
Abstract
Although the search for novel phytotherapeutics is an area of current research focus, man has always sought plant products in an effort to alleviate illness and infection in both humans and animals. During the latter part of the twentieth century, the emergence of the modern pharmaceutical industry and the development of a range of effective medical and veterinary treatments tended to focus attention away from these traditional resources. However the subsequent emergence of resistance amongst veterinary microbial, protozoal and metazoan pathogens, the high cost of veterinary products to resource poor farmers, and consumer interest in reducing chemical treatments in food producing animals have all served to re-awaken interest in bioactive plant products.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
3.1 Introduction
Although the search for novel phytotherapeutics is an area of current research focus, man has always sought plant products in an effort to alleviate illness and infection in both humans and animals. During the latter part of the twentieth century, the emergence of the modern pharmaceutical industry and the development of a range of effective medical and veterinary treatments tended to focus attention away from these traditional resources. However the subsequent emergence of resistance amongst veterinary microbial, protozoal and metazoan pathogens, the high cost of veterinary products to resource poor farmers, and consumer interest in reducing chemical treatments in food producing animals have all served to re-awaken interest in bioactive plant products.
Because of the threat helminths pose to the health and welfare of ruminants throughout the world, anthelmintics have for more than 40 years been the chief means of controlling these debilitating diseases. However, resistance has been reported against the three current broad-spectrum anthelmintic families and in some countries multiple anthelmintic resistances is now a common phenomenon.
The search for cheap, effective and safe plant based alternatives for the control of ruminant nematodes is being conducted in many countries. Plants and their products can not only have direct effects against parasite populations resident in the gastrointestinal tract but also by improving host nutrition can also serve to enhance immunity against these parasites. The search for local forages that optimise host immuno-regulatory capacity and/or have direct antiparasitic effects is particularly relevant for resource poor farming communities who would clearly stand to benefit from the availability of “nutraceutical” plants, i.e. plants that are used first for their beneficial effects on health rather than for their nutritive value. The techniques used to study effects upon host immunity of plant products are somewhat specialised and are not within the realms of this article which is focused on in vitro methods for screening plant products for direct antiparasitic effects.
Since it is reasonable to assume that local forages that ruminants currently consume have, at best, only modest direct effects on the hosts’ parasite burden, the search for novel phytotherapeutics has naturally tended to focus on other plant species that are not currently consumed in large quantities. Given the bewildering array of plants available for testing, the first imperative is to find some way of reducing the numbers entering the screening process to a feasible level. The initial screening process to exclude known toxic plants and those which may be unsuitable on agronomic grounds or to select plants using some knowledge of their biochemistry, use in ethno-veterinary medicine or selective animal feeding behaviour is not the focus of this article, suffice to say that the best results will be achieved through collaborative efforts involving a range of specialists. If only a small number of plants are to be screened then there is little doubt that the best approach is to screen them is in vivo; feeding or administering the plant products to infected ruminants. The reasons for this are very simple; the extent of presentation of complex bioactive compounds to the intended target parasite will be influenced largely by the physical and biochemical conditions prevailing at the site of infection. Since these conditions change throughout the gut and it is invariably impossible to duplicate them under in vitro conditions, testing in the host is the best way to determine efficacy. However where large numbers of plants are being examined it is clearly not feasible to test them all in animals and researchers will need to resort to the use of in vitro techniques to provide primary screening. The two key processes involved in primary screening are:
-
1.
Preparation of parasite material, the isolation of different pre-parasitic stages from faecal material.
-
2.
In vitro screening, using a range of different bioassays all of which measure efficacy in comparative terms, examining the disruptive effects of a plant product on some vital biological process.
The various methods used to prepare the plant products and extracts are described in Chapter 2 and within other chapters of this book. Some pre-screening experimentation is almost inevitable to decide on an appropriate concentration ranges. There are two main reasons for this. Firstly, the way in which the whole plant or some extract from it will be used is important. If for example forage is the sole food source, for a period then it would be appropriate to test it, or products from it, at a higher concentration range than if it were only being used to provide a fraction of the daily dry matter intake. Secondly, differences in the parasite species being subject to testing will also influence the choice of concentration used in the screening process simply because of between species differences in susceptibility to the bioactive products.
