the editing analyses that are currently available. In the case of the Physcomitrella PPR proteins, it is relatively simple to survey all 13 sites in both organellar genomes. The same holds true for the 30-some sites in the plastid genomes of angiosperms (Schmitz-Linneweber and Barkan 2007). When it comes to the 100s of editing sites known in Arabidopsis mitochondria and the many yet-unknown sites in rice, maize, etc., a conclusive screen for defects seems ambitious. Not even the modern multiplex-based approaches have attempted to screen all editing sites in a given organism (Takenaka and Brennicke 2009). Thus, it can be expected that most of the mitochondrial editing PPR proteins will eventually be found to serve a larger number of sites than they appear to at this point. Nevertheless, it cannot be disputed that the specificity displayed by these proteins is exquisite. To understand how this is achieved, we must take a closer look at the PPR protein family.
1. The Architecture of the PPR Proteins
Members of the PPR protein family had been already studied in yeast and maize by the late 1990s (Barkan et al. 1994; Manthey and McEwen 1995; Coffin et al. 1997; Fisk et al. 1999; Ikeda and Gray 1999; Lahmy et al. 2000). Each of these studies had implicated individual proteins in the gene expression of organelles, but the family had not yet been recognized as such. The credit for identifying the existence of a large protein family whose members play potential roles in the RNA processing of plant organelles goes to Ian Small’s (UWA Perth, Australia) and Alain Lecharny’s (CNRS-INRA Evry, France) groups, which described the PPR motif and annotated the family in Arabidopsis (Aubourg et al. 2000; Small and Peeters 2000; Lurin et al. 2004). The PPR motif belongs to the widespread helical-hairpin-repeat motifs. The motif is defined as a repeat, meaning that PPR proteins always have at least two PPR motifs (Lurin et al. 2004). Repeats are predominantly found in tandem, and it is unclear whether isolated motifs are actually functional. Both the structure of the repeat and the overall structure of the PPR tract (i.e., the entirety of all repeats) have been modeled based on the known crystal structures of the closely related tetratricopeptide repeat (TPR) proteins (Small and Peeters 2000; Delannoy et al. 2007). These studies suggest that each PPR repeat encodes two alpha-helical elements, termed A and B, which fold back onto each other and also interact with the helical elements of the two adjacent repeats. Thus, the tandem repeats are stacked on top of one another to form an oblong superstructure. The A helices form the front of this structure, while the B helices form the backside. The surface produced by the A helix displays a curious aggregation of charged and hydrophilic amino acids that are believed to make contacts with RNA. Unfortunately, we do not yet have either a detailed point-mutant-based analysis of PPR tracts or a crystal structure to support these models. In any case, it is clear that PPR proteins are major players in all aspects of chloroplast RNA metabolism. A wealth of genetic data on PPR proteins almost uniformly suggests that they play direct roles in the RNA metabolism of organelles, including functions in RNA splicing, cleavage, stabilization, translation and editing (Schmitz-Linneweber and Small 2008). Importantly, both in vitro and in vivo studies have suggested that there is a direct interaction between PPR proteins and RNA (Tsuchiya et al. 2002; Nakamura et al. 2003; Lurin et al. 2004; Schmitz-Linneweber et al. 2005a, 2006; Okuda et al. 2006; Gillman et al. 2007; Kobayashi et al. 2007; Beick et al. 2008; Kazama et al. 2008; Williams-Carrier et al. 2008; Tang et al. 2010).
2. The Editing PPR Proteins Belong to the PLS Subgroup
The PPR family has been subclassified into two major groups: the pure (or P-type) PPR proteins, which contain only repeat units of 35 amino acids in length; and the PLS PPR proteins, which have repeats of varying lengths (P = normal; L = long repeats; S = short repeats, Lurin et al. 2004). The normal, long and short domains typically follow each other in triplicates, leading to the name: P-L-S. The P-type PPR proteins generally do not contain any other known protein domains, and members of this group have been associated with RNA stabilization, translation and splicing (e.g., Barkan et al. 1994; Schmitz-Linneweber et al. 2006; Pfalz et al. 2009; Prikryl et al. 2010). Intriguingly, all but one of the PPR proteins that have been implicated in RNA editing belong to the PLS subgroup; in Arabidopsis, this subgroup contains slightly less than half of the annotated PPR proteins (Lurin et al. 2004). The PLS subgroup has been further subdivided based on the presence of C-terminal extensions of unknown function (Lurin et al. 2004), and almost all PLS PPR proteins contain a so-called E-domain of ∼90–120 amino acids. Eighty-seven of the 450 PPR proteins in Arabidopsis contain the DYW domain, which was named after three highly conserved C-terminal amino acid residues, and spans roughly 100 amino acids. The majority of the editing PPR proteins in Arabidopsis (22 of 33) have DYW domains (Table 13.1).
The only non-PLS type PPR that appears to be involved in RNA editing is the Arabidopsis PPR596 protein (Doniwa et al. 2010). PPR596 is essential when plants are germinated on soil; the phenotype can be partially rescued by a longer growth period on sugar-containing medium, but the plants still display a strong growth retardation and aberrant leaf development. Mutants of this PPR are unusual in that they show an increase in the RNA editing of a mitochondrial site that is only partially edited in wild-type plants (rps3eU1344SS). However, in the absence of conclusive data on the processing of the rps3 transcript, it is currently unclear whether the observed defect was a direct effect, or alternatively was caused by other PPR596-mediated alterations in RNA metabolism.
3. How Do PPR Proteins Recognize RNA?
Biochemical evidence suggests that PPR proteins can interact with the cis-elements upstream of RNA-editing sites, but the details of this interaction are not yet known. In the last few years, a handful of RNA-editing factors have been shown to serve more than one target site, allowing researchers to determine consensus sequences for site recognition (Hammani et al. 2009). A simple consensus of base identities was found to be insufficient to explain the observed protein specificity, but the combination of several characteristics of RNA bases into a consensus model allowed the experimentally determined editing sites to be identified with high specificity (Hammani et al. 2009). The employed characteristics were: purine versus pyrimidine bases; and double versus triple hydrogen bond-forming bases. For example, the base identity consensus of the three sites served by OTP84 (psbZ, ndhB, ndhF) is: U – – – – – – U A – U – – – – C (the hyphens stand for ambiguous bases). This consensus contains little information, and in fact corresponds to 444 sites in the chloroplast genome of Arabidopsis. The improved consensus reads UWRYWWYUAYUWYRYC (W = A or U; Y = C or U; R = A or G) and is found only four times in the genome. Of the four occurrences, one is not in a transcribed region (Hammani et al. 2009) and the other three correspond to the known target sites for OTP84. This suggests that PPR proteins recognize editing-site cis-elements by distinguishing bases by their purine/pyrimidine natures and/or Watson–Crick characteristics rather than uniquely distinguishing among the four bases. This model holds true for most of the editing factors analyzed to date, suggesting that the same protein surface recognizes multiple targets (Hammani et al. 2009). A detailed structural characterization of the binding surface of PPR proteins should be a goal for the near future.
4. How Do PPR Proteins Help Edit Organellar RNAs?
The mechanism behind base conversion is still a matter of debate, as is the role of PPR proteins in RNA editing. Based on the factors isolated to date, it seems clear that the PLS PPR proteins act as editing-specificity factors. These proteins emerged in land plants and have not been found in green algae or any non-green organism (which generally have much lower PPR gene counts); this distribution parallels the presence of RNA editing, which also has not been found in green algae (Lurin et al. 2004). Importantly, the DYW domain is restricted to land plants and has been shown to correlate with taxa that exhibit RNA editing (Salone et al. 2007). The green algae, from which the embryophytes arose, do not show organellar RNA editing and do not have DYW PPR proteins. In addition, the marchantiid liverworts that secondarily lost their RNA editing also lack DYW proteins, whereas the Jungermanniid liverworts, close relatives that show extensive organellar RNA editing, possess proteins with DYW domains (Salone et al. 2007; Rüdinger et al. 2008).
The DYW domain has some interesting similarities to the cytidine deaminases from various eukaryotic organisms (Salone et al. 2007). In humans, these deaminases are involved in zinc-dependent RNA editing (Navaratnam and Sarwar 2006). The highest similarity to the DYW-domain was observed for the zinc-binding domain of these deaminases. This includes the histidines and cysteines required to form the complex with zinc, which are found in the HxExnCxxC motif of the DYW domain (Salone et al. 2007). To date, efforts to show that recombinant DYW domains are involved with RNA editing in vitro have been unsuccessful (Nakamura and Sugita 2008; Okuda et al. 2009b). By contrast, all four recombinant DYW domains tested so far were found to be capable of degrading RNA in vitro with different efficiencies (Nakamura and Sugita 2008; Okuda et al. 2009b). One of the four DYW-PPR proteins tested was CRR2, which may be involved in intercistronic cleavage, but for which no RNA-editing function has been genetically assigned (Hashimoto et al. 2003). In fact, not all DYW-PPR proteins are necessarily editing factors. For example, a reverse genetic screen for editing defects in null mutants of DYW-PPRs found editing defects in only 5 of 9 plastid mutants, and only 2 of 25 mitochondrial mutants (Hammani et al. 2009; Takenaka et al. 2010).
