
Abstract
Protein kinase B/Akt plays a critical role in the regulation of cardiac hypertrophy, angiogenesis and apoptosis. The evidences that elevation of Akt in cardiomyocytes in vivo and in vitro protects against apoptosis after ischemia/reperfusion injury provide possibility that agents targeting Akt activation become a novel therapeutic strategy for limiting myocardial injury following ischemia. Vanadium compounds inhibiting protein tyrosine phosphatases are potent activator of the Akt signaling pathways and elicit cardioprotection in heart ischemia/reperfusion injury along with cardiac functional recovery in rats. In addition, vanadium compounds has strong anti-hypertrophic in the pressure overload-induced hypertrophy in ovariectomized and aortic-banded rats. The elevation of Akt activity and Akt-dependent eNOS phosphorylation are central roles on vanadium compound-induced anti-hypertrophy and heart failure in the ovariectomized and aortic-banded rats. Taken together, vanadium compounds are potential therapeutics for ischemia/reperfusion-induced myocardial injury and heart failure associated with hypertension in the postmenopausal women.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cardiovascular disease
- Hypertrophy
- Endothelial nitric oxide synthase
- Postmenopausal women
- Protein kinase B
- Protein tyrosine phosphatase
- Vanadium compounds
1 Introduction
In the cardiovascular system, protein kinase B (PKB)/Akt plays an important role in the regulation of cardiac hypertrophy, angiogenesis, and apoptosis [1–5]. The Akt subfamily comprises three mammalian isoforms, PKBα/Akt1, PKBβ/Akt2 and PKBγ/Akt3, which are products of distinct genes and share a conserved structure that includes three functional domains: an N-terminal pleckstrin homology domain, a central kinase domain, and a C-terminal regulatory domain containing the hydrophobic motif phosphorylation site [FxxF(S/T)Y] [6]. Among the three Akt genes, only Akt1 and Akt2 are highly expressed in the heart. Consistent with the general trophic function of Akt, the Akt1 whole-genome-knockout mice weigh approximately 20% less than wild-type littermates and have a proportional reduction in size of all somatic tissues, including the heart [7, 8]. In contrast, Akt2-knockout mice have only a modest reduction in organ size [9]. Thus, data from Akt-knockout mice support a critical role specifically for Akt1 in normal growth of the heart [10]. The observation that acute activation of Akt in cardiomyocytes in vivo and in vitro protects against apoptosis after ischemia/reperfusion injury provide possibility that agents targeting Akt activation become a novel therapeutic strategy for limiting myocardial injury following ischemia [11].
2 Cardioprotection with Vanadium Compound in Myocardial Infarction
2.1 Vanadium Compounds as Akt Activator
Vanadium compounds are potent activator of the Akt signaling pathways [12–15] and elicit cardioprotection in heart ischemia/reperfusion injury along with cardiac functional recovery in rats [1, 2, 15, 16]. Vanadium compounds inhibit protein tyrosine phosphatases [12, 13] and promote an increase in protein tyrosine phosphorylation, leading to the upregulation of Akt [12, 14]. An increase in tyrosine phosphorylation in the heart via increased tyrosine kinase activity has been implicated in the signal transduction pathway of cardioprotection by ischemic preconditioning [17–19] which is the most potent endogenous mechanism to limit myocardial infarct size. Among several oxidation states of vanadium II to V, vanadium ion in living cells exists exclusively as vanadyl (IV) cation and a small amount as vanadate (V) anion [20]. Furthermore, vanadyl (VO2+) compounds, of oxidation state IV, under physiological conditions are subject to oxidation by a variety of oxidants, including molecular oxidant and vanadate compounds, of oxidation state V are thought to undergo reduction to state IV in the cell [21–24]. To develop a novel therapeutic drug to protect cardiomyocytes from heart diseases, we selected a novel vanadyl (IV) compound having the VO2+ chelate, bis(1-oxy-2-pyridinethiolato) oxovanadium(IV), [VO(OPT)], which is a potent activator of the Akt signaling pathway both in vivo and in vitro [12–14, 25].
2.2 Pharmacotoxicity of Vanadium Compounds
The toxicity of vanadium compounds is low. The most common toxic effects reported for inorganic vanadium compounds are diarrhea, decreased food uptake, dehydration and reduced body weight gain [14] which can, however, be corrected by adding sodium chloride to drinking water, adjusting the pH of the solution to neutrality and by gradually increasing the dose of vanadium [26, 27]. Organic vanadium compounds were much safer than inorganic vanadium salts and do not cause any gastrointestinal discomfort, hepatic or renal toxicity [25, 28]. Along with the previous studies, we also did not found any gastrointestinal, hepatic or renal toxicity in the VO(OPT) treated rats [29, 30].
Recent work has demonstrated that vanadium compounds inhibited serum- and growth factor-stimulated mitogenesis [31, 32] and possess anti-tumor activity [33, 34]. Many other studies have, however, failed to detect any change in the levels of urea, creatinine, glutamic oxaloacetic transaminase and glutamic pyruvic transaminase, indices of kidney and liver functions [35–37]. Moreover, no significant changes in the histopathology of several tissues, including the liver, spleen, stomach, heart and lung, have been observed among control and vanadyl sulfate-treated animals [38]. Electron microscopic examination of ob/ob mice treated with vanadate for 47 days revealed no sign of hepatotoxicity [27].
In patients treated with vanadium salts, gastrointestinal discomfort was the most common toxic effect, which could be corrected by decreasing the dose level [39]. Moreover, clinical studies have been of short duration (up to 6 weeks) and utilized lower doses than those administered in animal experiments; thus, the long-term toxicity of vanadium in humans remains to be explored. Clearly, at present, there is no consensus on the toxic effects of vanadium compounds, and detailed and systematic investigations are needed to evaluate the toxicity of various vanadium compounds before undertaking long-term clinical trials in humans. It should be noted that use of chelating agents and organo-vanadium compounds, such as VO(OPT), have shown significantly reduced vanadium toxicity and may serve as more potent cardioprotective agents than inorganic vanadium salts.
2.3 Cardioprotection of VO(OPT) in Ischemia/Reperfusion Injury
We tested whether VO(OPT) treatment has cardioprotective effect against myocardial ischemia/reperfusion injuries in rats. Rats were subjected to 30 min ischemia followed by 24 h reperfusion to define the cytoprotective effect of VO(OPT) (0.5 and 1.25 mg V/kg) on myocardial infarct size. The infarct sizes in the VO(OPT) treated group (53 ± 7% and 37 ± 2% in 0.5 and 1.25 mg V/kg, respectively) were significantly smaller than that in the vehicle group (67 ± 4%) [1]. This observation indicated that VO(OPT) has cardioprotective effect on ischemia/reperfusion induced myocardial infarction.