Wherever possible it is useful to incorporate a series of controls. Negative control data provides the base line against which the effects of the plant product are measured. Positive control data obtained by using either chemical or known bioactive plant products is useful for not only confirming that the bioassay is working but can also help to indicate the type of bioactive substance(s) implicated in activity. Finally, it must be remembered that primary screening using in vitro bioassays will inevitably throw up a number of positives that will, due to the very different physicochemical conditions in the gut, have little or no effect in vivo.
3.2 Preparation of Parasite Material
The preparation of clean parasite material is important since dirty preparations are difficult to count and the presence of faecal debris can interfere with the action of some plant secondary metabolites. Eggs, first and third stage larvae and adult worms recovered from post mortem material can be used in in vitro bioassays.
3.2.1 Mass Extraction of Nematode Eggs
3.2.1.1 Description
Nematode eggs are isolated from fresh faecal material for use in egg hatch assays, or for further culture to first stage (L1) larvae. Faecal material is thoroughly dispersed in tap water before being passed through a series of sieves and parasite eggs are further cleaned by floatation in saturated salt solution.
3.2.1.2 Materials
-
Fresh ruminant faeces
-
Top pan balance
-
130 × 230 mm polythene bags
-
1 mm, 500, 212, 75 and 38 μm sieves
-
Beckmann polyallomer centrifuge tubes (Cat. No. 337986)
-
15 mL polystyrene or glass centrifuge tubes (Sterilin or similar)
-
Cover slips, glass slides (26 × 76 mm)
-
Saturated sodium chloride solution
-
Centrifuge
-
Artery forceps
-
Micro pipette and disposable tips (20–400 μL)
-
Stereo or compound microscope fitted with a mechanical stage
3.2.1.3 Procedure
-
1.
Collect fresh faeces directly from the donor animal’s rectum into polythene bags no more than 1 h prior to extraction.
-
2.
Add tap water and disperse the faecal material to give a smooth liquid suspension, water may be added as required.
-
3.
Wash suspension over sieves in order; 1 mm, 500, 212 and 75 μm, collecting filtrate in a bucket or large beaker.
-
4.
Pass the above filtrate over 38 μm sieve and collect retentate (material off the sieve). Transfer the retentate into centrifuge tubes and wash it with tap water, followed by centrifugation, as described below in steps 5–9 (polyallomer tube method) or 5a–9a (polystyrene/glass tube method)
3.2.1.4 Polyallomer Tube Method
-
5.
Polyallomertubes are deformable semi rigid tubes that can be clamped externally to isolate the upper reaches of the suspension to isolate nematode eggs. Fill the tubes with the retentate collected at step 4 and centrifuge at 203g for 2 min.
-
6.
Remove supernatant with vacuum line leaving approximately 1 mL pellet and water or alternatively tip off supernatant leaving a faecal pellet.
-
7.
Re-suspend the pellet with saturated sodium chloride solution (specific gravity 1.2), seal the top of the tube and invert gently several times to loosen the faecal pellet.
-
8.
Re-centrifuge at 203g for 2 min. Clamp tubes just below meniscus using forceps (eggs will be on top of the meniscus), pour off top layer into 250 mL beaker, wash onto 38 μm sieve and rinse thoroughly with tap water.
-
9.
Collect retentate and resuspend with tap water and centrifuge at 203g for 2 min, remove supernatant with vacuum line. Steps 6–9 may be repeated to remove further debris.
-
10.
Make volume up to 10 mL with tap water and count eggs in 100 μl of suspension by streaking this volume along the glass slide and examining using the stereo or compound microscope.
3.2.1.5 Polystyrene/Glass Tube Method
-
5a.
Fill the tube with the retentate collected at step 4 and centrifuge at 203g for 2 min.
-
6a.
Remove supernatant with a vacuum line leaving approximately 1 mL pellet and water or alternatively tip off supernatant leaving a faecal pellet.
-
7a.