It is not yet clear how RNA cleavage by the DYW domains fits into the catalysis of C-to-U conversion. The phosphate backbone of RNA has been shown to remain intact during RNA editing (Rajasekhar and Mulligan 1993). Furthermore, if the backbone were cleaved near editing sites we should be able to recover defined degradation products of edited transcripts, and these have not been found to date. It is also possible that the observed cleavage activity is just a misleading side effect that unfolds only under reaction-tube conditions in experiments using naked RNA. Future work will be required to examine these and other questions.
Thus, the in vitro data on the role of the DYW domain in RNA editing are inconclusive at this point. In addition, results from in vivo studies are rather confusing. A T-DNA insertion in the DYW domain of the mitochondrial MEF11 protein obliterated RNA editing at two sites, but a third site was still partially edited in this mutant, whereas no editing was seen for the MEF11 null allele (Verbitskiy et al. 2010). In MEF1-deficient protoplasts or plants, partial restoration of editing events was seen following transient or stable complementation with a MEF1 mutant lacking the DYW domain (Zehrmann et al. 2010). In plastid DYW-PPR editing mutants for CRR22, CRR28 and OTP83, however, complete restoration was achieved following complementation with PPR genes lacking the DYW domain (Okuda et al. 2009a, b). These findings seem to indicate that the DYW domain is not necessary for catalytic editing activity. When the DYW domains of CRR22 and CRR28 were replaced with their counterpart from the non-editing PPR protein CRR2, no complementation of null mutants occurred. However, RNA editing was still supported by proteins in which the DYW domains from CRR22 and CRR28 were swapped (Okuda et al. 2009b). Conversely, when the DYW domains from CRR22 and CRR28 were used to replace the DYW domain of CRR2, the latter failed to show RNA cleavage, indicating that the DYW of CRR2 appears to be essential for protein function (i.e., RNA cleavage; Okuda et al. 2009b). This suggests that there are two types of DYW domains: DYW type 1 is found in CRR2 and is required for RNA cleavage but cannot function in RNA editing, whereas DYW type 2 is found in the editing PPR proteins and neither inhibits nor is required for RNA-editing activity. Thus, although the phylogenetic distribution of DYW editing sites suggests that they may be required for editing, the initial genetic experiments indicate otherwise. However, before we try to form a model that explains these contradicting phylogenetic and genetic data, we will briefly discuss the E-domain.
All PPR protein editing factors isolated to date have E-domains, and 10 PPR editing factors have an E-domain but not a DYW-domain (Table 13.1). Similar to the DYW domain, the E-domain is highly conserved within and between plant species, but it does not bear homologies to any known protein domain. Loss of the E-domains from CRR22, CRR28 and CRR4 abolished RNA editing at their cognate sites (Okuda et al. 2007, 2009b) but did not affect their binding to RNA (Okuda et al. 2007). The experimental addition of a stop codon right at the border between the PPR tract and the E-domain blocked the editing activity of MEF9 (Takenaka 2010), but swapping the E-domains of CRR4 and CRR21 did not interfere with RNA editing (Okuda et al. 2007). Together, these data show that the E-domain is essential for the RNA-editing activity of the editing PPR proteins.
Based on this, it is at present still difficult to incorporate these findings on the C-terminal extensions of PLS-PPR proteins into a unifying model. One possibility would be that both an E-domain and a type 2 DYW-domain are required for editing, but that the latter can also be added in trans (Okuda et al. 2009b; Fig. 13.3). Or perhaps two or more PPR proteins can act together to process RNA-editing sites (Fig. 13.3). Ad extremo, any DYW type 2 PPR could possibly complement any E-domain PPR protein. This would be a convenient arrangement, as it would provide the chloroplast with a high cumulative concentration of (possibly catalytically active) DYW domains at any given time, while the concentration of individual PPR proteins could remain low (i.e., just sufficient for RNA detection). This would dispense with the need to express and regulate an additional deaminase enzyme, and the E-domain could function as a protein-interaction domain for the recruitment of DYW-containing PPR proteins. If this model is correct, various DYW-PPR proteins should co-purify with any given E-domain-containing PPR editing factor used as bait.

Models for the organellar editosome. All models are based on the well supported assumption that the PPR tract of PPR proteins contacts the core cis-element in front of editing sites (bold line). (a) PPR-DYW proteins could be solely responsible for RNA editing of their target sites if the proposition holds that the DYW domain has cytidine deaminase activity. (b) For E-type PPR proteins without a DYW domain, other DYW PPR proteins could provide the catalytic DYW domain in trans. (c) PPR proteins could be only required for site recognition, but not directly for catalysis. Such an activity would be provided by a hitherto unknown cytidine deaminase (CD) that would be recruited by the E:DYW domains or by E-domains alone. (d) For cis-elements part of RNA secondary structures, additional factors could be required that make the RNA accessible for PPR protein entry and thus subsequent catalysis. Such non-essential factors could be cpRNP proteins with their suggested RNA chaperone activity.
This and other related hypotheses will likely be tested in the near future, as several researchers are seeking to identify factors that interact with PPR proteins. At present, however, the model is still highly speculative. In particular, there is currently no evidence for the direct interaction of PPR proteins with each other aside of the finding that the PPR protein HCF152 might form homodimers in vitro (Nakamura et al. 2003). Genetically, it has been shown that two PPR proteins can be required for the editing of one site (Robbins et al. 2009; Yu et al. 2009): The loss of either AtECB2 or RARE1 abrogates the editing of a specific site in the plastid accD mRNA. Given that both of these PPR proteins contain a DYW motif, it will be interesting to see how AtECB2 and RARE1 share non-redundant responsibilities in processing the accD site. In this regard, it is also interesting that there are mutants of DYW-PPR proteins that support RNA editing at specific sites, but are not essential for it. This includes MEF1, which is essential for two sites but only supportive for the editing of a nad2 site (Zehrmann et al. 2009); REME1, which supports another nad2 site; and OGR1, which is essential for a number of sites but contributes only slightly to the editing of nad4eU433LF. Future work will be required to assess how these PPR proteins contribute together with putative partner PPRs and possibly other factors to achieve high editing levels of their cognate targets. It will be particularly interesting to understand whether these PPR proteins directly associate with the RNA and thus contribute to recognition, or whether their DYW domains are simply recruited for catalysis by protein-protein interactions.
B. The Enigmatic Catalytic Activity
Both the sugar-phosphate backbone and the nucleotide base remain intact during RNA editing, indicating that catalysis does not involve nucleotide excision or base exchange (Rajasekhar and Mulligan 1993; Yu and Schuster 1995). Instead, the experimental evidence collected in the years following the discovery of plant organellar RNA editing unequivocally indicated that C-to-U RNA editing proceeds by base deamination (Araya et al. 1992; Rajasekhar and Mulligan 1993; Yu and Schuster 1995). Transamination would be an alternative scenario, but the standard amino-group acceptors and a candidate enzyme tested for trans-amination did not seem to be involved in vitro for C-to-U editing (Takenaka et al. 2007). One long-held theory is that cytidine deaminases carry out the reaction, in a manner analogous to that seen for human C-to-U editing (Navaratnam and Sarwar 2006). However, the first cytidine deaminase identified in Arabidopsis was not found to associate with RNA (Faivre-Nitschke et al. 1999), and another candidate deaminase protein turned out to be required for A-to-I editing of plastid tRNA-R(ACG), but not for C-to-U RNA editing (Delannoy et al. 2009; Karcher and Bock 2009). The classical cytidine deaminases utilize zinc as a co-factor during catalysis, but in vitro experiments in which zinc was chelated from editing reactions delivered mixed results: Although zinc depletion did not affect mitochondrial RNA editing in vitro (Takenaka et al. 2007), detrimental effects were observed in comparable plastid systems (Hegeman et al. 2005). Several predicted organellar cytidine deaminases remain to be tested for functions in RNA editing, but we may find that the true activity has evolved from a very different background, such as from RNA modifying enzymes that act on rRNAs or tRNAs, or from enzymes involved in single-stranded DNA metabolism and repair. Importantly, it remains possible that the DYW domain may have editing activity. Certainly, the future identification of this editing activity will be a most exciting and important task.
C. Other Factors Involved in RNA Editing
Aside from the PPR proteins and the enigmatic editase discussed above, the list of additional RNA-editing factors is fairly short. Most of the factors implicated in RNA editing have been determined biochemically, such as by the cross-linking of proteins to editing sites. Among the proteins identified in this manner is a 91-kD protein associated with the rpoBeU113SF editing site in tobacco (Kobayashi et al. 2008). Most likely, this protein corresponds to the recently identified YS1 PPR protein responsible for editing this site in Arabidopsis (Zhou et al. 2009). Similarly, the 95-kD protein that cross-linked to ndhBeU494PL and ndhFeU21SL in tobacco (Kobayashi et al. 2008) could turn out to be homologous to OTP84, an Arabidopsis PPR serving these exact same sites (Hammani et al. 2009). A 25-kD factor associated with tobacco psbL will not have a similarly corresponding factor in Arabidopsis, which lacks this site (Hirose and Sugiura 2001). Two other tobacco proteins (56-kD and 70-kD) that cross-linked to sites in the petB and psbE mRNAs also remain unidentified at this time (Miyamoto et al. 2002, 2004).