Sodium orthovanadate restores ischemia-induced decrease in Akt phosphorylation on Ser-473 in the gerbil hippocampus, thereby rescuing hippocampal neurons from ischemia-induced cell death [40]. Akt activation has been shown to reduce cardiomyocyte apoptosis, thereby preventing myocardial injury after transient ischemia [11]. We confirmed that post-treatment with VO(OPT) significantly rescues decreased Akt activity after myocardial ischemia/reperfusion and the preserved Akt activity possibly accounts for the VO(OPT)-induced cytoprotective action in cardiomyocytes. Activated Akt is believed to suppress apoptosis through phosphorylation of several substrates, including the Bcl-2 family member, Bad [41] and Forkhead transcription factors (FOXO) [42, 43]. We confirmed downstream targets of Akt to mediate anti-apoptotic signaling in cardiomyocytes. Significant decrease in phosphorylation of Bad and Forkhead transcription factors (FKHR and FKHRL1) are closely correlated with decreased Akt activity following myocardial ischemia/reperfusion injury. Phosphorylation of Bad and Forkhead transcription factors were markedly potentiated in cardiomyocytes by treatment with VO(OPT), similar to its response to Akt activity. These results suggest that both Bad and FOXOs are Akt targets and mediate cardioprotective action by inhibiting them following treatment with VO(OPT). Like FKHR, FKHRL1 has been shown to induce apoptosis in the brain [44] and fibroblasts through up-regulation of the Fas ligand expression and activation of the death receptor pathway [45]. We also found significantly increased expression of both Fas ligand and Bim after myocardial ischemia/reperfusion and significant inhibition by treatment with VO(OPT) [1].
Recent findings showed that FKHRL1 is regulated by Akt activity in endothelial cells and that FKHRL1 dephosphorylation promotes apoptosis by negatively regulating FLIP expression [46]. Moreover, Akt was found to rescue endothelial cells from Fas/Fas ligand-mediated cell death through up-regulation of FLIP level [47]. Similar with the previous studies, we found significant FLIP degradation after ischemia/reperfusion [48, 49] and treatment with VO(OPT) significantly increased FLIP expression in cardiomyocytes. The increased FLIP expression was correlated with decreased Fas ligand expression in VO(OPT)-treated group compared with vehicle-treated group. Taken together, inactivation of FKHR and FKHRL1 by treatment with VO(OPT) after ischemia/reperfusion likely promotes expression of FLIP, thereby inhibiting cardiomyocyte [1] apoptosis
Cardiomyocyte apoptosis is one of the major contributors in the development of myocardial infarct [50], which is related to the pathogenesis of heart failure after ischemia. Accumulating evidences indicate that apoptosis, different type of cell death form necrosis, plays essential role in cardiomyocyte death after ischemia/reperfusion [50]. Therefore, we examined whether VO(OPT) has anti-apoptotic effects in cardiomyocytes by measuring Caspase 3, Caspase 7 and Caspase 9 processing as a marker of apoptosis. Interestingly, short-term infusion of VO(OPT) significantly reduced cardiomyocyte apoptosis after ischemia/reperfusion as indicated by dose-dependent inhibition of Caspase-3, -7 and -9 processing [3]. The results confirm anti-apoptotic effects of VO(OPT) against myocardial ischemia/reperfusion injury.
VO(OPT) treatment protects the heart form ischemia/reperfusion-induced cardiac injury, thereby improving cardiac contractile dysfunction in rats. VO(OPT) induced cardioprotection is mainly elicited by Akt activation after myocardial ischemia/reperfusion. Akt activation induces phosphorylation of proapoptotic protein Bad, thereby reducing mitochondria-dependent apoptosis. Moreover, VO(OPT) treatment abolished dephosphorylation of forkhead transcription factors after ischemia/reperfusion injury, thereby inhibiting expression of Fas ligand and Bim. Furthermore, VO(OPT) treatment after ischemia/reperfusion promoted expression of FLIP through Akt activation, thereby further inhibiting activation of Fas/Fas-ligand intracellular signal. Taken together, VO(OPT) treatment at reperfusion is likely beneficial as a cardioprotective drug in subjects undergoing reperfusion therapy following a myocardial infarction (Fig. 9.1).

Putative mechanism of myocardial protection by VO(OPT). Binding of trophic/survival factors to tyrosine kinase receptors activate Akt through PI3K and phosphatidylinositol-dependent kinase-1(PDK1) activation. Ischemia/reperfusion caused inactivation of Akt, thereby promoting apoptotic pathways including the Bad, Forkhead transcription factors (FOXO; FKHR and FKHRL-1) and FLIP via the Fas pathways. Treatment with VO(OPT) activates and/or preserves Akt activity after ischemia-reperfusion leading to the phosphorylation and thereby inactivation of Bad and Forkhead transcription factors thus preventing apoptosis by Fas ligand and Bim. VO(OPT) treatment also preserves ischemia-reperfusion induced breakdown of FLIP and thereby preventing cell death via Fas pathways. Therefore, the cytoprotective action of VO(OPT) is mediated by the Akt-Forkhead transcription factor-FLIP mediated pathway
3 Cardioprotection of Vanadium Compounds in the Postmenopause
3.1 Cardiovascular Diseases in the Postmenopausal Women
Menopause is the permanent cessation of menstruation and ovarian follicular production of estrogens and progesterone. The mean age at menopause is 51 years, and 95% of women reach menopause between the ages of 45 and 55 [51]. Over the past 20 years, numerous observational, retrospective, interventional, and meta-analytic studies [52] as well as studies using animal models have supported the hypothesis that ovarian steroids exert important protective actions in women and the absence of sex hormones after menopause makes postmenopausal women more vulnerable than younger premenopausal women to cardiovascular diseases (CVD). Indeed, CVD is the leading cause of morbidity and mortality among postmenopausal women in westernized societies [53] and accounts for nearly half of all deaths in women [54]. The Framingham study showed that the incidence of CVD is higher among postmenopausal than in premenopausal women, even among women of the same age [55]. Women who also experience early menopause, both naturally and surgically (bilateral oophorectomy), also have increased risk of coronary events [56].
Approximately 1 in 8 women above age 55 years has undergone bilateral oophorectomy before reaching natural menopause [56, 57]. Bilateral oophorectomy may be performed for a benign disease or for prophylaxis against ovarian cancer, and is usually performed along with hysterectomy (in nearly 90% of cases) [58]. Of the more than 600,000 hysterectomies performed annually in the United States, approximately half include bilateral oophorectomy [59]. In addition, the practice of prophylactic oophorectomy has increased over time and became more than doubled between 1965 and 1990 [60]. Meanwhile, reports now link premenopausal oophorectomy with serious health consequences including premature death, cardiovascular and neurologic disease, and osteoporosis in addition to menopausal symptoms, psychiatric symptoms, and impaired sexual function [61]. The preponderance of evidence suggests that bilateral oophorectomy is associated with increased cardiovascular risk and premature death, and that oophorectomy at a young age further increases this risk [62]. Estrogen therapy started early after surgical or natural menopause at a young age appears to reduce this risk [62–65].
Clinical and experimental studies have established that sex influences the patterns of LV hypertrophy [66]. In response to pressure overload, such as hypertension or aortic stenosis, human male hearts exhibit LV dilatation or eccentric hypertrophy, whereas female hearts tend to maintain normal chamber size but develop increased wall thickness, consistent more with concentric hypertrophy [67]. Evidences of sex hormones influence on the patterns of hypertrophy is revealed from studies demonstrating that physiological replacement of 17β-estradiol, the main circulating form of estrogen in premenopausal women, to ovariectomized female mice limits pressure overload–induced LV hypertrophy [68, 69].