Resuspend the pellet with saturated sodium chloride solution (specific gravity 1.2), shake gently to mix and then fill the tube completely to form a positive meniscus (see Fig. 3.1). Carefully place a cover slip on top of the tube and place in the centrifuge.
-
8a.
Centrifuge at 203g for 2 min, carefully remove the tube with its cover slip.
-
9a.
Lift off the cover slip (the eggs will be held in the surface film attached to it) and wash into a beaker with tap water. Pass the contents over 38 μm sieve and rinse with tap water. Collect the retentate into a beaker.
-
10.
Sediment contents of the beaker, and reduce the volume to 10 mL by removing upper liquid portion with a vacuum line and count the numbers of eggs in 100 μL by streaking this volume along the glass slide and examining using the stereo or compound microscope.


Re-suspended faecal material in a tube filled to a positive meniscus
Clean eggs collected in this way can be used in ovicidal assays or may be used to provide first stage larvae.
3.2.2 Culture of First Stage (L1) Larvae from Nematode Eggs
3.2.2.1 Description
Nematode eggs obtained using Method 1 are cultured at room temperature until hatched to first stage larvae, and then filtered using a Baermann apparatus to remove debris and unhatched eggs.
3.2.2.2 Materials
-
Nematode egg suspension
-
10 cm plastic Petri dishes (Sterilin Ltd., or similar)
-
Baermann apparatus and filter collar (made from plastic tubing or similar material plus plastic collar, with elastic band to hold filter material in place) (see Fig. 3.4 for general structure of Baermann apparatus)
-
Suitable high wet strength filter paper such as Cottom Bottoms nappy liners
-
(Boots Ltd., UK) or 20 μm nylon mesh (Nytal, Sefar Ltd or similar)
-
250 mL Beaker (Nalgene) or similar to support filter collar
-
Glass slides (26 × 76 mm)
-
Micro pipette and disposable tips (20–400 μL)
-
Stereo or compound microscope fitted with a mechanical stage


Steps 2–7 of the isolation of adult nematodes
3.2.2.3 Procedure
-
1.
Place freshly extracted egg suspension into a suitable culture vessel. This should ideally have a large liquid surface area to allow sufficient gas exchange for the eggs to hatch.
-
2.
Incubate at room temperature or in an incubator should the ambient room temperature is likely to fall below 10°C for 24 h.
-
3.
Examine the culture microscopically to ensure hatching has occurred.
-
4.
Prepare the Baermann filter; the filter consists of a 22 mm diameter plastic tube about 5 cm long 20-μm nytal mesh has been glued over one end of the tube.
-
5.
Fill the beaker with tepid water (22°C).
-
6.
Pour the eggs and larvae onto the mesh of the Baermannn apparatus, ensuring that the sample is distributed evenly over the mesh.
-
7.
Immerse the Baermann apparatus in the warm tap water in the beaker.
-
8.
Allow 1 h for the larvae to migrate through the fine mesh, and then carefully remove the filter collar. Allow the larval suspension in the beaker to settle and then reduce the volume using a vacuum line or centrifugally.
-
9.
Count the larvae present by examining microscopically a small sub-sample taken with a pipette (100 μL) by streaking this volume along a glass slide and counting on a stereo or compound microscope fitted with a mechanical stage.
First stage larvae obtained in this way can be used in larvicidal assays or those that measure the disturbance of normal behavioural activity such as the larval feeding inhibition assay.
3.2.3 Culture of Third Stage (L3) Nematode Eggs from Sheep Faeces
3.2.3.1 Description
Faeces from infected sheep are incubated to allow nematode eggs to hatch and develop into third stage larvae [12]. Faeces are flooded with water until larvae migrate out of pellets, and then the resulting suspension is cleaned using a Baermann filter.
3.2.3.2 Materials
-
Faeces from monospecifically infected donor animal
-
Culture trays
-
Polythene bags
-
22°C incubator
-
Sieve – approximately 1 mm pore size
-
Glass slides (26 × 76 mm)
-
Baermann apparatus (20 cm diameter 5 cm long plastic tube with two layers of high wet strength paper held over one end by a rubber band)
-
Micro pipette and disposable tips (20–400 μL)
-
Stereo or compound microscope fitted with a mechanical stage.