A set of proteins consistently identified in cross-linking experiments are the chloroplast ribonucleoproteins or short cpRNPs, which are highly abundant RNA-binding proteins found in the chloroplasts of angiosperms (Tillich et al. 2010). These proteins are related to the nucleo-cytosolic RNA-recognition motif (RRM)-containing proteins, which play roles in RNA processing and can act as RNA chaperones (Maruyama et al. 1999). They were initially believed to be mostly required for protecting RNAs against degradation (Nakamura et al. 2001). However, a specific role in RNA editing was shown for at least one of their members in vitro (Hirose and Sugiura 2001): Extracts that had been immuno-depleted of the tobacco cpRNP, CP31, were found to be incapable of processing two editing sites in the ndhB and psbL mRNAs. Other tested cpRNPs were not required for this job; instead a domain of CP31 rich in acidic amino acid residues was found to be essential for this editing activity (Hirose and Sugiura 2001). Recently, knockout mutants of CP31A and CP31B, two Arabidopsis paralogs of tobacco CP31, were tested for RNA-editing defects (Tillich et al. 2009b). Multiple editing sites exhibited decreased editing efficiencies in the CP31A mutant, whereas the defects in the CP31B mutants were comparatively minor. This may reflect the effects of an extended acidic domain (similar to that found in tobacco CP31), which is present in CP31A but not CP31B. The strongest defects were found in CP31A/B double mutants, but even these mutants did not show a complete loss of RNA editing. It is not yet clear why CP31 is essential for tobacco editing sites in vitro, but the Arabidopsis orthologs seem to be just auxiliary in vivo. It is also not yet known how cpRNPs impact RNA editing in such a specific manner. It seems possible that they could be required to prepare the RNA for PPR protein access. PPR proteins have been shown to prefer single-stranded over double-stranded RNA (Tsuchiya et al. 2002; Nakamura et al. 2003; Williams-Carrier et al. 2008), so the cpRNPs could perhaps act as chaperones by helping dissolve double-stranded elements that obscure PPR binding sites. Indeed, the nucleo-cytosolic RRM proteins display such activity (Dreyfuss et al. 2002). Alternatively, the acidic domain could be part of a platform for recruiting PPR proteins and/or the editase in a manner analogous to the use of such domains for protein-protein interactions by nuclear-splicing factors (Valcarcel and Green 1996).
Finally, there appears to be an overlap in the editing-site target ranges of the PPR proteins and CP31A. This includes, for example, sites in the ndhB and rpoB messages, which are served by both CRR22 and CP31A. In the future, it would be instructive to analyze how these two proteins act together to achieve base deamination. Protein interaction studies and detailed analyses of the structural changes induced in the RNA targets by both proteins will likely help us understand this issue.
V. The Why Behind RNA Editing
Science is wonderfully equipped to answer the question ‘How?’ but it gets terribly confused when you ask the question ‘Why?’ (Chargaff 1977)
The seeming futility of the RNA-editing process has puzzled researches since the early detection of RNA editing. Why aren’t editing sites removed by C-to-T point mutations in the organellar genome, thereby avoiding the need for elaborate RNA processing? Recent reviews have addressed this salient point (Maier et al. 2008; Zehrmann et al. 2008; Tillich et al. 2010), so we will therefore only briefly summarize the current explanations herein.
Two major competing models attempt to rationalize the existence of organellar RNA editing. The first one draws on knowledge gained from other editing systems, particularly those in humans, where base transitions are used to generate and regulate protein diversity. To give a famous example, C-to-U editing of the apoB mRNA distinguishes the two protein isoforms of a lipoprotein that is important for lipid transport in the bloodstream (for a recent review see Blanc and Davidson 2010). The two isoforms are differentially expressed; editing occurs only in epithelia of the small intestine, whereas the unedited mRNA gives rise to an isoform that is expressed in the liver. Importantly, the two isoforms are functionally distinct. Other well-studied cases of regulated RNA editing are found in the generation of human neuroreceptor isoforms by RNA editing (Gott and Emeson 2000; Bass 2001, 2002; Valente and Nishikura 2005). The isoforms of such receptors (e.g., those for glutamate or serotonin) result from differential A-to-I editing at multiple sites, and have different receptor kinetics and permeabilities compared to the unedited versions. Obviously, the generation of protein diversity would be an attractive explanation for the persistence of organellar RNA editing in plants. However, almost all plastid-editing sites and the majority of mitochondrial sites are fully edited. We know relatively little regarding the tissue- or condition-specific modulations of editing events for the few sites that show only partial editing, and even if such variation were found, its physiological relevance remains dubious (Grosskopf and Mulligan 1996; Karcher and Bock 1998, 2002a, b; Nakajima and Mulligan 2001).
A number of reports have speculated on the regulatory role of specific editing events. For example, editing of the rpoB and rpoA mRNAs has been proposed to impact the activity of the encoded RNA polymerase (Hirose et al. 1999; Zhou et al. 2009). In theory, this could impact chlorophyll production by altering the expression of tRNA-Glu (Zhou et al. 2009), which is required for the first step in chlorophyll synthesis. Detailed correlational studies comparing chlorophyll production, the expression of editing factors serving rpo mRNAs, and polymerase activity will be required to assess this hypothesis.
Very little is known about the presence of protein isoforms resulting from partially edited sites. In tobacco plastids, monocistronic ndhD mRNA was found to associate with polysomal fractions despite having an unedited start codon (i.e. remaining ACG instead of AUG; Zandueta-Criado and Bock 2004). The maize ribosomal S12 protein is present in mitochondria in at least two isoforms generated by incomplete RNA editing, but only the edited isoform is incorporated in mature ribosomes (Phreaner et al. 1996). Confusingly, the orthologous protein in petunia is found in cell fractions enriched for ribosomes (Phreaner et al. 1996). In contrast to the situation for S12, no protein corresponding to the unedited messages of the mitochondrial ribosomal S13 protein was detected even though there was a high frequency of unedited cDNAs (Williams et al. 1998). Similarly, sequencing of portions of the mitochondrial NAD9 protein failed to identify any sequences derived from unedited mRNAs (Grohmann et al. 1994). Finally, an “unedited” protein version of ATP9 has been directly tested for functionality in studies in which it was expressed from the nucleus with a mitochondrial import address fused to the open reading frame. Notably, the imported and unedited ATP9 was found to interfere with normal mitochondrial function, as manifested by male sterility (Hernould et al. 1993; Zabaleta et al. 1996).
In the future, these somewhat contradictory findings should be examined further by proteomic studies of organelles, including searches of mass spectrometric data with unedited versions of the organellar genomes. We cannot yet verify that a shift in the balance between “unedited” and “edited” proteins has any physiological role. Furthermore, it is not yet clear whether editing events that affect start or stop codons can affect the translation of mRNAs, thereby contributing to the regulation of protein production. This leaves very little to substantiate the hypothesis that plant organellar RNA editing plays a general regulatory role. We suspect that although individual editing events may be exploited to regulate gene expression, this will not be the case for the vast majority of editing events.