Despite well established models of myocardial hypertrophy and heart failure using rodents, there has been lack of suitable animal models to study postmenopausal hypertension and hypertrophic cardiac remodeling. Although nonhuman primates, sheep, rabbits, mice and rats have all been used as models of various menopausal changes [70], progress in elucidating the mechanisms responsible for postmenopausal hypertension and hypertrophic remodeling has been hampered by the lack of a suitable animal model. Therefore, despite impressive progress in the diagnosis and treatment of hypertrophy, more research on postmenopausal cardiac decompensation under stress is absolutely needed, and adequate animal models are critical to fill the gap between basic science discovery and clinics. An ideal model of postmenopausal cardiac hypertrophy and heart failure should meet various requirements to mimic a complex syndrome characterized by cardiac, hemodynamic and neurohumoral alterations as close as possible. We focused on the molecular mechanisms that contribute to cardioprotective effects of estrogen on LV hypertrophy in response to pressure overload in a rat model of postmenopausal phenotypes to develop novel therapeutic strategy instead of hormone replacement therapy in the postmenopausal women.
3.2 Development of Postmenopausal Cardiac Hypertrophy Models
There have been attempts to elucidate the cardioprotective effect of estrogen in pressure overload induced hypertrophy by using ovariectomized animals [71, 72], however, these have rarely taken into account the effect of menopause on cardiac adverse remodeling and the transition to heart failure. In any case, there is no normotensive animal model that exhibits hypertrophy and decompensation with postmenopausal phenotypes. Female spontaneously hypertensive rats (SHR) represent some postmenopausal phenotypes as they stop cycling at age 10–12 months and have low estradiol levels comparable to postmenopausal women [73]. However, the sex difference in blood pressure no longer exists because of the increase in blood pressure in old females, whereas blood pressure in male SHR remains fairly stable after age 8 months [73]. Dahl salt-sensitive (DS) hypertensive rats also represent some characteristics of postmenopause as they exhibit increases in blood pressure with aging [74]. When the young female DS rats are ovariectomized and fed a high-salt diet, the blood pressure increases to higher levels than in intact females [74]. Still it is not known at what age these animals cease cycling and what happens to their blood pressure after cessation of cycling. In addition to the rat models, the follicular-stimulating hormone receptor knockout mouse also exhibits some of the characteristics of postmenopausal women [75]. These mice have low plasma estradiol levels, have functionally active estrogen receptors, increased testosterone levels, hypertension, hypercholesterolemia, and weight gain when compared with their wild-type counterparts [75]. However, these animals did not exhibit increased oxidative stress or endothelial dysfunction at age of 14–16 weeks, factors common to postmenopausal women [75].
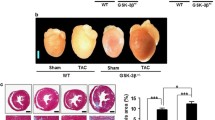
To produce left ventricular pressure overload in the ovariectomized rats, we introduced a model of transverse aortic constriction in the abdominal aorta between the right and left renal arteries [2, 4, 76]. Serum estrogen level was significantly decreased following ovariectomy (OVX) compared with the sham rats (sham: 56.6 ± 8.3 pg/ml; OVX: 3.6 ± 2.3 pg/ml). Pressure overload (PO)-induced hypertrophy had no effect on the serum estrogen level as seen both in the PO (60.8 ± 13.6 pg/ml) and OVX-PO (3.4 ± 1.2 pg/ml) group rats. Following ovariectomy, body weight (BW) was significantly increased compared to sham animals. Significant increases in LV weight were also seen in the OVX-PO group compared to sham, OVX and PO groups without significant changes in right ventricle weight.
3.3 Anti-hypertrophic Effect of VO(OPT)
Consistent with my previous observation, LV weight and LW significantly increased in the ovariectomized pressure overloaded (OVX-PO) group compared with the OVX group, without significant changes in right ventricle weight [2]. VO(OPT) (2.5 mgV/kg, p.o.) treatment on the sham and OVX rats have no effect on the morphometric parameters [4]. VO(OPT) treatment significantly and dose-dependently decreased the elevated LV weights and lung weight (LW). Notably, the ratio of HW to BW markedly increased in OVX-PO group compared with OVX group. VO(OPT) (2.5 mg/kg) treatment significantly inhibited the elevated the HW/BW ratio (P < 0.01 vs. OVX-PO) (Fig. 9.2). The LW/BW ratio was also significantly increased in OVX-PO group compared with OVX group. VO(OPT) treatment dose-dependently decreased the elevated LW/BW ratio. Moreover, oral treatment with VO(OPT) (2.5 mg V/kg) for 14 days have no effect on HW/BW ratio and HW/LW ratio in sham-operated animals [4].

Effect of VO(OPT) on heart weight (HW)/body weight (BW) (a) and lung weight (LW)/ body weight BW (b) ratio. Each bar represents the mean ± S.E.M.*, P < 0.05 and ***, P < 0.001 versus OVX group; #, P < 0.05 and ##, P < 0.01 versus OVX-PO-vehicle treated group (Modified from [4])
Since treatment with VO(OPT) (1.25 and 2.5 mg V/kg) restored HR and MABP, we evaluated LV functions in OVX-PO heart with or without treatment of VO(OPT). Consistent with my previous observation [2], left ventricular end diastolic pressure (LVEDP) was significantly increased in the OVX-PO group compared with OVX group. VO(OPT) treatment dose-dependently restored elevated LVEDP. Similarly, left ventricular developed pressure (LVDP) significantly increased in OVX-PO group and VO(OPT) treatment dose-dependently restored elevated LVDP. The rate of LV contraction (+dp/dt) and relaxation (−dp/dt) also significantly increased in OVX-PO [2] and VO(OPT) treatment dose-dependently restored the elevated LV contraction (+dp/dt) and relaxation (−dp/dt). In agreement with earlier studies [29, 30], the treatment with VO(OPT) containing 2.5 mg V/kg resulted in slight decrease in body weight and food intake compared to vehicle treated rats but these changes are not significant [4].
3.4 Impaired Cardiac Akt/eNOS Signaling in Postmenopause
Akt phosphorylation at Ser-473 was significantly reduced in the hearts of OVX-PO rats compared with the OVX group, indicating that Akt signaling is markedly impaired in pressure overload-induced heart failure concomitant with severe cardiac hypertrophy. Akt Thr-308 phosphorylation also decreases only in the OVX-PO group. To define the role of Akt activity in cardiac hypertrophy and heart failure, we evaluated the time course of cardiac hypertrophy, heart failure, and the left ventricular Akt activity. Heart weigh/body weight ratio is increased time dependently from 1 to 4 weeks following PO in OVX rats [4]. LV Akt phosphorylation at Ser 473 was increased 1 week after PO, thereafter decreased time dependently with significant decreased level observer 4 weeks after PO in OVX rats. On the contrary, no significant change was observed in the total Akt level [4]. Importantly, VO(OPT) treatment markedly and dose-dependently increased Akt activity as assessed by increased phosphorylation at Ser 473 and at Thr 308 (Fig. 9.3).

