
Abstract
Lung cancer is the leading cause of cancer death globally. Although molecularly targeted agents have made small advances in the treatment options for patients, the overall 5-year survival rate has changed little in the past several decades, necessitating a greater understanding of the biology driving tumor progression and metastasis. MicroRNAs (miRNAs) are a relatively recently discovered class of non-protein coding RNAs that modulate extremely important cellular functions via their post-transcriptional regulation of mRNAs. Recent evidence from multiple tumor types and model systems implicates miRNA dysregulation as a common mechanism of tumorigenesis and progression. This represents a rapidly emerging and changing field with new biological connections and applications being reported each month, which provide unique insights into miRNA functions and potential new approaches for diagnosis and therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Epidermal Growth Factor Receptor
- Epidermal Growth Factor Receptor Mutation
- Epidermal Growth Factor Receptor Activation
- Epidermal Growth Factor Receptor mRNA
- Human NSCLC Cell Line
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
2.1 Introduction
Lung cancer is the leading cause of cancer-related death in the United States and worldwide (Jemal et al. 2010; Kamangar et al. 2006). In the year 2010, it is estimated that approximately 222,520 new cases will be diagnosed and 157,300 deaths will occur in the United States alone. Only 16% of patients diagnosed with lung cancer are alive at 5 years (Jemal et al. 2010; Kamangar et al. 2006), because at the time of diagnosis more than 70% of patients are found to have advanced disease that is not amenable to curative therapy (Goldstraw et al. 2007) and among the remaining patients who undergo surgical resection with curative intent for early-stage disease, there is a high rate of recurrence. Given the generally poor prognosis of patients with this disease, better treatments are needed based upon the molecular events driving tumor progression and metastasis.
Lung cancer is broadly divided into non-small cell lung cancer (NSCLC), which arises from epithelial cells of the airways and accounts for 85% of cases, and small cell lung cancer (SCLC), which is a neuroendocrine tumor that comprises the remainder. Non-small cell lung cancer is further divided into several major histologic subtypes, including adenocarcinoma, squamous cell carcinoma, and large cell. Although significant work has gone into identifying and studying the oncogenes responsible for tumorigenesis and progression in NSCLC and SCLC, there has been limited success in translating these findings into the therapeutic setting or in using this information to better stratify patient risk and select treatments.
MicroRNAs (miRNAs) are a class of highly conserved non-coding RNAs, 19–25 nucleotides in length, that regulate gene expression by recognition and binding of the 3′ untranslated regions (3′UTRs) of mRNAs. Mature miRNAs form stem-loop structures with sequences partially complementary to their target mRNAs, which alter mRNA stability or the efficiency of translation. Based upon recent re-evaluation of the miRNAs in the mouse genome by high-throughput sequencing, there are ~500 confidently identified murine miRNAs, ~300 of which are conserved in mammals (Chiang et al. 2010), and it is estimated that approximately one-third of the genome is regulated by miRNAs (Lewis et al. 2005). Interestingly, since each miRNA can regulate the expression of large sets of targets and any one gene can be regulated by multiple miRNAs, a rich network of regulatory fine-tuning is at play that can go awry in pathologic states such as cancer. Given this powerful biology, various miRNAs have been implicated as either tumor suppressors or oncogenes (“oncomirs”) in many different tumor types. Genomically miRNAs are frequently found to be at fragile sites in the human genome (Calin et al. 2004), but there are myriad additional mechanisms by which the miRNAs can become dysregulated in cancer. This chapter will outline our understanding of miRNA biology in lung cancer development and metastasis.
2.2 MiRNA Profiling of Lung Cancer and Clinical Application
MiRNAs have the ability to post-transcriptionally regulate large sets of genes, and as such their expression levels should faithfully represent the overall biologic state of tumor cells. In fact, miRNA signatures segregate samples of different tumor types better than mRNA signatures (Lu et al. 2005), suggesting that they may be useful biomarkers of the unique underlying set of molecular events driving tumorigenesis and metastasis (Cho 2010). This idea has raised significant research interest in identification of oncogenic drivers of tumor progression, and in the development of miRNA signatures that would be clinically prognostic of outcome in early-stage lung cancer. Such signatures could be useful for stratification of patient risk for recurrence, helping to inform the decision of who should receive adjuvant treatment after surgery, or predictive of which tumors will respond to particular chemotherapeutic agents. Several different approaches have been utilized in these efforts, including comparison of tumor samples to matched non-cancerous lung tissue with the aim of identifying pathways driving tumorigenesis, and the use of miR profiling across a large panel of tumors to derive multi-miR signatures for classification of patient risk for recurrence from this heterogeneous group (Table 2.1).
Profiling of miRNA expression in 104 NSCLC tumors versus matching non-cancerous lung tissue demonstrated that 43 miRs were differentially expressed, including 15 up-regulated (e.g. miR-21, miR-191, miR-155, and miR-17-3p) and 28 down-regulated (e.g. let-7a-2 and miR-145) (Yanaihara et al. 2006). The authors specifically demonstrated that high miR-155 and low let-7a-2 levels correlated with survival in multiple independent sets of patients with adenocarcinoma. Many of these changes were similar in a separate study of multiple solid tumor types versus normal tissue, which included 123 NSCLC cases, along with breast, colon, gastric, endocrine pancreatic, and prostate cancers (Volinia et al. 2006). These data demonstrate the large number of differences seen in tumors versus normal tissue, likely representing both the biological heterogeneity of NSCLC and the different stages of cancer being analyzed. Despite this complexity, these analyses have clearly identified multiple interesting miRNAs involved in tumorigenesis and progression.