3.2.3.3 Procedure
-
1.
Place faeces collected from infected donor animal in a culture tray to a maximum depth of 30 mm and seal tray inside polythene bag. If the faeces are very loose (diarrhoeic), then it may be necessary to add vermiculite or washed peat or charcoal to provide better culture conditions. Sufficient material needs to be added to enable the mixture to be formed into balls. Puncture bag to allow airflow.
-
2.
Incubate tray at 22°C for 7 days.
-
3.
Flood tray with tepid tap water (22°C). Allow to soak for 1 h.
-
4.
Sieve fluid through a 1.0 mm sieve and collect filtrate.
-
5.
Sediment filtrate for 2 h at 4°C. Reduce volume using a vacuum line and filter using Baermann apparatus as described for L1 culture. Make the larval suspension up to a suitable volume and count the numbers of larvae present in 100-μL sub sample taken with a micropipette and streaking the sub sample along a glass slide and counting on a stereo or compound microscope fitted with a mechanical stage. Calculate the total numbers of larvae present.
-
6.
Larvae may be stored at 4–10°C until required.
Infective larvae may be used in larvicidal assays or behavioural assays such as the larval migration inhibition assay, which measures the effect of a test substance on locomotion.
3.2.3.4 Larval Identification
Where field (mixed species) infections are used to provide material for the tests, it can be useful to determine the prevalence of the different species simply because there can be large interspecies differences in the sensitivity of nematodes to bioactive substances.
The following references are the most appropriate sources of information for identification (speciation) of ruminant nematodes based on the morphology of third stage larvae [12, 17].
3.2.4 Isolation of Adult Nematodes
3.2.4.1 Description
Adult worms are isolated from gastrointestinal contents by combining the contents with agar and soaking mixture in water. Worms will migrate from the agar slab and can be collected from the water. Where only small numbers of adult worms are required they can be recovered directly by using a stereo microscope to examine the washings/contents collected from the abomasa, small intestines or large intestines.
3.2.4.2 Materials
-
Abomasum or small or large intestine from freshly killed animal
-
Physiological saline (0.85% w/v NaCl solution)
-
Technical grade agar (Sigma Aldrich Ltd, catalogue number -A7002)
-
10 L Funnel fitted with tap at base
-
Micro pipette (200–1000 μL) and tips
-
Stereo microscope


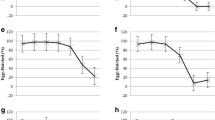
Graphical representation of egg hatch test results
3.2.4.3 Procedure
-
1.
Heat two batches of saline to 56°C and maintain at that temperature.
-
2.
Collect and cut open gut sample, empty contents into 10 L bucket containing 5 L saline kept at 39°C, wash gut surface in further 1 L saline, pool the contents.
-
3.
Sediment and remove the liquid portion to adjust volume.
-
4.
Dissolve 1.8 g agar per 100 mL saline (56°C), allowing a total volume of 500 mL per sample. Add the 500–200 mL of digesta stirring continuously throughout the mixing process. Cool it to 45°C.
-
5.
The Baermann mesh apparatus consists of the upper portion of a bucket that has a snap on lid. A 1 mm mesh support is fitted within the apparatus next to where the lid is attached. Carefully fit the lid and place lid down onto a cool surface (approximately 10°C).
-
6.
Pour the agar-digesta mix over mesh and allow to set for 15 min. The lid of the apparatus ensures that the mix does not leak out of the apparatus during the period when the agar/digesta mix solidifies.
-
7.
Remove lid from Baermann apparatus and place the apparatus in a Baermann funnel filled with warm 0.85% saline and maintained at 37–39°C. The Baermann funnel should have a wide bore (4 mm internal diameter) tap fitted to enable the collection of worms migrating out of the agar bed.
-
8.
Add saline to ensure contact between saline and agar slab.
-
9.