Effects of VO(OPT) on Akt and eNOS phosphorylation in the left ventricle. Representative Western blot analysis using cell extracts from OVX (n = 8), OVX-PO-vehicle (n = 8), OVX-PO-VO(OPT) containing 1.25 mg/kg vanadium (V 1.25) (n = 8) and OVX-PO-VO(OPT) containing 2.5 mg/kg vanadium (V 2.5) (n = 8) treated hearts probed with phospho-Akt (Ser-473) and phospho-Akt (Thr-308) (a) and eNOS and phospho-eNOS (Ser 1179) (b). Data are expressed as percentages of the value of OVX rats. Each column represents the mean ± S.E.M. **, P < 0.01 versus the OVX group; ## P < 0.01 and ### P < 0.001 versus the OVX-PO-vehicle treated group (Modified from [4])
Since eNOS is physiological substrate for Akt in human vascular endothelial cells [77], we determine whether VO(OPT)-induced Akt activation results in increased eNOS phosphorylation and its activity. The time course studies revealed that both eNOS and Akt mediated eNOS phosphorylation at Ser 1179 decreased time dependently following PO-treatment with significant decreased level observed 4 weeks after PO in OVX rats [4]. Consistent with our previous observation [2], we also observed severe impairment of eNOS expression following OVX-PO treatment. I here found a slight but not significant reduction of eNOS phosphorylation at Ser 1179. Notably, VO(OPT) treatment dose-dependently increased eNOS phosphorylation (Fig. 9.3). VO(OPT) also significantly increased eNOS expression.
3.5 Prevention of β-Adrenergic Induced Heart Attack with VO(OPT)
β-adrenoceptor (β-AR) agonists differently affect heart rate during the estrous cycle in female rats and in ovariectomized rats with or without estrogen replacement [78]. Ovariectomy also increases susceptibility to the effects of β-AR agonists [79]. Importantly, Kam et al. [80] demonstrated that β-AR stimulation with isoproterenol led to a significantly greater increase in electrical stimulation-induced Ca2+ elevation, Ca2+-uptake through cardiac L-type Ca2+ channels, heart rate and contractility in hearts of ovariectomized rats compared to sham rats. These responses were rescued by estrogen replacement. Several studies hypothesized that excessive adrenergic activation could initiate the progression from compensated LV hypertrophy in hypertension to cardiac dysfunction and that this effect is primarily through adverse LV remodeling.
Similar with aortic banding [2], chronic administration of β-AR agonist to rats led to adverse cardiac injury through impaired Akt-eNOS signaling pathways and ovariectomy further aggravated the cardiac injury suggesting the cardioprotective role of ovarian hormones [4]. Kaplan-Meier survival data in indicate that chronic β-adrenergic stimulation with isoproterenol has no effect of the survival of sham and pressure overloaded female rats (Fig. 9.4). Remarkably, treatment with isoproterenol on the ovariectomized rats tended to increase mortality with a survival rate of 84% at 28 days. Interestingly, the most significant mortality occurred in the OVX-PO rats with a survival rate of 0% at 21 days following chronic β-adrenergic stimulation (Fig. 9.4) [4]. These observations indicate that decompensation against cardiac stress such as isoproterenol treatment occurs only in OVX and OVX-PO rats. Treatment with the Akt activator VO(OPT) dose dependently increased survival following acute cardiac stress caused by chronic β-adrenergic stimulation [1, 3], suggesting that cardiac remodeling and recovered cardiac functions by VO(OPT) treatment contributes the reduced mortality (Fig. 9.4). Thus, simultaneous impairment of Akt-eNOS signaling by ovariectomy contributed to cardiac decompensation during PO-induced hypertrophy, deteriorated heart functions and thereby increased mortality during chronic β-adrenergic stimulation. Potentiation of the Akt and eNOS signaling pathways by treatment with VO(OPT) after OVX-PO likely contributes to increased survival following acute cardiac stress caused by chronic β-adrenergic stimulation. These results contribute to our understanding of the mechanisms underlying increased cardiac injuries in postmenopausal women by chronic β-adrenergic activation and bear relevance to further clinical management strategies to prevent adverse cardiac remodeling.

Kaplan-Meier survival analysis following chronic isoproterenol treatment. Five mg/kg isoproterenol (Isp 5) was administered daily to sham, sham-PO, OVX, OVX-PO-vehicle, OVX-PO-VO(OPT) containing 1.25 mg/kg vanadium (V 1.25) and OVX-PO-VO(OPT) containing 2.5 mg/kg vanadium (V 2.5) treated groups. Survival over 28 days was monitored. ***, P < 0.001 versus the sham group; ##, P < 0.01 and ###, P < 0.001 versus the OVX-PO-vehicle treated group (Modified from [4])
4 Crosstalk of Estrogen Receptor and Akt/eNOS Signaling
4.1 Cardioprotection Through Estrogen Receptor
Both in vivo and in vitro studies indicates that estrogen have cardioprotective effects. However, despite these positive results, hormone replacement therapy has not been shown to consistently lower blood pressure in postmenopausal women. Moreover, in women who have experienced surgical menopause, estrogen replacement therapy also did not result in significant sustained reductions in blood pressure. The mechanisms responsible for increased prevalence of cardiovascular diseases in postmenopausal women are complex and multifaceted. They are not nearly so simple as a reduction in estradiol. We focused on evaluating the molecular mechanisms responsible cardiac injuries in postmenopausal women and characterized the postmenopausal OVX-PO model to provide a suitable animal model to quantitatively evaluate some of the mechanisms and hoped that the information could be extrapolated to women.
Ovarian hormones are believed to possess cardiovascular protective effects, and they seem to play a role in the gender-related differences in the development of hypertension in experimental models [81, 82]. Naturally, ovariectomy causes a significant reduction in estradiol and progesterone levels [74, 83]. Comparison of estradiol levels with MABP in DS rats suggests that the OVX-induced increase in MABP is associated with decreased levels of plasma estrogen because estradiol replacement was able to prevent the OVX-induced hypertension [74]. Moreover, estradiol supplementation did not markedly alter circulating progesterone levels in both young and aged DS rats [74]. Furthermore, progesterone levels did not correlate with the difference in MABP between the OVX and OVX-estrogen groups in DS rats [74]. This lack of effect of progesterone supports previous reports in deoxycorticosterone salt hypertension showing that progesterone had no effect on the development of hypertension in OVX rats [82].
4.2 Stimulation of Akt/eNOS Signaling Through Estrogen Receptor
Signaling through phosphatidylinositol 3-kinase (PI3K)/Akt pathway is important for the physiological growth and inhibition of pathological hypertrophy [11, 84, 85]. Moreover, physiological hypertrophy induced by exercise training also requires the activation of myocardial Akt. By contrast, pathological hypertrophies induced by pressure overload cause an inactivation of Akt signaling pathway [86]. It is evident that the E2-activated PI3K/Akt pathway functions as one of the acute nongenomic actions of E2 in various types of cells [87]. Studies indicate that young women possess higher levels of nuclear-localized phospho-Akt (Serine 473) relative to comparably aged men or postmenopausal women [88] and Phospho-Akt (Serine 473) is also localized to the nucleus of cultured cardiomyocytes after exposure to E2 [88]. Previous studies have demonstrated that OVX-reduced Akt activation occurs with decreased myocyte contractile function and impaired intracellular calcium handling, whereas E2-upregulated Akt is associated with restored cardiac contractility and intracellular calcium homeostasis [89]. Moreover, activation of the PI3K/Akt pathway is required for E2-suppressed apoptosis and E2-protected myocardial function in the heart following ischemia [68]. Estrogen receptor a mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia [90]. Recent study also suggests that E2-mediated improvement in cardiac function following trauma-hemorrhage is mediated by an increase in cardiac Akt activation [91]. In endothelial cells, estrogen receptors localize to caveolae and their stimulation activate eNOS activity via PI3K/Akt signaling [92, 93]. Strikingly, Grasselli et al. [94] demonstrated that estrogen receptor alpha and eNOS form a complex and translocate to nucleus to bind to the estrogen response element in the promoter region of telomerase catalytic subunit gene, thereby leading to the increase telomerase activity. Since the telomerase activity is critical to determine the lifespan of a cell. This is the first report to define enhancement of telomerase activity by eNOS activation and NO production.