An alternative approach to miRNA profiling of tumor tissues is to measure miR levels in relation to outcome, without explicit comparison to normal or uninvolved tissue. This approach has principally been applied in lung cancer to address the question of whether miRNA signatures can be developed to stratify early-stage patients (those who have undergone surgical resection) into high or low risk for early recurrence or metastasis. By using either microarray or quantitative real-time PCR, multiple studies have demonstrated the ability to stratify patient risk for recurrence or survival based upon multi-miR classifiers, and often independently of the clinical staging (Patnaik et al. 2009; Yu et al. 2008). One study approached the specific question of whether there is a miR signature in squamous cell carcinoma of the lung (Raponi et al. 2009). Interestingly, this study identified several miRNA families that have also been found to be altered in other studies of NSCLC, in which there was strong representation of adenocarcinoma samples (Table 2.1). An alternative approach infers the miRNA networks involved from the changes in mRNA expression data (Liang 2008). Using this methodology, reduced expression of miR-34 family members was identified in multiple datasets. Currently it is unclear how to use these signatures clinically, and whether they are true biomarkers of the disease, linked with the underlying pathways driving the tumor biology, or simply epiphenomena. However, as our understanding of the miRNA/mRNA target biology advances, this approach may be clinically useful in understanding how to assign risk for recurrence in individual patients.
A final way in which tumor miR profiling data may become clinically useful is in the selection of chemotherapeutic agents for patients. A recent study correlated measured drug sensitivity across the panel of drugs in the NCI Developmental Therapeutics Program drug screen with both miRNA and mRNA expression in the NCI-60 panel of cell lines (Liu et al. 2010a). They demonstrated strong associations between the sensitivity to drugs with known mechanisms of action and the patterns of miRNA and mRNA expression. This type of analysis might provide a miRNA-based molecular pharmacologic classification for tumors that would be helpful in either trial design or personalized selection of drugs for individual patients.
In thinking about how to incorporate these predictive or prognostic signatures into standard clinical practice or investigational trial design, certain practical considerations must be kept in mind. MiRNAs may be ideal for these types of studies, especially in comparison to mRNA-based studies, owing to their relative stability against degradation in formalin-fixed paraffin-embedded tissues. However, there is currently no gold standard methodology for the evaluation of miRNA levels from samples. Some investigators have reported poor overall correlation between high-throughput microarray-based techniques and quantitative real-time reverse transcription-PCR (qRT-PCR), which is more commonly used in clinical laboratories (Koshiol et al. 2010; Liu et al. 2010b). Alternate methods, such as cloning or in situ hybridization are technically challenging, time consuming and/or expensive for adoption by clinical laboratories. As the availability of high-throughput sequencing technology continues to spread, with the attendant decrease in cost, this may well become an appropriate standard that could be adopted by clinical labs. Finally, consideration must be given to the fact that diagnostic biopsy specimens, either from bronchoscopy or from fine needle biopsies, frequently provide relatively small amounts of tissue. These may be insufficient for standard pathologic analysis of stained slides, newer molecular tests such as epidermal growth factor receptor (EGFR) or KRAS mutational status, and miRNA analysis. One potential solution to this issue would be the measurement of miRNAs from patient serum or plasma, which would provide the added clinical advantage of being able to measure repeatedly during the course of treatment or in post-treatment surveillance. Several studies have reported the differential expression of miRNAs in patient serum that correlates with disease or outcome, including one study in a group of 303 early-stage NSCLC patients (Chen et al. 2008; Ng et al. 2009; Hu et al. 2010). Using high-throughput sequencing on miRNAs derived from serum in a cohort of Chinese patients versus healthy donor controls, a four-miRNA signature (miR-486, miR-30d, miR-1, and miR-499) was derived that was prognostic for patients with a shorter median survival and increased risk for death (Hu et al. 2010). Such studies provide a starting point for trials of how to incorporate these potential biomarkers into clinical decision-making, but much work still remains to be done before circulating miRNA levels become a standard clinical tool in the diagnosis and management of cancer patients.
2.3 Pathogenesis
Lung tumorigenesis is frequently related to changes induced in the epithelium of the airway by tobacco or carcinogen exposure. Recent studies to identify the link between tobacco exposure and changes in miRNA expression have been performed in rodents (both mouse and rat) and in chronic smokers (Izzotti et al. 2009a, b; Schembri et al. 2009). In all three studies it was observed that the majority of changes involved down-regulation of miRNAs, which for the rodent studies correlated strongly with the changes in mRNA and protein documented from prior work using the same model system (Izzotti et al. 2009a, b). Additionally, measurement of the miRNA levels proved to be an extremely sensitive marker of tobacco exposure, producing down-regulation in 126 out of 484 measured miRNAs (26%). Comparison of the results from the mouse and rat studies found involvement of the same miR pathways, with 13 of 15 miRs down-regulated at least twofold in the mouse lungs also significantly down-regulated in rat (including the let-7 family members, miR-34b, miR-30b, miR-30c, and miR-125). Although the overlap between the rodent samples and human were more limited (e.g. miR-30, miR-99, and miR-125), there are likely differences related to the pathology of cells analyzed (mixed cell population of whole animal lungs versus relatively pure human bronchial epithelial cell population), chronicity of exposure (4 week exposure in animals versus chronic human smokers with an average of 18.8 pack years) and species. The human data also clearly indicates that changes in a relatively small number of miRs could account for a large percentage of the documented smoking-associated mRNA changes (Schembri et al. 2009).