The second (more recent) hypothesis accounting for the existence of RNA editing, which draws on our understanding of plant organelles as having descended from endosymbiotic bacteria, is called the “genome debugging hypothesis” (Maier et al. 2008). Obligate endosymbionts are prone to accumulating deleterious point mutations in a phenomenon called Müller’s ratchet, and there is no reason to think that chloroplasts and mitochondria (i.e., direct descendants of endosymbiotic bacteria) would not also face this problem. Unlike more recent endosymbiotic descendants (e.g., the endosymbiotic gut bacteria of insects), however, plant organelles can draw on the nuclear genome, which is a source of genetic information that evolves rapidly and recombines sexually. Nuclear factors can be imported into chloroplasts and mitochondria to mitigate problems arising from fixed point mutations. Conceptually, such factors could be involved on all levels during the realization of organelle genetic information. A striking example of the nuclear-based repair of organellar mutations comes from plant breeding. Plant breeders have long taken advantage of cytoplasmic male sterility (CMS), a phenomenon caused by mitochondrial mutations that arise in various plant species (reviewed in Chase 2007). Such mutations are of agronomical interest because they avoid the need for the labor-intensive emasculation of plants to prevent selfing, but these mutations must be suppressed to allow the later mass production of seeds in the field. Strikingly, suppressor mutations have been isolated that map to the nuclear genome and nearly all of them affect RNA binding proteins that belong to the PPR protein family (reviewed in Schmitz-Linneweber and Small 2008). These PPR proteins “repair” the CMS-specific mutational problems in the mitochondrial genome. The repair is not carried out on the DNA level, but rather works on the RNA that are derived from the defective genetic information. Specifically, the PPR proteins either help degrade unwanted, aberrant mRNAs that would otherwise give rise to toxic proteins (Wang et al. 2006), or prevent the translation of such RNAs (Uyttewaal et al. 2008). As PPR proteins are also major players in RNA editing, it could be speculated that they evolved to suppress deleterious U-to-C point mutations arising in plant organelles. It should be noted that such rescue of organellar mutations by nuclear factors makes sense given that the plant organellar genomes evolve more slowly than the nuclear genome. In metazoans, in contrast, mitochondrial genomes evolve much more rapidly than the nuclear genomes; thus, back mutations are a much more likely and rapid response to mutational problems than the evolution of nuclear-encoded antidotes (Maier et al. 2008). In fact, recent studies found an inverse correlation between the editing frequencies and overall substitution rates of mitochondrial genomes, suggesting that a slowly evolving genome tends not to jettison its RNA-editing sites at the DNA level (Parkinson et al. 2005; Cuenca et al. 2010). An important prediction of the genome debugging hypothesis is that the removal of an editing site (i.e., the repair of the site on the genomic level) would not interfere with plant viability. Consistent with this hypothesis, when an edited C in the plastid atpA mRNA was turned into a T, rendering RNA editing obsolete at this site, there was no detrimental effect on the resulting plants grown under standard conditions (Schmitz-Linneweber et al. 2005b). Also, editing sites evolve rapidly (Shields and Wolfe 1997) and loss of a site in one lineage, while the same site is maintained in a closely related sister group seems to be tolerated (Hayes and Hanson 2008; Tillich et al. 2009a). In the future, by constructing an editing-site-free organellar genome and substituting it for the wild-type genome, researchers should be able to examine whether editing sites are used to regulate gene expression or have some other function, or whether RNA editing is truly an unnecessary freak of evolution. Notably, the results from studies on the evolutionary behavior of editing sites across large time scales (from the beginning of land plant evolution or encompassing at least angiosperm evolution) have suggested that RNA-editing sites tend to be lost over time at least in plastids (Tillich et al. 2006, 2009a; Hayes and Hanson 2008). Perhaps, we are studying a process on the brink of extinction.
VI. Perspectives
Unlike other editing systems, such as the C-to-U and A-to-I editing in humans or the rampant and excessive RNA editing in trypanosome mitochondria, relatively little is known about RNA editing in plant mitochondria. Whereas we know details on the machinery and catalysis of RNA editing in humans, this information is lacking in plant organelles. At present, there are four major questions in the field. First, we need to elucidate which factors carry the catalytic activity for base deamination and what makes up the editosome (if there is one). Second, we should examine how PPR proteins recognize the cis-elements in front of editing sites (i.e., how is site-specificity generated?). The answers to these two mechanistic questions should be obtainable within the next few years, as techniques to determine structures of PPR proteins associated with their target RNAs are at hand along with proteomic methods for detailed characterization of the editosome using PPR proteins as bait. The second pair of questions addresses the still elusive function of RNA editing, and will be much harder to answer. First, are there editing events that distinguish two functional proteins from each other and, on a grander scale, is any RNA-editing event rate-limiting for the production of the correct protein? Second, does the lack of specific RNA editing in many PPR protein mutants truly not have an effect on the corresponding proteins, as suggested by the absence of any macroscopic phenotype? Considerable experimental efforts will be required to address these questions. Cryptic phenotypic alterations could be unveiled by applying various stresses to the editing mutants. Ideally (although rarely done due to the immense space and time requirements), competition experiments between mutant and wild-type plants could be used to determine possible fitness deficits under field conditions. In terms of assessing regulation, recent studies on RNA processing factors in Chlamydomonas could light the way. In this case, hypomorphic mutant series with ever-decreasing amounts of PPR proteins showed clear correlations between the amount of PPR proteins and the amount of proteins generated from the PPR-target message, providing a clear sign that the PPR proteins are true regulators of gene expression (Raynaud et al. 2007). In this example, the PPR protein was required to stabilize the target RNA; however, similar approaches could also be applied to PPR proteins as editing factors. Ultimately, it would be a dream to harness the RNA-editing machinery and use it to switch proteins on or off at will in plant organelles; this would be particularly useful in plastids, which are important sites for biotechnological expression of transgenes (Bock 2007). Possibly, RNA-editing factors could also be used to manipulate RNAs in vitro or perhaps even to fight detrimental RNAs and/or viral RNAs in humans. In any case, and even without daydreaming about possible applications, plant organellar RNA editing – with its curious origins, uncertain functions and enigmatic machinery – remains a formidable and exciting challenge for future research.
Abbreviations
- 3D –:
-
Three-dimensional;
- CMS –:
-
Cytoplasmic male sterility;
- cpRNPs –:
-
Chloroplast ribonucleoproteins;
- CRR –:
-
Chloroplast respiratory reduction;
- CURE –:
-
Cytidine-to-uridine recognizing editor;
- EMS –:
-
Ethyl methane sulfonate;
- GOBASE–:
-
The organelle genome database;
- MEF –:
-
Mitochondrial editing factor;
- NDH –:
-
NAD(P)H dehydrogenase;
- OGR1 –:
-
Opaque and growth retardation 1;
- PPR –:
-
Pentatricopeptide repeat;
- PREP –:
-
Predictive RNA editors for plants;
- PREPACT –:
-
Plant RNA editing prediction and analysis computer tool;
- REGAL –:
-
RNA Editing site prediction by Genetic Algorithm Learning;
- RESOPS –:
-
RNA editing sites of land plant organelles on protein three-dimensional structures;
- RRM –:
-
RNA-recognition motif;
- TPR –:
-
Tetratricopeptide repeat;
- WT –:
-
Wild type
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Araya A, Domec C, Begu D, Litvak S (1992) An in vitro system for the editing of ATP synthase subunit 9 mRNA using wheat mitochondrial extracts. Proc Natl Acad Sci USA 89:1040–1044