Incubate for 4 h at 37°C (or 39°C), remove any worms from top surface using forceps, draw off the remaining worms using the tap fitted at the bottom of the funnel, concentrate the adult worms and store in 0.85% saline at 37°C (or 39°C), until required. Adjust to a suitable volume (between 100 mL for small numbers of worms and 1000 mL for large numbers of worms. Take a 1 mL sub sample (remember to clip the tip off the pipette tip to give an internal diameter of about 4 mm to allow the adult worms to be sucked into the tip) and count the numbers of worms in 1 mL using the stereo microscope.
Adult worms can be used in adulticide trials or those measuring disruption in locomotory behaviour because of exposure to plant material.
3.3 In Vitro Screening
A number of different bioassays using either the pre-parasitic stages of nematodes or adult nematodes can be used to screen plant extracts. A number of plant products and plant secondary metabolites have been shown to have direct anthelmintic effects in these assays. These include condensed tannins, saponins, flavonols, lectins and various proteases. However, since most of these assays operate best within a narrow pH range around pH 7 it cannot be assumed that activity observed in vitro will be seen under the very different conditions that exist in vivo.
The principal assays and their target stages are shown in Table 3.1 below. Wherever possible it is useful to incorporate positive controls using an appropriate anthelmintic (a compound or forage) to confirm that the assay is working correctly.
Although all of these assays can be used for primary screening, the simple and robust assays such as the EHT, LMIT and LFIT have tended to be used in the first instance. The more time consuming or expensive assays such as the LEA, LDT and AMT are generally used with products that are known to have some efficacy. Inevitably, laboratories may need to adapt the methodologies described here to suit to their facilities and budgets.
3.3.1 Preparation of Stock Solutions of Plant Material
3.3.1.1 Description
Since plant material for use in the bioassays can come in a variety of different forms including whole plants, freeze dried, finely ground (milled) material, semi or fully purified (following solvent extraction) it is not possible to provide details of the full range of stock solutions that might be required.
For simplicity, the stock solution concentrations mentioned with the following assays are those based around a possible 10% inclusion rate of whole plant material and represent a concentration range that might be expected to occur in the gut under those circumstances. Again for simplicity the stock solutions are also aqueous solutions, not based on the use of organic solvents and represent a range of concentrations that are effective against the drug sensitive populations of Teladorsagia, Haemonchus and Trichostrongylus maintained at Moredun Research Institute (Pentland Science Park, Edinburgh, Scotland, EH26 0PZ), or in UMR 1225 INRA DGER (Ecole Nationale Veterinaire de Toulouse 23 Chemin des Capelles, 31076 Toulouse France).
The use of a phosphate buffer saline (PBS; 0.1 M phosphate containing 0.05 M NaCl, pH 7.2) to prepare the stock solution might also be recommended to avoid any non specific effects due to acidic or basic pH sometimes related to dilution of plant extracts.
Using ruminal fluids collected from animals being fed bioactive forages has also been proposed as a means of mimicking in vivo conditions. However, due to the difficulties of maintaining, defining and standardizing this medium, this approach has not found favour in many laboratories.
3.3.1.2 Materials
-
Top pan balance
-
10 mL volumetric flask
-
0.2 μm syringe filters
-
Micro pipettes and tips
-
Bench top micro centrifuge
-
2 mL eppendorf tubes
-
Plastic universal tubes
3.3.1.3 Preparation of a 10 mg/mL Aqueous Plant Material Stock Solution
-
1.
Weigh 0.3 g of freeze dried finely milled plant product into a 10 mL volumetric flask.
-
2.
Add distilled water to the required level and mix with a vortex mixer for 2 min.
-
3.
Pipette the suspension into labelled 2 mL eppendorf tubes.
-
4.
Microfuge the sample for 3 min at 18,000g. Transfer the supernatant to a syringe and pass through a 0.20 μm syringe filter into a plastic universal bottle.
-
5.
Serially dilute the stock solution in PBS or distilled water as required to produce working solutions of 10, 5, 2.5, 1.25 mg/mL, etc.
-
6.
Maintain these stock solutions at 4°C and use within 24 h.