4.3 Stimulation of Nitric Oxide Production Through Estrogen Receptor
Men and postmenopausal women may have less endogenous nitric oxide (NO) production, shown by less vasoconstriction following inhibition of NO synthase by L-N-monomethyl-arginine (L-NMMA), than premenopausal women. After estrogen therapy, L-NMMA resulted in greater constriction consistent with estrogen restoring vascular NO activity to levels seen in premenopausal women [95]. Other studies have shown improvement in endothelial function with estrogen therapy with a greater improvement in hypertensive postmenopausal women [96]. Acute estrogen deprivation after oophorectomy in healthy women results in impaired endothelium-dependent vasodilatation as a result of reduced NO availability [97]. Estrogen therapy improves endothelium-dependent vasodilatation after oophorectomy and also natural menopause [98]. However, among a broader sample of postmenopausal women, HRT results in improved flow-mediated dilation only among women with no cardiovascular risk factors [99, 100]. Studies indicate that estrogen activates eNOS activity through Akt pathway, thereby promoting NO production in heart [87]. Moreover, estrogen mediated Akt activation results in eNOS activation in cultured human endothelial cells [77] and in intact elastic and muscular arteries in vivo [101]. Upregulation of PI3K/Akt by administration of E2 results in endothelial nitric oxide synthase activation via a transcription-independent mechanism [77]. Localization and activity of eNOS are regulated by making a complex with a chaperone protein, heat shock protein 90 and caveolin-3 in cardiomyocytes, especially in caveolae [102]. I also found significantly increased expression of the eNOS regulatory proteins like HSP-90 and caveolin-3 in the hearts of the OVX rats and pressure overload downregulated their expression on the OVX-PO rats [2]. More extensive studies are required to determine temporal changes and immunohistochemical localization of HSP-90 and eNOS after OVX-PO.
Under physiological conditions, myosin light chain (MLC) phosphorylation correlates with increased maximum tension or dp/dt values [103]. In the postmenopausal hypertrophy rat model, I found that phosphorylation was markedly increased only in OVX-PO rats. By contrast, total MLC content was significantly decreased in OVX-PO rats. The increased MLC phosphorylation/total MLC ratio was likely associated in part with an increase in ± dP/dtmax in heart contractile function [2]. Nitric oxide in vascular smooth muscle activates soluble guanylyl cyclase (sGC) to increase cGMP formation, thereby leading to a decrease in [Ca2+]i with subsequent inhibition of myosin light chain phosphorylation and contraction [104, 105]. My data suggest that ovariectomy followed by pressure overload subsequently increases MLC phosphorylation accompanied by increased cardiac contractility, as observed particularly in OVX-PO rats (Fig. 9.5).

Putative mechanism of VO(OPT) mediated cardioprotection. In normal female heart, eNOS is localized to the caveolae through its interaction with caveolin 3 and compartmentalized with L-type Ca2+ channel and b-aderenergic receptor. Activation of Akt signaling ultimately leads to eNOS phosphorylation, eNOS activation and thereby activates nitric oxide signaling pathways. The resulting combination of these effects subsequently confers ventricular dilation and cardioprotection. Pressure overload-induced hypertrophy severely impairs eNOS and Akt signaling pathways, thereby imbalances the NO mediated cardioprotective action. Treatment with VO(OPT) activate the Akt activity and enhances Akt mediated eNOS activity and subsequently confers cardioprotection against myocardial hypertrophy
Detailed in vivo and in vitro studies suggest that underlying mechanisms of estrogen mediated cardioprotection against myocardial hypertrophy are multifold. Estradiol-mediated activation of PI3K signaling blocks angiotensin II- or endothelin-1-initiated cardiomyocyte hypertrophy in vitro via upregulation of the gene encoding modulatory calcineurin-interacting protein (MCIP), a calcineurin antagonist. 17b-estradiol stimulates both the transcriptional transactivation and mRNA stability of the MCIP gene. Interestingly, Estradiol-induced PI3K signaling to MCIP up-regulation does not occur through AKT, as indicated by silencing Akt [106]. As the MCIP1 gene utilizes four different promoters, in a cell context-specific fashion, and so a detailed in vivo analysis is required to further clarify this effect.
5 Conclusion
Despite numerous studies on postmenopausal cardiac remodeling in hypertension and hypertrophy, we are just beginning to understand the pathophysiology of postmenopausal cardiovascular diseases. Particularly in light of the disappointing cardioprotective results obtained in several clinical trials with hormone replacement therapy, the mechanisms underlying estrogen-mediated cardioprotective action merit further study. In normal female heart, estrogen-mediated increased Akt-eNOS signaling restores the impaired nitric oxide signaling pathways and subsequently confers cardioprotection. In our postmenopausal model of cardiac hypertrophy by ovariectomy and pressure overload, impaired Akt-eNOS signaling imbalances the physiological protective mechanism by nitric oxide. Therefore, menopause likely accounts for cardiac decompensation against chronic stress through impairment of functions of eNOS and Akt signaling, and showed increased mortality following acute cardiac stress caused by chronic β-adrenergic stimulation. The novel model of postmenopausal cardiac decompensation using OVX-PO rats and elucidation of the mechanisms of detrimental cardiac remodeling are attractive for testing cardioprotective drugs in hypertension-induced cardiac injury in postmenopausal women. Furthermore, Akt/eNOS signaling is central dogma of cardiac remodeling following ischemia and hypertension and vanadium compound therapy instead of HRT is clinically relevant for cardioprotection not only in ischemic injury but also postmenopausal heart disease.