Aubourg S, Boudet N, Kreis M, Lecharny A (2000) In Arabidopsis thaliana, 1% of the genome codes for a novel protein family unique to plants. Plant Mol Biol 42:603–613
Barkan A, Walker M, Nolasco M, Johnson D (1994) A nuclear mutation in maize blocks the processing and translation of several chloroplast mRNAs and provides evidence for the differential translation of alternative mRNA forms. EMBO J 13:3170–3181
Bass BL (2001) RNA editing. Oxford University Press, Oxford
Bass BL (2002) RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 71:817–846
Beick S, Schmitz-Linneweber C, Williams-Carrier R, Jensen B, Barkan A (2008) The pentatricopeptide repeat protein PPR5 stabilizes a specific tRNA precursor in maize chloroplasts. Mol Cell Biol 28:5337–5347
Benne R, Van den Burg J, Brakenhoff JP, Sloof P, Van Boom JH, Tromp MC (1986) Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell 46:819–826
Bentolila S, Elliott LE, Hanson MR (2008) Genetic architecture of mitochondrial editing in Arabidopsis thaliana. Genetics 178:1693–1708
Bentolila S, Knight W, Hanson M (2010) Natural variation in Arabidopsis leads to the identification of REME1, a pentatricopeptide repeat-DYW protein controlling the editing of mitochondrial transcripts. Plant Physiol 154:1966–1982
Blanc V, Davidson NO (2010) APOBEC-1-mediated RNA editing. Wiley Interdiscip Rev Syst Biol Med 2:594–602
Bock R (2000) Sense from nonsense: how the genetic information of chloroplasts is altered by RNA editing. Biochimie 82:549–557
Bock R (2007) Plastid biotechnology: prospects for herbicide and insect resistance, metabolic engineering and molecular farming. Curr Opin Biotechnol 18:100–106
Bock R, Kössel H, Maliga P (1994) Introduction of a heterologous editing site into the tobacco plastid genome: the lack of RNA editing leads to a mutant phenotype. EMBO J 13:4623–4628
Bock R, Hermann M, Kössel H (1996) In vivo dissection of cis-acting determinants for plastid RNA editing. EMBO J 15:5052–5059
Bolle N, Kempken F (2006) Mono- and dicotyledonous plant-specific RNA editing sites are correctly edited in both in organello systems. FEBS Lett 580:4443–4448
Cai W, Ji D, Peng L, Guo J, Ma J, Zou M, Lu C, Zhang L (2009) LPA66 is required for editing psbF chloroplast transcripts in Arabidopsis. Plant Physiol 150:1260–1271
Chargaff E (1977) Voices in the Labyrinth: nature, man and science. Seabury Press, New York
Chase CD (2007) Cytoplasmic male sterility: a window to the world of plant mitochondrial-nuclear interactions. Trends Genet 23:81–90
Chateigner-Boutin AL, Hanson MR (2002) Cross-competition in transgenic chloroplasts expressing single editing sites reveals shared cis elements. Mol Cell Biol 22:8448–8456
Chateigner-Boutin AL, Hanson MR (2003) Developmental co-variation of RNA editing extent of plastid editing sites exhibiting similar cis-elements. Nucleic Acids Res 31:2586–2594
Chateigner-Boutin AL, Ramos-Vega M, Guevara-Garcia A, Andres C, de la Luz Gutierrez-Nava M, Cantero A, Delannoy E, Jimenez LF, Lurin C, Small I, Leon P (2008) CLB19, a pentatricopeptide repeat protein required for editing of rpoA and clpP chloroplast transcripts. Plant J 56:590–602
Chaudhuri S, Maliga P (1996) Sequences directing C to U editing of the plastid psbL mRNA are located within a 22 nucleotide segment spanning the editing site. EMBO J 15:5958–5964
Chaudhuri S, Carrer H, Maliga P (1995) Site-specific factor involved in the editing of the psbL mRNA in tobacco plastids. EMBO J 14:2951–2957
Choury D, Farre JC, Jordana X, Araya A (2004) Different patterns in the recognition of editing sites in plant mitochondria. Nucleic Acids Res 32:6397–6406
Coffin JW, Dhillon R, Ritzel RG, Nargang FE (1997) The Neurospora crassa cya-5 nuclear gene encodes a protein with a region of homology to the Sacharomyces cerevisiae PET309 protein and is required in a post-transcriptional step for the expression of the mitochondrially encoded COXI protein. Curr Genet 32:273–280
Covello PS, Gray MW (1989) RNA editing in plant mitochondria. Nature 341:662–666
Cuenca A, Petersen G, Seberg O, Davis JI, Stevenson DW (2010) Are substitution rates and RNA editing correlated? BMC Evol Biol 10:349
Cui L, Veeraraghavan N, Richter A, Wall K, Jansen RK, Leebens-Mack J, Makalowska I, dePamphilis CW (2006) ChloroplastDB: the chloroplast genome database. Nucleic Acids Res 34:D692–D696
Cummings MP, Myers DS (2004) Simple statistical models predict C-to-U edited sites in plant mitochondrial RNA. BMC Bioinformatics 5:132
Delannoy E, Stanley WA, Bond CS, Small ID (2007) Pentatricopeptide repeat (PPR) proteins as sequence-specificity factors in post-transcriptional processes in organelles. Biochem Soc Trans 35:1643–1647
Delannoy E, Le Ret M, Faivre-Nitschke E, Estavillo GM, Bergdoll M, Taylor NL, Pogson BJ, Small I, Imbault P, Gualberto JM (2009) Arabidopsis tRNA adenosine deaminase arginine edits the wobble nucleotide of chloroplast tRNAArg (ACG) and is essential for efficient chloroplast translation. Plant Cell 21:2058–2071
Doniwa Y, Ueda M, Ueta M, Wada A, Kadowaki K, Tsutsumi N (2010) The involvement of a PPR protein of the P subfamily in partial RNA editing of an Arabidopsis mitochondrial transcript. Gene 454:39–46
Dreyfuss G, Kim VN, Kataoka N (2002) Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol 3:195–205
Du P, Li Y (2008) Prediction of C-to-U RNA editing sites in plant mitochondria using both biochemical and evolutionary information. J Theor Biol 253:579–586
Du P, Jia L, Li Y (2009) CURE-Chloroplast: a chloroplast C-to-U RNA editing predictor for seed plants. BMC Bioinformatics 10:135
Duff RJ, Moore FB (2005) Pervasive RNA editing among hornwort rbcL transcripts except Leiosporoceros. J Mol Evol 61:571–578
Faivre-Nitschke SE, Grienenberger JM, Gualberto JM (1999) A prokaryotic-type cytidine deaminase from Arabidopsis thaliana gene expression and functional characterization. Eur J Biochem 263:896–903
Farre JC, Araya A (2001) Gene expression in isolated plant mitochondria: high fidelity of transcription, splicing and editing of a transgene product in electroporated organelles. Nucleic Acids Res 29:2484–2491
Farre JC, Leon G, Jordana X, Araya A (2001) cis Recognition elements in plant mitochondrion RNA editing. Mol Cell Biol 21:6731–6737
Fiebig A, Stegemann S, Bock R (2004) Rapid evolution of editing sites in a small non-essential plastid gene. Nucleic Acids Res 7:3615–3622
Fisk DG, Walker MB, Barkan A (1999) Molecular cloning of the maize gene crp1 reveals similarity between regulators of mitochondrial and chloroplast gene expression. EMBO J 18:2621–2630
Freyer R, Kiefer-Meyer MC, Kössel H (1997) Occurrence of plastid RNA editing in all major lineages of land plants. Proc Natl Acad Sci USA 94:6285–6290
Giegé P, Brennicke A (1999) RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc Natl Acad Sci USA 96:15324–15329
Gillman JD, Bentolila S, Hanson MR (2007) The petunia restorer of fertility protein is part of a large mitochondrial complex that interacts with transcripts of the CMS-associated locus. Plant J 49:217–227
Gott JM, Emeson RB (2000) Functions and mechanisms of RNA editing. Annu Rev Genet 34:499–531
Gray MW (1996) RNA editing in plant organelles: a fertile field. Proc Natl Acad Sci USA 93:8157–8159
Grohmann L, Thieck O, Herz U, Schroder W, Brennicke A (1994) Translation of nad9 mRNAs in mitochondria from Solanum tuberosum is restricted to completely edited transcripts. Nucleic Acids Res 22:3304–3311
Grosskopf D, Mulligan RM (1996) Developmental- and tissue-specificity of RNA editing in mitochondria of suspension-cultured maize cells and seedlings. Curr Genet 29:556–563
Groth-Malonek M, Pruchner D, Grewe F, Knoop V (2005) Ancestors of trans-splicing mitochondrial introns support serial sister group relationships of hornworts and mosses with vascular plants. Mol Biol Evol 22:117–125
Groth-Malonek M, Wahrmund U, Polsakiewicz M, Knoop V (2007) Evolution of a pseudogene: exclusive survival of a functional mitochondrial nad7 gene supports Haplomitrium as the earliest liverwort lineage and proposes a secondary loss of RNA editing in Marchantiidae. Mol Biol Evol 24:1068–1074
Gualberto JM, Lamattina L, Bonnard G, Weil JH, Grienenberger JM (1989) RNA editing in wheat mitochondria results in the conservation of protein sequences. Nature 341:660–662
Halter CP, Peeters NM, Hanson MR (2004) RNA editing in ribosome-less plastids of iojap maize. Curr Genet 45:331–337
Hammani K, Okuda K, Tanz SK, Chateigner-Boutin AL, Shikanai T, Small I (2009) A study of new Arabidopsis chloroplast RNA editing mutants reveals general features of editing factors and their target sites. Plant Cell 21:3686–3699