Smoking is the single most important risk factor for the development of lung cancer. However, 10–15% of cases of NSCLC occur in never smokers, corresponding to approximately 20,000 deaths annually and making this subcategory one of the top 10 causes of cancer mortality (Samet et al. 2009). This is an incompletely understood class of patients that has received increasing focus in recent years, especially given the findings that never-smokers have a higher incidence of activating mutation in the EGFR and are therefore more likely to benefit from the approved tyrosine kinase inhibitors (Engelman and Janne 2008). To assess the role of miRNA changes in this unique group of patients, one study compared the global miRNA expression profile from tumors in smokers versus never-smokers. Among the differences identified, one of the most significant changes was the increase in levels of miR-21, which strongly correlated with mutation in EGFR (Seike et al. 2009). The authors further demonstrated that EGFR activation drives miR-21 expression and that antisense targeting of miR-21 enhanced the apoptotic response induced by EGFR tyrosine kinase inhibition. A separate study by Cho and coworkers found similar results for the levels of miR-21 in a small panel of non-smoking patients with lung adenocarcinoma, along with changes in several other miRs, including miR-145, miR-126 *, miR-182, miR-183, and miR-210 (Cho et al. 2009).
2.4 Regulation of Known Oncogenes in Lung Cancer
2.4.1 Let-7, RAS, c-Myc and HMGA2
The RAS proto-oncogene family plays a central role in the growth factor receptor signaling pathways and is found to have an activating mutation in many epithelial tumor types. Approximately 30% of human NSCLC cases have mutation of the KRAS gene, frequently associated with a history of tobacco exposure. In genetic mouse models an activating mutation in KRAS produces lung adenocarcinoma, the most common histologic subtype of lung cancer, with differing propensities to invade and metastasize (Fisher et al. 2001; Jackson et al. 2001; 2005; Johnson et al. 2001; Liu et al. 2000; Olive et al. 2004; Zheng et al. 2007). One of the first miRNAs to be identified and one of the best studied to date is let-7, which was originally identified as a gene responsible for regulating temporally-specific developmental changes in C. elegans (Reinhart et al. 2000). The let-7 family contains at least nine members and is highly conserved across species (Pasquinelli et al. 2000). In C. elegans it was demonstrated that let-7 regulates let-60/RAS and inversely correlates with the altered RAS levels in human NSCLC tumors, providing clear evidence that miRNAs can act as tumor suppressors (Johnson et al. 2005) (Table 2.2). Several studies also demonstrated that loss of let-7 expression in surgically resected tumor specimens of NSCLC is prognostic of survival, regardless of the pathologic staging (Takamizawa et al. 2004; Yanaihara et al. 2006). Using multiple in vivo models of NSCLC development, re-expression of let-7 suppressed tumorigenesis in a manner that was largely dependent upon modulation of the Ras levels (Kumar et al. 2008). In addition, let-7 has been demonstrated to down-regulate MYC and revert MYC-induced growth in Burkitt’s lymphoma cells (Sampson et al. 2007), a finding that may have relevance to NSCLC and which further demonstrates how the loss in expression of a single miRNA can activate multiple cooperative oncogenic pathways.
As mentioned in the section on miRNA changes in response to tobacco exposure, let-7 suppression can occur with relatively acute exposure to tobacco smoke, but multiple potential mechanisms have been proposed based upon analysis of human cancer cell lines, tissue specimens, and developmental studies (Boyerinas et al. 2010). One of the most intriguing mechanisms was described in a study of single nucleotide polymorphisms (SNPs) in the 3′UTR of KRAS, which found that the prevalence of a variant allele containing an altered let-7 complementary site was higher in a cohort of patients with NSCLC (20.3%) than in the general population (7.6% in a population of European descent) (Chin et al. 2008). Interestingly, the variant allele predicted increased risk for NSCLC in patients with a moderate smoking history (< 40 pack years) in multiple independent patient cohorts. This data suggests a synergy in miR regulation of oncogenes between the host genetic background and exposure history up to an exposure threshold, past which the exposure becomes the dominant tumorigenic factor.
Finally, from analyses of the NCI-60 cell line panel and ovarian cancer patient samples, it was demonstrated that let-7 loss correlated with the expression of markers for less differentiated tumors, such as HMGA2 (Shell et al. 2007). Analysis of survival data from the patient samples clearly indicated that tumors with loss of let-7 were more likely to metastasize or be poorly responsive to treatment, producing striking differences in patient survival. HMGA2 is also an established epithelial-mesenchymal transition-inducing gene (Thuault et al. 2006), which would provide a reasonable link between the observed histological differences and a mechanism of tumor progression.
2.4.2 EGFR
The epidermal growth factor receptor, EGFR (ErbB1/Her-1), is a member of the ErbB family of growth factor receptor tyrosine kinases that is frequently found to be activated by mutation or amplification in epithelial malignancies. In lung cancer it is mutated in approximately 10% of patients in the United States and 30% of Asian patients, but is also frequently over-expressed at the protein level and/or has increased gene copy number (Gazdar 2010). Overall, blockade of EGFR activation has been the focus of tremendous efforts to develop oral tyrosine kinase inhibitors, e.g. erlotinib and gefitinib. Currently such medications are approved for use in NSCLC patients who have failed prior chemotherapy, and their use has been found to be of greatest benefit in patients whose tumors carry an activating EGFR mutation.