References
Bhuiyan MS, Shibuya M, Shioda N, Moriguchi S, Kasahara J, Iwabuchi Y, Fukunaga K (2007) Cytoprotective effect of bis(1-oxy-2-pyridinethiolato)oxovanadium (IV) on myocardial ischemia/reperfusion injury elicits inhibition of Fas ligand and Bim expression and elevation of FLIP expression. Eur J Pharmacol 571:180–188
Bhuiyan MS, Shioda N, Fukunaga K (2007) Ovariectomy augments pressure overload-induced hypertrophy associated with changes in Akt and nitric oxide synthase signaling pathways in female rats. Am J Physiol Endocrinol Metab 293:E1606–E1614
Bhuiyan MS, Takada Y, Shioda N, Moriguchi S, Kasahara J, Fukunaga K (2008) Cardioprotective effect of vanadyl sulfate on ischemia/reperfusion-induced injury in rat heart in vivo is mediated by activation of protein kinase B and induction of FLICE-inhibitory protein. Cariovasc Ther 26:10–23
Bhuiyan MS, Shioda N, Shibuya M, Iwabuchi Y, Fukunaga K (2009) Activation of endothelial nitric oxide synthase by a vanadium compound ameliorates pressure overload-induced cardiac injury in ovariectomized rats. Hypertension 53(1):57–63
Oudit GY, Sun H, Kerfant BG, Crackower MA, Penninger JM, Backx PH (2004) The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J Mol Cell Cardiol 37:449–471
Hanada M, Feng J, Hemmings BA (2004) Structure, regulation and function of PKB/AKT: a major therapeutic target. Biochim Biophys Acta 1697:3–16
Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ (2001) Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem 276:38349–38352
Yang ZZ, Tschopp O, Hemmings-Mieszczak M, Feng J, Brodbeck D, Perentes E, Hemmings BA (2003) Protein kinase B alpha/Akt1 regulates placental development and fetal growth. J Biol Chem 278:32124–32131
Woulfe D, Jiang H, Morgans A, Monks R, Birnbaum M, Brass LF (2004) Defects in secretion, aggregation, and thrombus formation in platelets from mice lacking Akt2. J Clin Invest 113:441–450
Dorn LLGW, Force T (2005) Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest 115:527–537
Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard M, Force TL, Franke TF, Hajjar RJ, Rosenzweig A (2001) Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 104:330–335
Ou H, Yan L, Mustafi D, Makinen MW, Brady MJ (2005) The vanadyl (VO2+) chelate bis(acetylacetonato)oxovanadium(IV) potentiates tyrosine phosphorylation of the insulin receptor. J Biol Inorg Chem 10:874–886
Theberge JF, Mehdi MZ, Pandey SK, Srivastava AK (2003) Prolongation of insulin-induced activation of mitogen-activated protein kinases ERK 1/2 and phosphatidylinositol 3-kinase by vanadyl sulfate, a protein tyrosine phosphatase inhibitor. Arch Biochem Biophys 420:9–17
Mehdi MZ, Srivastava AK (2005) Organo-vanadium compounds are potent activators of the protein kinase B signaling pathway and protein tyrosine phosphorylation: mechanism of insulinomimetics. Arch Biochem Biophys 440:158–164
Takada Y, Hashimoto M, Kasahara J, Aihara K, Fukunaga K (2004) Cytoprotective effect of sodium orthovanadate on ischemia/reperfusion-induced injury in the rat heart involves Akt activation and inhibition of fodrin breakdown and apoptosis. J Pharmacol Exp Ther 311: 1249–1255
Liem DA, Gho CC, Gho BC, Kazim S, Manintveld OC, Verdouw DD, Duncker DJ (2004) The tyrosine phosphatase inhibitor Bis(Maltolato)oxovanadium attenuates myocardial reperfusion injury by opening ATP-sensitive potassium channels. J Pharmacol Exp Ther 309:1256–1262
Fryer RM, Schultz JE, Hsu AK, Gross GJ (1999) Importance of PKC and tyrosine kinase in single or multiple cycles of preconditioning in rat hearts. Am J Physiol 276:1229–1235
Przyklenk K, Kloner RA (1998) Ischemic preconditioning: exploring the paradox. Prog Cardiovasc Dis 40:517–547
Vahlhaus C, Schulz R, Post H, Rose J, Heusch G (1998) Prevention of ischemic preconditioning only by combined inhibition of protein kinase C and protein tyrosine kinase in pigs. J Mol Cell Cardiol 30:197–209
Sakurai H, Shimomura S, Fukuzawa K, Ishizu K (1980) Detection of oxovanadium (IV) and characterization of its ligand environment in subcellular fractions of the liver of rats treated with pentavalent vanadium (V). Biochem Biophys Res Commun 96:293–298
Shi X, Dalal SN (1992) Superoxide-independent reduction of vanadate by rat liver microsomes/NAD(P)H: vanadate reductase activity. Arch Biochem Biophys 295:70–75
Elberg G, Li J, Shechter Y (1994) Vanadium activates or inhibits receptor and non-receptor protein tyrosine kinases in cell-free experiments, depending on its oxidation state. J Biol Chem 269:9521–9527
Willsky GR, White DA, McCabe BC (1984) Metabolism of added orthovanadate to vanadyl and high-molecular-weight vanadates by Saccharomyces cerevisiae. J Biol Chem 259: 13273–13281
Huyer G, Liu S, Kelly J, Moffat J, Payette P, Kennedy B, Tsaprailis G, Gresser MJ, Ramachandran C (1997) Mechanism of inhibition of protein-tyrosine phosphatases by Vanadate and pervanadate. J Biol Chem 272:843–851
Reul BA, Amin SS, Buchet JP, Ongemba LN, Crans DC, Brichard SM (1999) Effect of vanadium complexes with organic ligands on glucose metabolism: a comparison study in diabetic rats. Br J Pharmacol 126:467–477
Strout HV, Vicario PP, Saperstein R et al (1989) The insulin-mimetic effect of vanadate is not correlated with insulin receptor tyrosine kinase activity or phosphorylation in mouse diaphragm in vivo. Endocrinology 124:1918–1924
Meyerovitch J, Farfel Z, Sack J et al (1987) Oral administration of vanadate normalizes blood glucose levels in streptozotocin-treated rats: characterization and mode of action. J Biol Chem 262:6658–6662
Srivastava AK (2000) Anti-diabetic and toxic effects of vanadium compounds. Mol Cell Biochem 206:177–182
Sakurai H, Sano H, Takino T, Yasui H (2000) An orally active antidiabetic vanadyl complex, bis(1-oxy-2-pyridinethiolato)oxovanadium(IV), with VO(S2O2) coordination mode; in vitro and in vivo evaluations in rats. J Inorg Biochem 80:99–105
Takeshita S, Kawamura I, Yasuno T et al (2001) Amelioration of insulin resistance in diabetic ob/ob mice by a new type of orally active insulin-mimetic vanadyl complex: bis(1-oxy-2-pyridinethiolato) oxovanadium (IV) with VO(S2O2) coordination mode. J Inorg Biochem 85:179–186
Fantus IG, Tsiani E (1998) Multifunctional actions of vanadium compounds on insulin signaling pathways: evidence for preferential enhancement of metabolic versus mitogenic effects. Mol Cell Biochem 182:109–119
Faure R, Vincent M, Dufour M et al (1995) Arrest at the G2/M transition of the cell cycle by protein-tyrosine phosphatase inhibition: studies on a neuronal and a glial cell line. J Cell Biochem 59:389–401
Djordjevic C (1995) Antitumor activity of vanadium compounds. Met Ions Biol Syst 31: 595–616
Liasko R, Kabanos TA, Karkabounas S et al (1998) Beneficial effects of a vanadium complex with cysteine, administered at low doses on benzo (alpha) pyrene-induced leiomyosarcomas in Wistar rats. Anticancer Res 18:3609–3613