Hanson M, Sutton C, Lu B (1996) Plant organelle gene expression: altered by RNA editing. Trends Plant Sci 1:57–64
Hashimoto M, Endo T, Peltier G, Tasaka M, Shikanai T (2003) A nucleus-encoded factor, CRR2, is essential for the expression of chloroplast ndhB in Arabidopsis. Plant J 36:541–549
Hayes ML, Hanson MR (2007) Identification of a sequence motif critical for editing of a tobacco chloroplast transcript. RNA 13:281–288
Hayes ML, Hanson MR (2008) High conservation of a 5′ element required for RNA editing of a C target in chloroplast psbE transcripts. J Mol Evol 67:233–245
Hayes ML, Reed ML, Hegeman CE, Hanson MR (2006) Sequence elements critical for efficient RNA editing of a tobacco chloroplast transcript in vivo and in vitro. Nucleic Acids Res 34:3742–3754
He T, Du P, Li Y (2007) dbRES: a web-oriented database for annotated RNA editing sites. Nucleic Acids Res 35:D141–D144
Hecht J, Grewe F, Knoop V (2011b) Extreme RNA editing in coding islands and abundant microsatellites in repeat sequences of Selaginella moellendorffii mitochondria: the root of frequent plant mtDNA recombination in early tracheophytes. Genome Biol Evol 3:344–358
Hegeman CE, Hayes ML, Hanson MR (2005) Substrate and cofactor requirements for RNA editing of chloroplast transcripts in Arabidopsis in vitro. Plant J 42:124–132
Hernould M, Suharsono S, Litvak S, Araya A, Mouras A (1993) Male-sterility induction in transgenic tobacco plants with an unedited atp9 mitochondrial gene from wheat. Proc Natl Acad Sci USA 90:2370–2374
Hiesel R, Wissinger B, Schuster W, Brennicke A (1989) RNA editing in plant mitochondria. Science 246:1632–1634
Hirose T, Sugiura M (2001) Involvement of a site-specific trans-acting factor and a common RNA-binding protein in the editing of chloroplast mRNAs: development of a chloroplast in vitro RNA editing system. EMBO J 20:1144–1152
Hirose T, Kusumegi T, Tsudzuki T, Sugiura M (1999) RNA editing sites in tobacco chloroplast transcripts: editing as a possible regulator of chloroplast RNA polymerase activity. Mol Gen Genet 262:462–467
Hoch B, Maier RM, Appel K, Igloi GL, Kössel H (1991) Editing of a chloroplast mRNA by creation of an initiation codon. Nature 353:178–180
Ikeda TM, Gray MW (1999) Characterization of a DNA-binding protein implicated in transcription in wheat mitochondria. Mol Cell Biol 19:8113–8122
Karcher D, Bock R (1998) Site-selective inhibition of plastid RNA editing by heat shock and antibiotics: a role for plastid translation in RNA editing. Nucleic Acids Res 26:1185–1190
Karcher D, Bock R (2002a) The amino acid sequence of a plastid protein is developmentally regulated by RNA editing. J Biol Chem 277:5570–5574
Karcher D, Bock R (2002b) Temperature sensitivity of RNA editing and intron splicing reactions in the plastid ndhB transcript. Curr Genet 41:48–52
Karcher D, Bock R (2009) Identification of the chloroplast adenosine-to-inosine tRNA editing enzyme. RNA 15:1251–1257
Kazama T, Nakamura T, Watanabe M, Sugita M, Toriyama K (2008) Suppression mechanism of mitochondrial ORF79 accumulation by Rf1 protein in BT-type cytoplasmic male sterile rice. Plant J 55(4):619–628
Kim SR, Yang JI, Moon S, Ryu CH, An K, Kim KM, Yim J, An G (2009) Rice OGR1 encodes a pentatricopeptide repeat-DYW protein and is essential for RNA editing in mitochondria. Plant J 59:738–749
Knoop V (2004) The mitochondrial DNA of land plants: peculiarities in phylogenetic perspective. Curr Genet 46:123–139
Knoop V (2010) When you can’t trust the DNA: RNA editing changes transcript sequences. Cell Mol Life Sci 68(4):567–586
Kobayashi K, Suzuki M, Tang J, Nagata N, Ohyama K, Seki H, Kiuchi R, Kaneko Y, Nakazawa M, Matsui M, Matsumoto S, Yoshida S, Muranaka T (2007) Lovastatin insensitive 1, a Novel pentatricopeptide repeat protein, is a potential regulatory factor of isoprenoid biosynthesis in Arabidopsis. Plant Cell Physiol 48:322–331
Kobayashi Y, Matsuo M, Sakamoto K, Wakasugi T, Yamada K, Obokata J (2008) Two RNA editing sites with cis-acting elements of moderate sequence identity are recognized by an identical site-recognition protein in tobacco chloroplasts. Nucleic Acids Res 36:311–318
Kotera E, Tasaka M, Shikanai T (2005) A pentatricopeptide repeat protein is essential for RNA editing in chloroplasts. Nature 433:326–330
Kubo N, Kadowaki K (1997) Involvement of 5′ flanking sequence for specifying RNA editing sites in plant mitochondria. FEBS Lett 413:40–44
Kugita M, Yamamoto Y, Fujikawa T, Matsumoto T, Yoshinaga K (2003) RNA editing in hornwort chloroplasts makes more than half the genes functional. Nucleic Acids Res 31:2417–2423
Lahmy S, Barneche F, Derancourt J, Filipowicz W, Delseny M, Echeverria M (2000) A chloroplastic RNA-binding protein is a new member of the PPR family. FEBS Lett 480:255–260
Lenz H, Rudinger M, Volkmar U, Fischer S, Herres S, Grewe F, Knoop V (2010) Introducing the plant RNA editing prediction and analysis computer tool PREPACT and an update on RNA editing site nomenclature. Curr Genet 56:189–201
Lippok B, Brennicke A, Wissinger B (1994) Differential RNA editing in closely related introns in Oenothera mitochondria. Mol Gen Genet 243:39–46
Lurin C, Andres C, Aubourg S, Bellaoui M, Bitton F, Bruyere C, Caboche M, Debast C, Gualberto J, Hoffmann B, Lecharny A, Le Ret M, Martin-Magniette ML, Mireau H, Peeters N, Renou JP, Szurek B, Taconnat L, Small I (2004) Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 16:2089–2103
Maier RM, Hoch B, Zeltz P, Kössel H (1992a) Internal editing of the maize chloroplast ndhA transcript restores codons for conserved amino acids. Plant Cell 4:609–616
Maier RM, Neckermann K, Hoch B, Akhmedov NB, Kössel H (1992b) Identification of editing positions in the ndhB transcript from maize chloroplasts reveals sequence similarities between editing sites of chloroplasts and plant mitochondria. Nucleic Acids Res 20:6189–6194
Maier RM, Zeltz P, Kössel H, Bonnard G, Gualberto JM, Grienenberger JM (1996) RNA editing in plant mitochondria and chloroplasts. Plant Mol Biol 32:343–365
Maier U-G, Bozarth A, Funk H, Zauner S, Rensing S, Schmitz-Linneweber C, Börner T, Tillich M (2008) Complex chloroplast RNA metabolism: just debugging the genetic programme? BMC Biol 6:36
Malek O, Lättig K, Hiesel R, Brennicke A, Knoop V (1996) RNA editing in bryophytes and a molecular phylogeny of land plants. EMBO J 15:1403–1411
Manthey GM, McEwen JE (1995) The product of the nuclear gene PET309 is required for translation of mature mRNA and stability or production of intron-containing RNAs derived from the mitochondrial COX1 locus of Saccharomyces cerevisiae. EMBO J 14:4031–4043
Maruyama K, Sato N, Ohta N (1999) Conservation of structure and cold-regulation of RNA-binding proteins in cyanobacteria: probable convergent evolution with eukaryotic glycine-rich RNA-binding proteins. Nucleic Acids Res 27:2029–2036
Miyamoto T, Obokata J, Sugiura M (2002) Recognition of RNA editing sites is directed by unique proteins in chloroplasts: biochemical identification of cis-acting elements and trans-acting factors involved in RNA editing in tobacco and pea chloroplasts. Mol Cell Biol 22:6726–6734
Miyamoto T, Obokata J, Sugiura M (2004) A site-specific factor interacts directly with its cognate RNA editing site in chloroplast transcripts. Proc Natl Acad Sci USA 101:48–52
Miyata Y, Sugita M (2004) Tissue- and stage-specific RNA editing of rps 14 transcripts in moss (Physcomitrella patens) chloroplasts. J Plant Physiol 161:113–115
Miyata Y, Sugiura C, Kobayashi Y, Hagiwara M, Sugita M (2002) Chloroplast ribosomal S14 protein transcript is edited to create a translation initiation codon in the moss Physcomitrella patens. Biochim Biophys Acta 1576:346–349
Mower JP (2005) PREP-Mt: predictive RNA editor for plant mitochondrial genes. BMC Bioinformatics 6:96
Mower JP (2009) The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res 37:W253–W259
Mulligan RM, Williams MA, Shanahan MT (1999) RNA editing site recognition in higher plant mitochondria. J Hered 90:338–344
Nakajima Y, Mulligan RM (2001) Heat stress results in incomplete C-to-U editing of maize chloroplast mRNAs and correlates with changes in chloroplast transcription rate. Curr Genet 40:209–213
Nakajima Y, Mulligan RM (2005) Nucleotide specificity of the RNA editing reaction in pea chloroplasts. J Plant Physiol 162:1347–1354
Nakamura T, Sugita M (2008) A conserved DYW domain of the pentatricopeptide repeat protein possesses a novel endoribonuclease activity. FEBS Lett 582:4163–4168