Multiple miRNAs have been implicated in the regulation of EGFR, including miR-128b, let-7 and miR-21 (as discussed in the previous section on pathogenesis). MiR-128b exists on chromosome 3p22, which is a region frequently deleted in lung cancer. A recent study evaluated whether miR-128b levels in human NSCLC cell lines regulated EGFR levels and demonstrated that miR-128b directly recognizes the 3′UTR of EGFR (Weiss et al. 2008). In an analysis of patient samples, the same group also showed that loss-of-heterozygosity at the locus containing miR-128b was frequent and strongly correlated with improved disease control with gefitinib treatment, producing improved overall survival (23.4 vs. 10.5 months, p = 0.02). These findings suggest that the loss of miR-128b may be useful as a predictive marker of response to treatment with EGFR inhibitors. Similarly, let-7 was found to regulate EGFR mRNA and protein levels in multiple different cancer cell lines, including lung, and to subsequently reduce signaling through the Akt pathway and decrease cell viability by an apoptosis-independent process (Webster et al. 2009).
2.4.3 p53 and MiR-34
The miR-34 family (miR-34a, miR-34b, and miR-34c) is frequently decreased in expression in solid tumors, including NSCLC (He et al. 2007b). The two genomic loci encoding the three family members each have a p53 binding site in their promoter, and their expression is induced in a p53-dependent fashion by oncogenic stress or DNA damage (He et al. 2007a), demonstrating that miR-34 is an effector in the p53 tumor suppressor network. In a cohort of 70 patients who underwent surgical resection for NSCLC, miR-34a and miR-34b levels were significantly repressed versus paired normal tissue, and mutations in p53 were much more frequent in cases with low miR-34a expression (Gallardo et al. 2009). The expression level of miR-34 in the patient samples was independently prognostic for relapse, while the combination of p53 mutational status and miR-34 expression level was an even more powerful prognosticator. This study also confirmed prior observations that miR-34 expression is frequently regulated by the methylation status of the promoter region (Lodygin et al. 2008).
2.4.4 Fus-1 and the 3p21.3 Deletion
The 3p21.3 region in the human genome has been associated with inhibition of tumor growth and progression, and within this locus the FUS1 gene (or tumor suppressor candidate 2, TUSC2) is a tumor suppressor that is lost in expression in 90–100% of cases (Zabarovsky et al. 2002). It is hypothesized that hemizygous deletion, coupled with additional epigenetic regulation, may account for the complete loss of expression of this region in lung tumors. In fact, it was recently demonstrated that expression levels were reduced or absent in 82% of non-small cell and 100% of small cell lung cancer specimens (Prudkin et al. 2008). This gene has been recently shown to be under the regulation of four different miRNAs, miR-93, miR-98, miR-197, and miR-378 (Du et al. 2009a; Lee et al. 2007).
2.4.5 MiR-17-92
The miR-17-92 cluster (also called oncomir-1) contains six miRNAs located at 13q31.3, a region that is frequently amplified in lymphoma and solid tumors (Mendell 2008), with concordant up-regulation of expression in many solid tumors, including lung (Volinia et al. 2006). Additionally, expression of the cluster is regulated by c-Myc and subsequently targets the transcription factor E2F1 (O’Donnell et al. 2005). This cluster is therefore proposed to act as a regulator of tumor cell growth/proliferation and apoptosis, dysregulation of which produces a phenotype of hyperproliferation. In fact, when the miR-17-92 locus was expressed from an early time point during development in a transgenic murine model, the animals developed hyperplasia of the lung epithelium, along with a block in epithelial cell differentiation, producing few primitive alveoli in the distal airways (Lu et al. 2007). Conversely, mice with a homozygous knockout of the miR-17-92 locus have significant hypoplasia of the lung, along with a ventricular septal defect, which produced 100% neonatal lethality (Ventura et al. 2008). Besides E2F1-3, the miR-17-92 cluster modulates other downstream targets such as PTEN, CDK4, and BIM (He et al. 2007a; Ventura et al. 2008; Xiao et al. 2008), providing multiple potential mechanistic explanations for a proliferative phenotype. Finally, targeting of miR-17-5p and miR-20a in lung cancer cell lines over-expressing the miR-17-92 locus induced apoptosis (Matsubara et al. 2007).
2.4.6 MiR-155
Despite the association of high miR-155 levels with poor outcome in NSCLC patients, it is currently unclear how miR-155 is involved in NSCLC pathogenesis. MiR-155 has been postulated to provide a pivotal link between chronic inflammatory states and cancer development (Tili et al. 2009). It has also been demonstrated to play an important role in the cellular reactivation of oncogenic viruses such as Epstein-Barr virus (EBV) and in modulating the anti-tumor effects of the bone morphogenetic protein pathway signaling (Yin et al. 2010). Most recently miR-155 has been shown to regulate components of the mismatch repair machinery in colon cancer (including MLH1, MSH2, and MSH6), with high expression resulting in a mutator phenotype and genomic instability (Valeri et al. 2010). Each of these biologic functions could conceivably play a role in lung cancer biology, but further studies will be necessary to elucidate the particular mechanisms at work.