Blondel O, Bailbe D, Portha B (1989) In vivo insulin resistance in streptozotocin diabetic rats – evidence for reversal following oral vanadate treatment. Diabetologia 32:185–190
Al-Bayati MA, Giri SN, Raah OG (1990) Time and dose response study of the effects of vanadate in rats: changes in blood cells, serum enzymes, protein, cholesterol, glucose, calcium and inorganic phosphate. J Environ Pathol Toxicol Oncol 10:206–213
Bishayee A, Cecchin F (1995) Time course effects of vanadium supplements on cytosolic reduced glutathione level and glutathione S-transferase activity. Biol Trace Elem Res 48: 275–285
Mongold JJ, Cros GH, Vian L et al (1990) Toxicological aspects of vanadyl sulphate on diabetic rats: effects on vanadium levels and pancreatic B-cell morphology. Pharmacol Toxicol 67:192–198
Goldfine AB, Simonson DC, Folli F et al (1995) In vivo and in vitro studies of vanadate in human and rodent diabetes mellitus. Mol Cell Biochem 153:217–231
Kawano T, Fukunaga K, Takeuchi Y, Morioka M, Yano S, Hamada J, Ushio Y, Miyamoto E (2001) Neuroprotective effect of sodium orthovanadate on delayed neuronal death after transient forebrain ischemia in gerbil hippocampus. J Cereb Blood Flow Metab 21: 1268–1280
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231–241
Burgering BMT, Kops GJPL (2002) Cell cycle and death control: long live Forkheads. Trends Biochem Sci 27:352–360
Fukunaga K, Ishigami T, Kawano T (2005) Transcriptional regulation of neuronal genes and its effect on neural functions: expression and function of forkhead transcription factors in neurons. J Pharmacol Sci 98:205–211
Shioda N, Han F, Moriguchi S, Fukunaga K (2007) Constitutively active calcineurin mediates delayed neuronal death through Fas-ligand expression via activation of NFAT and FKHR transcriptional activities in mouse brain ischemia. J Neurochem 102:1506–1517
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857–868
Skurk C, Maatz H, Kim HS, Yang J, Abid MR, Aird WC, Walsh K (2004) The Akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J Biol Chem 279:1513–1525
Suhara T, Mano T, Oliveira BE, Walsh K (2001) Phosphatidylinositol 3-Kinase/Akt signaling controls endothelial cell sensitivity to fas-mediated apoptosis via regulation of FLICE-inhibitory protein (FLIP). Circ Res 89:13–19
Rasper DM, Vaillancourt JP, Hadano S, Houtzager VM, Seiden I, Keen SL, Tawa P, Xanthoudakis S, Nasir J, Martindale D, Koop BF, Peterson EP, Thornberry NA, Huang J, MacPherson DP, Black SC, Hornung F, Lenardo MJ, Hayden MR, Roy S, Nicholson DW (1998) Cell death attenuation by ‘Usurpin’, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ 5:271–288
Imanishi T, Murry CE, Reinecke H, Hano T, Nishio I, Liles WC, Hofsta L, Kim K, O’Brien KD, Schwartz SM, Han DK (2000) Cellular FLIP is expressed in cardiomyocytes and down-regulated in TUNEL-positive grafted cardiac tissues. Cardiovasc Res 48:101–110
Gottilieb RA, Engler RL (1999) Apoptosis in myocardial ischemia–reperfusion. Ann NY Acad Sci 874:412–426
Belchetz PE (1994) Hormonal treatment of postmenopausal women. N Engl J Med 330: 1062–1071
Turgeon JL, McDonnell DP, Martin KA, Wise PM (2004) Hormone therapy: physiological complexity belies therapeutic simplicity. Science 304:1269–1273
Gorodeski GI (2002) Update on cardiovascular disease in post-menopausal women. Best Pract Res Clin Obstet Gynaecol 16:329–355
Mosca L, Manson JE, Sutherland SE, Langer RD, Manolio T, Barrett-Connor E (1997) Cardiovascular disease in women: a statement for healthcare professionals from the American Heart Association. Writing Group. Circulation 96:2468–2482
Kannel WB (1983) Prevalence and natural history of electrocardiographic left ventricular hypertrophy. Am J Med 3:4–11
Howe HL (1984) Age-specific hysterectomy and oophorectomy prevalence rates and the risks for cancer of the reproductive system. Am J Public Health 74:560–563
Howard BV, Kuller L, Langer R, Manson JE, Allen C, Assaf A, Cochrane BB, Larson JC, Lasser N, Rainford M, Van Horn L, Stefanick ML, Trevisan M (2005) Risk of cardiovascular disease by hysterectomy status, with and without oophorectomy: the Women’s Health Initiative Observational Study. Circulation 111: 1462–1470
Melton LJ 3rd, Bergstralha QEJ, Malkasian GD, O’Fallon WM (1991) Bilateral oophorectomy trends in Olmsted County, Minnesota, 1950–1987. Epidemiology 2:149–152
Whiteman MK, Hillis SD, Jamieson DJ, Morrow B, Podgornik MN, Brett KM, Marchbanks PA (2008) Inpatient hysterectomy surveillance in the United States, 2000–2004. Am J Obstet Gynecol 198(34):e1–e7
Keshavarz H, Hillis SD, Kieke BA, Marchbanks PA (2002) Hysterectomy surveillance-United States, 1994–1999. MMWR Surveill Summ 51:1–8
Shuster LT, Gostout BS, Grossardt BR, Rocca WA (2008) Prophylactic oophorectomy in pre-menopausal women and long term health- a review. Menopause Int 14:111–116
Lobo RA (2007) Surgical menopause and cardiovascular risks. Menopause 14:562–566
Lokkegaard E, Jovanovic Z, Heitmann BL, Keiding N, Ottesen B, Pedersen AT (2006) The association between early menopause and risk of ischaemic heart disease: influence of hormone therapy. Maturitas 53:226–233
Rossouw JE, Prentice RL, Manson JE, Wu L, Barad D, Barnabei VM, Ko M, LaCroix AZ, Margolis KL, Stefanick ML (2007) Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA 297:1465–1477
Dubey RK, Imthurn B, Barton M, Jackson EK (2005) Vascular consequences of menopause and hormone therapy: importance of timing of treatment and type of estrogen. Cardiovasc Res 66:295–306
Leinwand LA (2003) Sex is a potent modifier of the cardiovascular system. J Clin Invest 112:302–307
Hayward CS, Kelly RP, Collins P (2000) The roles of gender, the menopause and hormone replacement on cardiovascular function. Cardiovasc Res 46:28–49
Patten RD, Pourati I, Aronovitz MJ, Baur J, Celestin F, Chen X, Michael A, Haq S, Nuedling S, Grohe C, Force T, Mendelsohn ME, Karas RH (2004) 17beta-estradiol reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signaling. Circ Res 95:692–699
Van Eickels M, Grohe C, Cleutjens JP, Janssen BJ, Wellens HJ, Doevendans PA (2001) 17beta-estradiol attenuates the development of pressure-overload hypertrophy. Circulation 104:1419–1423
Thorndike EA, Turner AS (1998) In search of an animal model for postmenopausal diseases. Front Biosci 3:c17–c26
Fang Z, Carlson S, Chen Y, Oparil S, Wyss J (2001) Estrogen depletion induces NaCl-sensitive hypertension in female spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol 281:R1934–R1939
Peng N, Clark J, Wei C, Wyss J (2003) Estrogen depletion increases blood pressure and hypothalamic norepinephrine in middle-aged spontaneously hypertensive rats. Hypertension 41:1164–1167
Fortepiani LA, Zhang H, Racusen LC, Roberts LJ II, Reckelhoff JF (2003) Characterization of an animal model of postmenopausal hypertension in SHR. Hypertension 41:640–645
Hinojosa-Laborde C, Craig T, Zheng W, Ji H, Haywood JR, Sandberg K (2004) Ovariectomy augments hypertension in aging female Dahl salt sensitive rats. Hypertension 44:405–409