Nakamura T, Ohta M, Sugiura M, Sugita M (2001) Chloroplast ribonucleoproteins function as a stabilizing factor of ribosome-free mRNAs in the stroma. J Biol Chem 276:147–152
Nakamura T, Meierhoff K, Westhoff P, Schuster G (2003) RNA-binding properties of HCF152, an Arabidopsis PPR protein involved in the processing of chloroplast RNA. Eur J Biochem 270:4070–4081
Navaratnam N, Sarwar R (2006) An overview of cytidine deaminases. Int J Hematol 83:195–200
Neuwirt J, Takenaka M, van der Merwe JA, Brennicke A (2005) An in vitro RNA editing system from cauliflower mitochondria: editing site recognition parameters can vary in different plant species. RNA 11:1563–1570
O’Brien EA, Zhang Y, Wang E, Marie V, Badejoko W, Lang BF, Burger G (2009) GOBASE: an organelle genome database. Nucleic Acids Res 37:D946–D950
Ohtani S, Ichinose M, Tasaki E, Aoki Y, Komura Y, Sugita M (2010) Targeted gene disruption identifies three PPR-DYW proteins involved in RNA editing for five editing sites of the moss mitochondrial transcripts. Plant Cell Physiol 51:1942–1949
Okuda K, Nakamura T, Sugita M, Shimizu T, Shikanai T (2006) A pentatricopeptide repeat protein is a site recognition factor in chloroplast RNA editing. J Biol Chem 281:37661–37667
Okuda K, Myouga F, Motohashi R, Shinozaki K, Shikanai T (2007) Conserved domain structure of pentatricopeptide repeat proteins involved in chloroplast RNA editing. Proc Natl Acad Sci USA 104:8178–8183
Okuda K, Hammani K, Tanz SK, Peng L, Fukao Y, Myouga F, Motohashi R, Shinozaki K, Small I, Shikanai T (2009a) The pentatricopeptide repeat protein OTP82 is required for RNA editing of plastid ndhB and ndhG transcripts. Plant J 61:339–349
Okuda K, Chateigner-Boutin AL, Nakamura T, Delannoy E, Sugita M, Myouga F, Motohashi R, Shinozaki K, Small I, Shikanai T (2009b) Pentatricopeptide repeat proteins with the DYW motif have distinct molecular functions in RNA editing and RNA cleavage in Arabidopsis chloroplasts. Plant Cell 21:146–156
Parkinson CL, Mower JP, Qiu YL, Shirk AJ, Song K, Young ND, DePamphilis CW, Palmer JD (2005) Multiple major increases and decreases in mitochondrial substitution rates in the plant family Geraniaceae. BMC Evol Biol 5:73
Pfalz J, Bayraktar OA, Prikryl J, Barkan A (2009) Site-specific binding of a PPR protein defines and stabilizes 5′ and 3′ mRNA termini in chloroplasts. EMBO J 28:2042–2052
Phreaner CG, Williams MA, Mulligan RM (1996) Incomplete editing of rps12 transcripts results in the synthesis of polymorphic polypeptides in plant mitochondria. Plant Cell 8:107–117
Picardi E, Regina TM, Brennicke A, Quagliariello C (2007) REDIdb: the RNA editing database. Nucleic Acids Res 35:D173–D177
Picardi E, Regina TM, Verbitskiy D, Brennicke A, Quagliariello C (2010) REDIdb: an upgraded bioinformatics resource for organellar RNA editing sites. Mitochondrion 11(2):360–365
Prikryl J, Rojas M, Schuster G, Barkan A (2010) Mechanism of RNA stabilization and translational activation by a pentatricopeptide repeat protein. Proc Natl Acad Sci USA 108:415–420
Rajasekhar VK, Mulligan RM (1993) RNA editing in plant mitochondria: [alpha]-phosphate is retained during C-to-U conversion in mRNAs. Plant Cell 5:1843–1852
Raynaud C, Loiselay C, Wostrikoff K, Kuras R, Girard-Bascou J, Wollman FA, Choquet Y (2007) Evidence for regulatory function of nucleus-encoded factors on mRNA stabilization and translation in the chloroplast. Proc Natl Acad Sci USA 104:9093–9098
Reed ML, Peeters NM, Hanson MR (2001a) A single alteration 20 nt 5′ to an editing target inhibits chloroplast RNA editing in vivo. Nucleic Acids Res 29:1507–1513
Reed ML, Lyi SM, Hanson MR (2001b) Edited transcripts compete with unedited mRNAs for trans-acting editing factors in higher plant chloroplasts. Gene 272:165–171
Robbins JC, Heller WP, Hanson MR (2009) A comparative genomics approach identifies a PPR-DYW protein that is essential for C-to-U editing of the Arabidopsis chloroplast accD transcript. RNA 15:1142–1153
Rudinger M, Polsakiewicz M, Knoop V (2008) Organellar RNA editing and plant-specific extensions of pentatricopeptide repeat proteins in jungermanniid but not in marchantiid liverworts. Mol Biol Evol 25:1405–1414
Rudinger M, Funk HT, Rensing SA, Maier UG, Knoop V (2009) RNA editing: only eleven sites are present in the Physcomitrella patens mitochondrial transcriptome and a universal nomenclature proposal. Mol Genet Genomics 281:473–481
Salone V, Rudinger M, Polsakiewicz M, Hoffmann B, Groth-Malonek M, Szurek B, Small I, Knoop V, Lurin C (2007) A hypothesis on the identification of the editing enzyme in plant organelles. FEBS Lett 581:4132–4138
Sasaki T, Yukawa Y, Miyamoto T, Obokata J, Sugiura M (2003) Identification of RNA editing sites in chloroplast transcripts from the maternal and paternal progenitors of tobacco (Nicotiana tabacum): comparative analysis shows the involvement of distinct trans-factors for ndhB editing. Mol Biol Evol 20:1028–1035
Sasaki T, Yukawa Y, Wakasugi T, Yamada K, Sugiura M (2006) A simple in vitro RNA editing assay for chloroplast transcripts using fluorescent dideoxynucleotides: distinct types of sequence elements required for editing of ndh transcripts. Plant J 47:802–810
Schmitz-Linneweber C, Barkan A (2007) RNA splicing and RNA editing in chloroplasts. In: Barkan A (ed) Cell and molecular biology of plastids, vol 19. Springer, Berlin/Heidelberg, pp 213–248
Schmitz-Linneweber C, Small I (2008) Pentatricopeptide repeat proteins: a socket set for organelle gene expression. Trends Plant Sci 13:663–670
Schmitz-Linneweber C, Tillich M, Herrmann RG, Maier RM (2001) Heterologous, splicing-dependent RNA editing in chloroplasts: allotetraploidy provides trans-factors. EMBO J 20:4874–4883
Schmitz-Linneweber C, Regel R, Du TG, Hupfer H, Herrmann RG, Maier RM (2002) The plastid chromosome of Atropa belladonna and its comparison with that of Nicotiana tabacum: the role of RNA editing in generating divergence in the process of plant speciation. Mol Biol Evol 19:1602–1612
Schmitz-Linneweber C, Williams-Carrier R, Barkan A (2005a) RNA immunoprecipitation and microarray analysis show a chloroplast Pentatricopeptide repeat protein to be associated with the 5¢ region of mRNAs whose translation it activates. Plant Cell 17:2791–2804
Schmitz-Linneweber C, Kushnir S, Babiychuk E, Poltnigg P, Herrmann RG, Maier RM (2005b) Pigment deficiency in nightshade/tobacco cybrids is caused by the failure to edit the plastid ATPase alpha-subunit mRNA. Plant Cell 17:1815–1828
Schmitz-Linneweber C, Williams-Carrier R, Williams P, Kroeger T, Vichas A, Barkan A (2006) A pentatricopeptide repeat protein binds to and facilitates the trans-splicing of the maize chloroplast rps12 pre-mRNA. Plant Cell 18:2650–2663
Shields DC, Wolfe KH (1997) Accelerated evolution of sites undergoing mRNA editing in plant mitochondria and chloroplasts. Mol Biol Evol 14:344–349
Small I, Peeters N (2000) The PPR motif – a TPR-related motif prevalent in plant organellar proteins. Trends Biochem Sci 25:46–47
Smith H, Gott J, Hanson M (1997) A guide to RNA editing. RNA 3:1105–1123
Staudinger M, Kempken F (2003) Electroporation of isolated higher-plant mitochondria: transcripts of an introduced cox2 gene, but not an atp6 gene, are edited in organello. Mol Genet Genomics 269:553–561
Staudinger M, Bolle N, Kempken F (2005) Mitochondrial electroporation and in organello RNA editing of chimeric atp6 transcripts. Mol Genet Genomics 273:130–136
Steinhauser S, Beckert S, Capesius I, Malek O, Knoop V (1999) Plant mitochondrial RNA editing. J Mol Evol 48:303–312
Sugita M, Miyata Y, Maruyama K, Sugiura C, Arikawa T, Higuchi M (2006) Extensive RNA editing in transcripts from the PsbB operon and RpoA gene of plastids from the enigmatic moss Takakia lepidozioides. Biosci Biotechnol Biochem 70:2268–2274
Sung TY, Tseng CC, Hsieh MH (2010) The SLO1 PPR protein is required for RNA editing at multiple sites with similar upstream sequences in Arabidopsis mitochondria. Plant J 63:499–511
Takenaka M (2010) MEF9, an E-subclass pentatricopeptide repeat protein, is required for an RNA editing event in the nad7 transcript in mitochondria of Arabidopsis. Plant Physiol 152:939–947
Takenaka M, Brennicke A (2009) Multiplex single-base extension typing to identify nuclear genes required for RNA editing in plant organelles. Nucleic Acids Res 37:e13
Takenaka M, Neuwirt J, Brennicke A (2004) Complex cis-elements determine an RNA editing site in pea mitochondria. Nucleic Acids Res 32:4137–4144
Takenaka M, Verbitskiy D, van der Merwe JA, Zehrmann A, Plessmann U, Urlaub H, Brennicke A (2007) In vitro RNA editing in plant mitochondria does not require added energy. FEBS Lett 581:2743–2747