With increasing frequency, other miRNAs are being identified as regulators of various oncogenic functions in human samples, cell lines and mouse models of non-small cell lung cancer, adding to the growing list of potential ways in which miRs regulate tumor initiation or progression. Some examples include miR-31-mediated down-regulation of the tumor suppressors LATS2 and PPP2R2A (Liu et al. 2010b) and the loss of regulation of the anti-apoptotic protein PED/PEA-15 by miR-212 (Incoronato et al. 2010). This later report highlights the fact that some of the changes in miR expression may directly affect the sensitivity of malignant cells to therapeutic intervention, which would clearly be useful in formulating treatment plans. It will be exciting to monitor the increasing list of roles for miRNA function in lung tumors and how this information can be incorporated into ways to personalize treatment for patients.
2.5 Invasion and Metastasis Progression
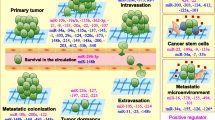
Tumorigenesis and metastasis are two inter-locking, multi-step processes. Because of their ability to simultaneously modulate many targets, miRNA dysregulation could certainly affect many of the independent steps necessary in the transformation to a tumorigenic and metastatic cell. Using the well-characterized RIP-Tag2 murine model of pancreatic neuroendocrine carcinoma, Hanahan’s group recently documented the miRNA changes associated with each of the discrete steps in carcinogenesis, from normal islets to hyperplasia, development of the angiogenic switch, followed by encapsulated tumor development, then invasive carcinoma and metastasis (Olson et al. 2009). This work highlighted that the observed miRNA changes correlate closely with the hallmark capabilities acquired by tumor cells during progression, and that pharmacologic anti-angiogenic therapy could alter the angiogenesis signature while invoking some of the changes observed in the metastatic signature. Several groups have also reported elegant mechanistic work in human breast cancer and murine model systems, demonstrating the role of multiple miRs during progression and metastasis, including miR-10b, miR-31, miR-126, and miR-335 (Ma et al. 2007; Tavazoie et al. 2008; Valastyan et al. 2009). However, in lung cancer much less is known about the role of miRNAs in invasion and metastasis.
Work by our group has demonstrated that a mutant p53 allele (p53 R172HΔG) confers metastatic potential to lung adenocarcinomas arising in mice due to a latent, somatically-activated Kras G12D allele (Zheng et al. 2007). mRNA expression profiling of metastatic versus matched lung tumors from this model revealed a signature of differentially expressed genes, that when applied to patient cohorts identified a subset of early-stage lung cancer patients with poor prognosis (Gibbons et al. 2009b). Subsequent work with this model identified epithelial-mesenchymal transition (EMT) as a critical step in metastasis, regulated by the expression level of the miR-200 family (including miR-141, miR-200a, miR-200b, miR-200c, and miR-429) and the EMT-inducing transcription factor ZEB1 (Gibbons et al. 2009a). Human NSCLC cell lines also displayed a strong correlation between the miR-200 family levels and the EMT markers, suggesting this mechanism as a potential driver of the biology of these cells. As noted in the section on miRNA profiling of human NSCLC specimens, miR-200 family members are part of a multi-miRNA signature defining high risk for recurrence after resection (Patnaik et al. 2009). Our work also demonstrated that the miR-200/ZEB1 balance is under epigenetic regulation from interactions between the tumor cells and their microenvironment, including cell-matrix interactions and interactions with morphogens such as TGFβ (Gibbons et al. 2009a). It has also been reported that expression of the miR-200 family in murine and human cells (including normal human mammary epithelial cells, breast cancer cell lines and prostate cancer cell lines) is regulated by DNA methylation of the promoter (Vrba et al. 2010).
The association between altered miR-200 levels and EMT or metastasis has been demonstrated in other epithelial tumor types, including breast, ovarian and gastric (Du et al. 2009b; Gregory et al. 2008; Hu et al. 2009; Park et al. 2008). Additionally, several studies have demonstrated the role of the miR-200 family in regulation or maintenance of normal and tumor stem-cell features in breast, colon and pancreatic cancer (Shimono et al. 2009; Wellner et al. 2009). These data support the concept that EMT links the acquisition of stem cell features with metastasis, suggesting that during metastasis tumor cells might acquire certain properties of progenitor cells, either transiently or permanently.
2.6 Therapeutics
Because miRNAs control many known oncogenes, while also acting on their own as either oncogenes or tumor suppressors, greater insight into their biology also carries great promise for this class of molecules as potential therapeutic agents or targets. Techniques to directly target their expression or modulate their levels in tumor cells are an intense area of investigation. In general the inhibitors are single-stranded oligonucleotides complementary to the mature miRNAs, chemically modified with phosphorothioate, 2′-O-methyl or locked nucleic acid (LNA) substitutions to improve their resistance to nuclease-mediated degradation (Krutzfeldt et al. 2007). Targeted tissue delivery of either antisense oligonucleotides complementary to specific mature miRNAs, or of precursor or mature miRNAs to replace loss of expression, is still a significant challenge and has not been translated to the clinical cancer setting. However, significant work is being conducted in pre-clinical animal models to demonstrate the in vivo biology of the miRNAs during the complex progression of tumors and to test the ability to target these processes.