3.3.2 Egg Hatch Test
3.3.2.1 Description
Freshly extracted eggs are incubated in varying concentrations of thiabendazole for 48 hr, after which they are fixed and stained and the numbers of hatched larvae and unhatched eggs are counted [6, 10, 11]. The concentration of extracts that is required to inhibit 50% of the eggs from hatching (ED50) is calculated to determine the ED50. Ideally, the assay should incorporate a solvent control (solvent used for preparing plant extract) to confirm that the solvent has no effect on the bioassay and a positive control (thiabendazole) to confirm the bioassay is working. The solvent control well should contain the same amount of solvent that is present in the highest plant product concentration. All of the test and control wells should be run in duplicate.
3.3.2.2 Materials
-
Fresh nematode egg suspension
-
15 mg/mL plant extract stock solution in PBS or distilled water
-
1000 μg/mL thiabendazole (TBZ) stock solution
-
1000 μg TBZ/mL of di-methyl sulphoxide [DMSO]
-
10 mL volumetric flasks
-
24 well culture plate
-
Helminthological iodine (50 g iodine and 250 g potassium iodide in 500 mL distilled water)
-
Micro pipettes (200–1000, 40–200, 1–10 μL) and disposable tips
3.3.2.3 Procedure
-
1.
Prepare the 15 mg/mL stock solution of plant material as described in the previous section. Prepare working solutions of 15, 10, 5, 1 and 0.1 mg/mL in distilled water. When used in the assay these will give final concentrations of 14.25, 9.5, 4.75, and 0.95 mg/mL.
-
2.
Add 10 μl of the TBZ stock solution (1000 μg/mL) to each positive control well (in a total volume of 2 mL this will give a final concentration of 5.0 μg/mL of TBZ, sufficient to prevent hatching of nematode eggs).
-
3.
Count eggs present in suspension and adjust volume to provide a concentration of 1000 eggs/mL.
-
4.
Add 100 μl of the egg suspension to each well, ensuring that the suspension is thoroughly mixed prior to every aliquot being taken as the eggs will settle very rapidly.
-
5.
Add 1900 μl of working plant stock solution to the wells to give a total volume of 2000 μl. Mix by pipetting in and out several times. For the control wells simply use 1900 μl of distilled water. Both the test and control wells should be run in duplicate.
-
6.
Place the culture plate in a sealed polythene container on some wet paper towel to ensure a high relative humidity and incubate at 25°C for 48 h.
-
7.
Following incubation, add 50 μl of helminthological iodine to each well. This both fixes and stains the nematode egg and larvae.
-
8.
Count the number of eggs and first stage larvae using an inverted or stereo microscope (Table 3.2).
-
9.
Calculate mean numbers of eggs and larvae at each concentration and the percentage hatch using the formula:
${\rm{Percentage\,hatch = [(numbers\,of\,larvae)/(number\,of\,eggs + numbers\,of\,larvae)] }} \times {\rm{ 100}} $ -
10.
Plot the percentage hatch at each concentration on a graph (Fig. 3.3), and using a suitable statistical technique such as probit analysis calculate the ED50 estimate – the concentration at which 50% of the eggs fail to hatch.


Illustration of the fitting of the mesh and collar to the well
3.3.3 Larval Migration Inhibition Test
3.3.3.1 Description
The larval migration inhibition assay measures effects upon locomotion [4, 8, 15]. Ensheathed or exsheathed third stage larvae can be used. They are incubated in a range of concentrations of the test substance and then their capacity to migrate through 20-μm nylon mesh is determined. The anthelmintic levamisole can be used to provide positive control data. Although the technique as described uses multiwell plates and requires an inverted stereo microscope, it is also possible to use centrifuge tubes to collect migrating larvae and to subsequently determine larval numbers using a standard stereo microscope.
3.3.3.2 Materials
-
15 mL centrifuge tube
-
Sodium hypochlorite solution (2% w/v)
-
Filter collars to fit wells of 24 well cluster plates
-
20 μm nylon mesh (Nytal, Sefar Ltd or similar)
-
24 well culture plates
-
Levamisole stock solution (1000 μg/mL distilled water)
-
Micro pipette and disposable tips (20–200 μL)
-
Helminthological iodine (50 g iodine and 250 g potassium iodide in 500 mL distilled water)
-
Inverted stereo microscope
3.3.3.3 Procedures
Rapid Exsheathment of infective nematode larvae
-
1.