Javeshghani D, Touyz R, Sairam M, Virdis A, Neves M, Schiffrin E (2003) Attenuated responses to angiotensin II in follitropin receptor knockout mice, a model of menopause-associated hypertension. Hypertension 42:761–767
Bhuiyan MS, Fukunaga K (2010) Characterization of an animal model of post menopausal cardiac hypertrophy and novel mechanisms responsible for cardiac decompensation using ovariectomized pressure-overloaded rats. Menopause 17:213–221
Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, Sessa WC, Bender JR (2000) Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res 87:677–682
Ciric O, Susic D (1980) Effect of isoprenaline on blood pressure and heart rate in different phases of the oestrous cycle. Endokrinologie 76:274–278
Thawornkaiwong A, Preawnim S, Wattanapermpool J (2003) Upregulation of beta 1-adrenergic receptors in ovariectomized rat hearts. Life Sci 72:1813–1824
Kam KW, Kravtsov GM, Liu J, Wong TM (2005) Increased PKA activity and its influence on isoprenaline-stimulated L-type Ca2+ channels in the heart from ovariectomized rats. Br J Pharmacol 144:972–981
Cambotti LJ, Cole FE, Gerall AA, Frolich ED, Macphee AA (1984) Neonatal gonadal hormones and blood pressure in the spontaneously hypertensive rat. Am J Physiol 247: E258–E264
Crofton JT, Share L (1997) Gonadal hormones modulate deoxycorticosterone-salt hypertension in male and female rats. Hypertension 29:494–499
David FL, Carvalho MHC, Cobra ALN, Nigro D, Fortes ZB, Rebouças NA, Tostes RCA (2001) Ovarian hormones modulate endothelin-1 vascular reactivity and mRNA expression in DOCA-salt hypertensive rats. Hypertension 38:692–696
Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K (2000) The HMG-CoA reductase inhibitor simvastatin activates the protein-kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med 6:1004–1010
Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K (2000) Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation 101:660–667
Kemi OJ, Celi M, Wisloff U, Grimaldi S, Gallo P, Smith GL, Condorelli G, Ellingsen O (2008) Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy. J Cell Physiol 214:316–321
Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK (2000) Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3′-OH kinase. Nature 407:538–541
Camper-Kirby D, Welch S, Walker A, Shiraishi I, Setchell KD, Schaefer E, Kajstura J, Anversa P, Sussman MA (2001) Myocardial Akt activation and gender: increased nuclear activity in females versus males. Circ Res 88:1020–1027
Ren J, Hintz KK, Roughead ZK, Duan J, Colligan PB, Ren BH, Lee KJ, Zeng H (2003) Impact of estrogen replacement on ventricular myocyte contractile function and protein kinase B/Akt activation. Am J Physiol Heart Circ Physiol 284:H1800–H1807
Wang M, Wang Y, Weil B, Abarbanell A, Herrmann J, Tan J, Kelly M, Meldrum DR (2009) Estrogen receptor beta mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am J Physiol Regul Integr Comp Physiol 296:R972–R978
Hsu JT, Kan WH, Hsieh CH, Chowdhry MA, Bland KI, Chaudry IH (2009) Mechanism of salutary effects of estrogen on cardiac function following trauma-hemorrhage: Akt-dependent HO-1 up-regulation. Crit Care Med 37:1–7
Chen Z, Yuhanna IS, Galcheva-Gargova Z, Karas RH, Mendelsohn ME, Shaul PW (1999) Estrogen receptor alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J Clin Invest 103:401–406
Chambliss KL, Yuhanna IS, Mineo C, Liu P, German Z, Sherman TS, Mendelsohn ME, Anderson RG, Shaul PW (2000) Estrogen receptor alpha and endothelial nitric oxide synthase are organized into a functional signaling module in caveolae. Circ Res 87:e44–e52
Grasselli ANS, Colussi C, Aiello A, Benvenuti V, Moretti F, Sacchi A, Bacchtt S, Gaetano C, Capogrossi MC, Pontecrvi A, Farsetti A (2008) Estrogen receptor alpha and endothelial nitric oxide synthase nuclear complex regulates transcription of human teromerase. Circ Res 103:34–42
Majmudar NG, Robson SC, Ford GA (2000) Effects of the menopause, gender, and estrogen replacement therapy on vascular nitric oxide activity. J Clin Endocrinol Metab 85:1577–1583
Higashi Y, Sanada M, Sasaki S, Nakagawa K, Goto C, Matsuura H, Ohama K, Chayama K, Oshima T (2001) Effect of estrogen replacement therapy on endothelial function in peripheral resistance arteries in normotensive and hypertensive postmenopausal women. Hypertension 37:651–657
Virdis A, Ghiadoni L, Pinto S, Lombardo M, Petraglia F, Gennazzani A, Buralli S, Taddei S, Salvetti A (2000) Mechanisms responsible for endothelial dysfunction associated with acute estrogen deprivation in normotensive women. Circulation 101:2258–2263
Sanada M, Higashi Y, Nakagawa K, Tsuda M, Kodama I, Kimura M, Chayama K, Ohama K (2002) Hormone replacement effects on endothelial function measured in the forearm resistance artery in normocholesterolemic and hypercholesterolemic postmenopausal women. J Clin Endocrinol Metab 87:4634–4641
Herrington DM (1995) Dehydroepiandrosterone and coronary atherosclerosis. Ann NY Acad Sci 774:271–280
Kelemen M, Vaidya D, Waters DD, Howard BV, Cobb F, Younes N, Tripputti M, Ouyang P (2005) Hormone therapy and antioxidant vitamins do not improve endothelial vasodilator function in postmenopausal women with established coronary artery disease: a substudy of the Women’s Angiographic Vitamin and Estrogen (WAVE) trial. Atherosclerosis 179: 193–200
Guo X, Razandi M, Pedram A, Kassab G, Levin ER (2005) Estrogen induces vascular wall dilation: mediation through kinase signaling to nitric oxide and estrogen receptors a and b. J Biol Chem 280:19704–19710
Fulton D, Gratton J-P, Sessa WC (2001) Post-translational control of endothelial nitric oxide synthase: why isn’t calcium/calmodulin enough? J Pharmacol Exp Ther 299:818–824
Solaro RJ (1986) Protein phosphorylation and cardiac myofilaments. In: Solaro RJ (ed) Protein phosphorylation in heart muscle. CRC, Boca Raton, pp 129–156
Pabla R, Curtis MJ (1996) Effect of endogenous nitric oxide on cardiac systolic and diastolic function during ischemia and reperfusion in the rat isolated perfused heart. J Mol Cell Cardiol 28:2111–2121
Schmidt HHHW, Lohmann SM, Walter U (1993) The nitric oxide and cGMP signal transduction system: regulation and mechanism of action. Biochim Biophys Acta 1178: 153–175
Pedram A, Razandi M, Aitkenhead M, Levin ER (2005) Estrogen inhibits cardiomyocyte hypertrophy in vitro. Antagonism of calcineurin-related hypertrophy through induction of MCIP1. J Biol Chem 280:26339–26348
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Fukunaga, K., Bhuiyan, M.S. (2012). Cardiovascular Protection with Vanadium Compounds. In: Michibata, H. (eds) Vanadium. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-0913-3_9
Download citation
DOI: https://doi.org/10.1007/978-94-007-0913-3_9
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-0912-6
Online ISBN: 978-94-007-0913-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)