In human and murine models of lung adenocarcinoma, expression of the let-7 family is frequently suppressed. Exogenous in vivo delivery by inhalation of adenoviral or lentiviral vectors expressing let-7 genes at the time of mutant KRAS activation was able to halt tumor progression (Esquela-Kerscher et al. 2008; Kumar et al. 2008), while delivery after the establishment of tumors halted tumor proliferation, producing a significant reduction in tumor burden after only a few weeks of treatment (Trang et al. 2010). One advantage in the treatment of lung cancer versus other solid tumor types may be the ability to deliver these reagents by inhalation or tracheal instillation, providing an appropriate therapeutic effect to the primary tumors, without the potential systemic side effects. Obviously this route of administration would not provide any advantage in patients who have developed metastatic disease.
Similarly, an adeno-associated virus-mediated delivery system was used in a murine model of hepatocellular carcinoma to re-express miR-26a. Even with pre-existing disease, re-expression of miR-26a had significant effect to induce apoptosis and halt tumor progression in the treated animals (Kota et al. 2009). Of equal importance, there was no evidence of systemic or liver toxicity, despite the high levels of expression of the exogenously-delivered miRNA vector.
Metastasis prevention or targeting of occult tumor metastases, rather than primary tumor growth, is emerging as a potential design for adjuvant trials of targeted biological agents. In a recent study on the miR-10b, which is frequently up-regulated in metastatic tumors, Ma and colleagues demonstrated selective inhibition of the metastatic process upon targeting of orthotopic mammary tumors by the systemic administration of an antagomir to miR-10b (Ma 2010). Antagomirs are antisense oligonucleotides containing a 2′-O-methyl on the ribose moieties, partial replacement of the phosphodiester backbone with phosphorothioate bonds, and conjugation of a cholesterol moiety to the 3′ end (Krutzfeldt et al. 2007). Despite a pronounced treatment effect on the development of metastases, there was no effect on the growth of primary tumors. Interestingly, this effect was due to suppression of the early steps in metastasis, rather than the later stages of the metastatic cascade such as colonization. Again, this study was notable for no evidence of systemic toxicity in the animals.
Currently there are no human trials targeting over-expression or loss of miRNA function in cancer, but the clinical application and the necessary technology are progressing in another patient setting. In chronic hepatitis C virus (HCV) infection, viral replication is dependent upon the cellular host factor miR-122 and in in vitro experiments can be readily suppressed by inhibition of miR-122 expression. Following the systemic administration of a LNA-anti-miR recognizing miR-122 in healthy African green monkeys, the miR-122 levels were repressed with concordant decrease of the serum cholesterol levels, demonstrating delivery of the anti-miR to the liver, but without evidence of toxicity (Elmen 2008). In chimpanzees chronically infected with HCV, use of the agent (now termed SPC3649) decreased serum HCV RNA levels by 2.3 orders of magnitude, with no evidence for the emergence of viral resistance to the therapy (Lanford 2010). The company developing this LNA-anti-miR, Santaris Pharma, currently has two Phase I clinical trials underway in healthy volunteers and reportedly plans to open a Phase II clinical trial in late 2010 for patients with HCV infection. The progress of this agent through clinical development will be closely monitored by those in the field of cancer biology and trial development, for as the biology of miRNAs in cancer becomes more thoroughly understood, there will be increasing efforts to translate those findings into the clinical trial setting for cancer patients.
2.7 Conclusions
The current revolution in non-coding RNAs, especially in the field of miRNAs, provides tremendous opportunity for cancer biologists to further define and refine our understanding of the basis for carcinogenesis and tumor progression. This discovery and enhanced understanding may by itself produce additional insights into better clinical decision making, patient risk stratification and potential therapeutic options. However, the greater promise of miRNAs is in their pleiotrophic biology, which may provide for uniquely innovative strategies to target the spectrum of heterogeneity in cancer that arises from the cumulative changes in multiple pathways. Finally, these insights will likely provide some unifying strategies against multiple cancers, as many of the specifics outlined in this chapter probably have counterparts in the biology of other epithelial and non-epithelial tumor types.
References
Boyerinas B, Park SM, Hau A, et al. The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer. 2010;17:F19–36.
Burk U, Schubert J, Wellner U, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582–9.
Calin GA, Sevignani C, Dumitru CD, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA. 2004;101:2999–3004.
Chen X, Ba Y, Ma L, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18:997–1006.
Chiang HR, Schoenfeld LW, Ruby JG, et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24:992–1009.
Chin LJ, Ratner E, Leng S, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008;68:8535–40.
Cho WC. MicroRNAs: potential biomarkers for cancer diagnosis, prognosis and targets for therapy. Int J Biochem Cell Biol. 2010;42:1273–81.
Cho WC, Chow AS, Au JS. Restoration of tumour suppressor hsa-miR-145 inhibits cancer cell growth in lung adenocarcinoma patients with epidermal growth factor receptor mutation. Eur J Cancer. 2009;45:2197–206.
Du L, Schageman JJ, Subauste MC, et al. MiR-93, miR-98, and miR-197 regulate expression of tumor suppressor gene fus1. Mol Cancer Res. 2009a;7:1234–43.
Du Y, Xu Y, Ding L, et al. Down-regulation of miR-141 in gastric cancer and its involvement in cell growth. J Gastroenterol. 2009b;44:556–61.
Elmén J, Lindow M, Schütz S, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896–9.
Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14:2895–99.
Esquela-Kerscher A, Trang P, Wiggins JF, et al. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. 2008;7:759–64.