Takenaka M, Verbitskiy D, van der Merwe JA, Zehrmann A, Brennicke A (2008) The process of RNA editing in plant mitochondria. Mitochondrion 8:35–46
Takenaka M, Verbitskiy D, Zehrmann A, Brennicke A (2010) Reverse genetic screening identifies five E-class PPR proteins involved in RNA editing in mitochondria of Arabidopsis thaliana. J Biol Chem 285:27122–27129
Tang J, Kobayashi K, Suzuki M, Matsumoto S, Muranaka T (2010) The mitochondrial PPR protein LOVASTATIN INSENSITIVE 1 plays regulatory roles in cytosolic and plastidial isoprenoid biosynthesis through RNA editing. Plant J 61:456–466
Tasaki E, Hattori M, Sugita M (2010) The moss pentatricopeptide repeat protein with a DYW domain is responsible for RNA editing of mitochondrial ccmFc transcript. Plant J 62:560–570
Thompson J, Gopal S (2006) Genetic algorithm learning as a robust approach to RNA editing site prediction. BMC Bioinformatics 7:145
Thompson J, Higgins D, Gibson T (1994) Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tillich M, Funk HT, Schmitz-Linneweber C, Poltnigg P, Sabater B, Martin M, Maier RM (2005) Editing of plastid RNA in Arabidopsis thaliana ecotypes. Plant J 43:708–715
Tillich M, Lehwark P, Morton BR, Maier UG (2006) The evolution of chloroplast RNA editing. Mol Biol Evol 23:1912–1921
Tillich M, Le Sy V, Schulerowitz K, von Haeseler A, Maier UG, Schmitz-Linneweber C (2009a) Loss of matK RNA editing in seed plant chloroplasts. BMC Evol Biol 9:201
Tillich M, Hardel SL, Kupsch C, Armbruster U, Delannoy E, Gualberto JM, Lehwark P, Leister D, Small ID, Schmitz-Linneweber C (2009b) Chloroplast ribonucleoprotein CP31A is required for editing and stability of specific chloroplast mRNAs. Proc Natl Acad Sci USA 106:6002–6007
Tillich M, Beick S, Schmitz-Linneweber C (2010) Chloroplast RNA-binding proteins: repair and regulation of chloroplast transcripts. RNA Biol 7:172–178
Tseng CC, Sung TY, Li YC, Hsu SJ, Lin CL, Hsieh MH (2010) Editing of accD and ndhF chloroplast transcripts is partially affected in the Arabidopsis vanilla cream1 mutant. Plant Mol Biol 73:309–323
Tsuchiya N, Fukuda H, Sugimura T, Nagao M, Nakagama H (2002) LRP130, a protein containing nine pentatricopeptide repeat motifs, interacts with a single-stranded cytosine-rich sequence of mouse hypervariable minisatellite Pc-1. Eur J Biochem 269:2927–2933
Tsudzuki T, Wakasugi T, Sugiura M (2001) Comparative analysis of RNA editing sites in higher plant chloroplasts. J Mol Evol 53:327–332
Uyttewaal M, Arnal N, Quadrado M, Martin-Canadell A, Vrielynck N, Hiard S, Gherbi H, Bendahmane A, Budar F, Mireau H (2008) Characterization of Raphanus sativus pentatricopeptide repeat proteins encoded by the fertility restorer locus for Ogura cytoplasmic male sterility. Plant Cell 20:3331–3345
Valcarcel J, Green MR (1996) The SR protein family: pleiotropic functions in pre-mRNA splicing. Trends Biochem Sci 21:296–301
Valente L, Nishikura K (2005) ADAR gene family and A-to-I RNA editing: diverse roles in posttranscriptional gene regulation. Prog Nucleic Acid Res Mol Biol 79:299–338
van der Merwe JA, Takenaka M, Neuwirt J, Verbitskiy D, Brennicke A (2006) RNA editing sites in plant mitochondria can share cis-elements. FEBS Lett 580:268–272
Verbitskiy D, van der Merwe JA, Zehrmann A, Brennicke A, Takenaka M (2008) Multiple specificity recognition motifs enhance plant mitochondrial RNA editing in vitro. J Biol Chem 283:24374–24381
Verbitskiy D, Zehrmann A, van der Merwe JA, Brennicke A, Takenaka M (2009) The PPR protein encoded by the LOVASTATIN INSENSITIVE 1 gene is involved in RNA editing at three sites in mitochondria of Arabidopsis thaliana. Plant J 61(3):446–455
Verbitskiy D, Zehrmann A, Brennicke A, Takenaka M (2010) A truncated MEF11 protein shows site-specific effects on mitochondrial RNA editing. Plant Signal Behav 5(5):558–560
Wakasugi T, Tsudzuki T, Sugiura M (2001) The genomics of land plant chloroplasts: gene content and alteration of genomic information by RNA editing. Photosynth Res 70:107–118
Walbot V (1991) RNA editing fixes problems in plant mitochondrial transcripts. Trends Genet 7:37–39
Wang Z, Zou Y, Li X, Zhang Q, Chen L, Wu H, Su D, Chen Y, Guo J, Luo D, Long Y, Zhong Y, Liu YG (2006) cytoplasmic male sterility of rice with boro II cytoplasm is caused by a cytotoxic peptide and is restored by two related PPR motif genes via distinct modes of mRNA silencing. Plant Cell 18:676–687
Williams MA, Tallakson WA, Phreaner CG, Mulligan RM (1998) Editing and translation of ribosomal protein S13 transcripts: unedited translation products are not detectable in maize mitochondria. Curr Genet 34:221–226
Williams-Carrier R, Kroeger T, Barkan A (2008) Sequence-specific binding of a chloroplast pentatricopeptide repeat protein to its native group II intron ligand. RNA 14:1930–1941
Wintz H, Hanson MR (1991) A termination codon is created by RNA editing in the petunia atp9 transcript. Curr Genet 19:61–64
Wolf PG, Rowe CA, Hasebe M (2004) High levels of RNA editing in a vascular plant chloroplast genome: analysis of transcripts from the fern Adiantum capillus-veneris. Gene 339:89–97
Yoshinaga K, Iinuma H, Masuzawa T, Uedal K (1996) Extensive RNA editing of U to C in addition to C to U substitution in the rbcL transcripts of hornwort chloroplasts and the origin of RNA editing in green plants. Nucleic Acids Res 24:1008–1014
Yoshinaga K, Kakehi T, Shima Y, Iinuma H, Masuzawa T, Ueno M (1997) Extensive RNA editing and possible double-stranded structures determining editing sites in the atpB transcripts of hornwort chloroplasts. Nucleic Acids Res 25:4830–4834
Yu W, Schuster W (1995) Evidence for a site-specific cytidine deamination reaction involved in C to U RNA editing of plant mitochondria. J Biol Chem 270:18227–18233
Yu QB, Jiang Y, Chong K, Yang ZN (2009) AtECB2, a pentatricopeptide repeat protein, is required for chloroplast transcript accD RNA editing and early chloroplast biogenesis in Arabidopsis thaliana. Plant J 59:1011–1023
Yura K, Sulaiman S, Hatta Y, Shionyu M, Go M (2009) RESOPS: a database for analyzing the correspondence of RNA editing sites to protein three-dimensional structures. Plant Cell Physiol 50:1865–1873
Zabaleta E, Mouras A, Hernould M, Suharsono AA (1996) Transgenic male-sterile plant induced by an unedited atp9 gene is restored to fertility by inhibiting its expression with antisense RNA. Proc Natl Acad Sci USA 93:11259–11263
Zandueta-Criado A, Bock R (2004) Surprising features of plastid ndhD transcripts: addition of non-encoded nucleotides and polysome association of mRNAs with an unedited start codon. Nucleic Acids Res 32:542–550
Zehrmann A, van der Merwe JA, Verbitskiy D, Brennicke A, Takenaka M (2008) Seven large variations in the extent of RNA editing in plant mitochondria between three ecotypes of Arabidopsis thaliana. Mitochondrion 8:319–327
Zehrmann A, Verbitskiy D, van der Merwe JA, Brennicke A, Takenaka M (2009) A DYW domain-containing pentatricopeptide repeat protein is required for RNA editing at multiple sites in mitochondria of Arabidopsis thaliana. Plant Cell 21:558–567
Zehrmann A, Verbitskiy D, Hartel B, Brennicke A, Takenaka M (2010) RNA editing competence of trans-factor MEF1 is modulated by ecotype-specific differences but requires the DYW domain. FEBS Lett 584:4181–4186
Zhou W, Cheng Y, Yap A, Chateigner-Boutin AL, Delannoy E, Hammani K, Small I, Huang J (2009) The Arabidopsis gene YS1 encoding a DYW protein is required for editing of rpoB transcripts and the rapid development of chloroplasts during early growth. Plant J 58:82–96
Zito F, Kuras R, Choquet Y, Kössel H, Wollman FA (1997) Mutations of cytochrome b6 in Chlamydomonas reinhardtii disclose the functional significance for a proline to leucine conversion by petB editing in maize and tobacco. Plant Mol Biol 33:79–86
Acknowledgements
The authors apologize for the many studies on organellar RNA editing that were not discussed and cited here due to space limitations. Support by the DFG to CSL (Emmy Noether stipend) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Finster, S., Legen, J., Qu, Y., Schmitz-Linneweber, C. (2012). Land Plant RNA Editing or: Don’t Be Fooled by Plant Organellar DNA Sequences. In: Bock, R., Knoop, V. (eds) Genomics of Chloroplasts and Mitochondria. Advances in Photosynthesis and Respiration, vol 35. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-2920-9_13
Download citation
DOI: https://doi.org/10.1007/978-94-007-2920-9_13
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-2919-3
Online ISBN: 978-94-007-2920-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)