Fisher GH, Wellen SL, Klimstra D, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a k-ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249–62.
Gallardo E, Navarro A, Vinolas N, et al. MiR-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis. 2009;30:1903–9.
Gazdar AF. Epidermal growth factor receptor inhibition in lung cancer: the evolving role of individualized therapy. Cancer Metastasis Rev. 2010;29:37–48.
Gibbons DL, Lin W, Creighton CJ, et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009a;23:2140–51.
Gibbons DL, Lin W, Creighton CJ, et al. Expression signatures of metastatic capacity in a genetic mouse model of lung adenocarcinoma. PLoS One. 2009b;4:e5401.
Goldstraw P, Crowley J, Chansky K, et al. The IASLC lung cancer staging project: proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM classification of malignant tumours. J Thorac Oncol. 2007;2:706–14.
Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and sip1. Nat Cell Biol. 2008;10:593–601.
He L, He X, Lim LP, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007a;447:1130–4.
He L, He X, Lowe SW, et al. MicroRNAs join the p53 network–another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007b;7:819–22.
Hu Z, Chen X, Zhao Y, et al. Serum microRNA signatures identified in a genome-wide serum microRNA expression profiling predict survival of non-small-cell lung cancer. J Clin Oncol. 2010;28:1721–6.
Hu X, Macdonald DM, Huettner PC, et al. A miR-200 microRNA cluster as prognostic marker in advanced ovarian cancer. Gynecol Oncol. 2009;114:457–64.
Incoronato M, Garofalo M, Urso L, et al. MiR-212 increases tumor necrosis factor-related apoptosis-inducing ligand sensitivity in non-small cell lung cancer by targeting the antiapoptotic protein ped. Cancer Res. 2010;70:3638–46.
Izzotti A, Calin GA, Arrigo P, et al. Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J. 2009a;23:806–12.
Izzotti A, Calin GA, Steele VE, et al. Relationships of microRNA expression in mouse lung with age and exposure to cigarette smoke and light. FASEB J. 2009b;23:3243–50.
Jackson EL, Olive KP, Tuveson DA, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–8.
Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic k-ras. Genes Dev. 2001;15:3243–8.
Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300.
Johnson SM, Grosshans H, Shingara J, et al. Ras is regulated by the let-7 microRNA family. Cell. 2005;120:635–47.
Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the k-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–6.
Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol. 2006;24:2137–50.
Koshiol J, Wang E, Zhao Y, et al. Strengths and limitations of laboratory procedures for microRNA detection. Cancer Epidemiol Biomarkers Prev. 2010;19:907–11.
Kota J, Chivukula RR, O’Donnell KA, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–17.
Krutzfeldt J, Kuwajima S, Braich R, et al. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007;35:2885–92.
Kumar MS, Erkeland SJ, Pester RE, et al. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci USA. 2008;105:3903–8.
Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201.
Lee DY, Deng Z, Wang CH, et al. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc Natl Acad Sci USA. 2007;104:20350–5.
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20.
Liang Y. An expression meta-analysis of predicted microRNA targets identifies a diagnostic signature for lung cancer. BMC Med Genomics. 2008;1:61.
Liu H, D’Andrade P, Fulmer-Smentek S, et al. mRNA and microRNA expression profiles of the NCI-60 integrated with drug activities. Mol Cancer Ther. 2010a;9:1080–91.
Liu G, McDonnell TJ, Montes de Oca Luna R, et al. High metastatic potential in mice inheriting a targeted p53 missense mutation. Proc Natl Acad Sci USA. 2000;97:4174–9.
Liu X, Sempere LF, Ouyang H, et al. MicroRNA-31 functions as an oncogenic microRNA in mouse and human lung cancer cells by repressing specific tumor suppressors. J Clin Invest. 2010b;120:1298–309.
Lodygin D, Tarasov V, Epanchintsev A, et al. Inactivation of miR-34a by aberrant CPG methylation in multiple types of cancer. Cell Cycle. 2008;7:2591–600.
Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8.
Lu Y, Thomson JM, Wong HY, et al. Transgenic over-expression of the microRNA miR-17-92 cluster promotes proliferation and inhibits differentiation of lung epithelial progenitor cells. Dev Biol. 2007;310:442–53.
Ma L, Reinhardt F, Pan E, et al. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat Biotechnol. 2010;28:341–7.
Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–8.
Matsubara H, Takeuchi T, Nishikawa E, et al. Apoptosis induction by antisense oligonucleotides against miR-17-5p and miR-20a in lung cancers overexpressing miR-17-92. Oncogene. 2007;26:6099–105.
Mendell JT. Miriad roles for the miR-17-92 cluster in development and disease. Cell. 2008;133:217–22.
Ng EK, Chong WW, Jin H, et al. Differential expression of microRNAs in plasma of patients with colorectal cancer: a potential marker for colorectal cancer screening. Gut. 2009;58:1375–81.
Olive KP, Tuveson DA, Ruhe ZC, et al. Mutant p53 gain of function in two mouse models of li-fraumeni syndrome. Cell. 2004;119:847–60.
Olson P, Lu J, Zhang H, et al. MicroRNA dynamics in the stages of tumorigenesis correlate with hallmark capabilities of cancer. Genes Dev. 2009;23:2152–65.
O’Donnell KA, Wentzel EA, Zeller KI, et al. C-myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–43.
Park SM, Gaur AB, Lengyel E, et al. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907.
Pasquinelli AE, Reinhart BJ, Slack F, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408:86–9.
Patnaik SK, Kannisto E, Knudsen S, et al. Evaluation of microRNA expression profiles that may predict recurrence of localized stage I non-small cell lung cancer after surgical resection. Cancer Res. 2009;70:36–45.
Prudkin L, Behrens C, Liu DD, et al. Loss and reduction of fus1 protein expression is a frequent phenomenon in the pathogenesis of lung cancer. Clin Cancer Res. 2008;14:41–7.
Raponi M, Dossey L, Jatkoe T, et al. MicroRNA classifiers for predicting prognosis of squamous cell lung cancer. Cancer Res. 2009;69:5776–83.
Reinhart BJ, Slack FJ, Basson M, et al. The 21-nucleotide let-7 RNA regulates developmental timing in caenorhabditis elegans. Nature. 2000;403:901–6.
Samet JM, Avila-Tang E, Boffetta P, et al. Lung cancer in never smokers: clinical epidemiology and environmental risk factors. Clin Cancer Res. 2009;15:5626–45.
Sampson VB, Rong NH, Han J, et al. MicroRNA let-7a down-regulates myc and reverts myc-induced growth in burkitt lymphoma cells. Cancer Res. 2007;67:9762–70.
Schembri F, Sridhar S, Perdomo C, et al. MicroRNAs as modulators of smoking-induced gene expression changes in human airway epithelium. Proc Natl Acad Sci USA. 2009;106:2319–24.
Seike M, Goto A, Okano T, et al. MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc Natl Acad Sci USA. 2009;106:12085–90.
Shell S, Park SM, Radjabi AR, et al. Let-7 expression defines two differentiation stages of cancer. Proc Natl Acad Sci USA. 2007;104:11400–5.
Shimono Y, Zabala M, Cho RW, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138:592–603.
Takamizawa J, Konishi H, Yanagisawa K, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–6.
Talotta F, Cimmino A, Matarazzo MR, et al. An autoregulatory loop mediated by miR-21 and pdcd4 controls the ap-1 activity in ras transformation. Oncogene. 2009;28:73–84.
Tavazoie SF, Alarcon C, Oskarsson T, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–52.
Thuault S, Valcourt U, Petersen M, et al. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol. 2006;174:175–83.
Tili E, Croce CM, Michaille JJ. MiR-155: on the crosstalk between inflammation and cancer. Int Rev Immunol. 2009;28:264–84.
Trang P, Medina PP, Wiggins JF, et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene. 2010;29:1580–7.
Valastyan S, Reinhardt F, Benaich N, et al. A pleiotropically acting microRNA, miR-31, inhibits breast cancer metastasis. Cell. 2009;137:1032–46.
Valeri N, Gasparini P, Fabbri M, et al. Modulation of mismatch repair and genomic stability by miR-155. Proc Natl Acad Sci USA. 2010;107:6982–7.
Ventura A, Young AG, Winslow MM, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–6.
Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103:2257–61.
Vrba L, Jensen TJ, Garbe JC, et al. Role for DNA methylation in the regulation of miR-200c and miR-141 expression in normal and cancer cells. PLoS One. 2010;5:e8697.
Webster RJ, Giles KM, Price KJ, et al. Regulation of epidermal growth factor receptor signaling in human cancer cells by microRNA-7. J Biol Chem. 2009;284:5731–41.
Weiss GJ, Bemis LT, Nakajima E, Sugita M, et al. EGFR regulation by microRNA in lung cancer: correlation with clinical response and survival to gefitinib and EGFR expression in cell lines. Ann Oncol. 2008;19:1053–9.
Wellner U, Schubert J, Burk UC, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–95.
Xiao C, Srinivasan L, Calado DP, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–14.
Yanaihara N, Caplen N, Bowman E, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–98.
Yin Q, Wang X, Fewell C, et al. MiR-155 inhibits bone morphogenetic protein (BMP) signaling and BMP mediated Epstein-Barr virus reactivation. J Virol. 2010;84:6318–27.
Yu SL, Chen HY, Chang GC, et al. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell. 2008;13:48–57.
Zabarovsky ER, Lerman MI, Minna JD. Tumor suppressor genes on chromosome 3p involved in the pathogenesis of lung and other cancers. Oncogene. 2002;21:6915–35.
Zheng S, El-Naggar AK, Kim ES, et al. A genetic mouse model for metastatic lung cancer with gender differences in survival. Oncogene. 2007;26:6896–904.
Acknowledgments
This work was supported by NIH R01 grants CA132608 and CA117965 (J.M.K.), a Howard Hughes Medical Institute Medical Scholar Fellowship to Z.H.R., an American Society for Clinical Oncology Young Investigator Award, an International Association for the Study of Lung Cancer Fellowship, and NIH 1K08CA151651 and NCI 5 T32 CA009666 (D.L.G.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer Netherlands
About this chapter
Cite this chapter
Gibbons, D.L., Rizvi, Z.H., Kurie, J.M. (2011). The Role of MicroRNAs in Lung Cancer Development, Progression, and Metastasis. In: Cho, W. (eds) MicroRNAs in Cancer Translational Research. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-0298-1_2
Download citation
DOI: https://doi.org/10.1007/978-94-007-0298-1_2
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-0297-4
Online ISBN: 978-94-007-0298-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)