Dispense 10 mL larval suspension into centrifuge tube and add 1 mL 2% sodium hypochlorite solution.
-
2.
Remove 100 μL aliquot and place onto a microscope slide. Examine slide under microscope and watch while larvae shed their protective sheath.
-
3.
When all the larvae have exsheathed, centrifuge at 203g for 2 min and remove supernatant. Wash the larvae by resuspending them in tap water and centrifuging. Repeat washings and centrifuging two more times to remove sodium hypochlorite solution.
Larval migration inhibition assay
-
1.
Fit mesh to collars as per Fig. 3.4.
-
2.
For the negative control wells, use distilled water and for the positive control wells use levamisole at a final concentration of 40 μg/mL of solution in the well.
-
3.
Count the exsheathed larvae and adjust the concentration to approximately 1000 larvae/mL.
-
4.
Dispense 100 μl of the larval suspension (approximately 100 larvae) into eppendorf tubes and add 1 mL of each of the working plant stock solutions (40, 20, 10 and 5 mg/mL). Incubate for 2 h at 37°C.
-
5.
Centrifuge at 3000g for 2 min and remove the supernatant, reducing the volume to approximately 200 μl.
-
6.
Add 1800 μl of each dilution of the plant extract to the appropriate wells on the culture plate and place a filter into the well ensuring that the mesh is fully submerged and that there are no air bubbles trapped beneath the mesh.
-
7.
Mix each larval suspension thoroughly and add 200 μl to the matching filter.
-
8.
Place a cover over the plates and incubate for 2 h at 37°C.
-
9.
Carefully remove the filters and wash any remaining larvae into individual labelled petri dishes and stain with a few drops of helminthological iodine.
-
10.
Add 50 μL of helminthological iodine to each well on the culture plate.
-
11.
Using an inverted microscope at × 100 magnification, count the number of larvae in each well and in each Petri dish.
-
12.
Calculate the percentage migration for each concentration using the standard formula:
$ \hbox{Percentage migration = (Nm)} \times \hbox{100/(Nm + Nr)} $Where: Nm = number of larvae migrating through mesh (i.e. found in the well) Nr = number of larvae retained by the mesh (i.e. washed off from mesh).
-
13.
Plot a graph of drug concentration against percentage migration and using the data in probit analysis calculate the LM50 value (concentration at which 50% of larvae fail to migrate).
3.3.4 Larval Feeding Inhibition Assay (LFIA)
This assay determines the effect of plant products on the feeding behaviour of first stage larvae [1, 7]. First stage larvae that have been exposed to different concentrations of the test (plant) product are subsequently offered lyophilised Escherichia coli that are labelled with fluorescein isothiocyanate (FITC). Larvae that have fed can be readily identified using an inverted fluorescence microscope by the presence of the labelled E. coli in their gut.
3.3.4.1 Materials
-
Lyophilised E. coli
-
Fluorescein isothiocyanate FITC
-
Bicarbonate buffer (pH 9.6)
-
Phosphate Buffered Saline (PBS)
-
Inverted fluorescence microscope (Blue Filter 475–490 nm)
3.3.4.2 Procedures
Fluorescein isothiocyanate (FITC) labelling of lyophilised Escherichia coli for use in larval feeding inhibition assay (LFIA)
-
1.
Incubate 1 mL of concentrated E. coli (2250 μg E. coli in 1 mL bicarbonate buffer containing 1 mg of FITC) in a 2 mL microcentrifuge tube at 20°C for 2 h.
-
2.
Centrifuge E. coli suspension at 18,000g for 2 min.
-
3.
Remove supernatant using a vacuum line. Re-suspend E. coli pellet in 1 mL of PBS.
-
4.
Repeat steps 2 and 3 twice.
-
5.
Re-suspend E. coli in 1 mL of PBS.
-
6.
Aliquot in 500 μL portions and store at –20°C for subsequent use.
Larval Feeding Inhibition Assay (LFIA)
-
1.
Add 100 first stage larvae in 100 μL of distilled water to 500 μL of the plant extract (at 10, 5, 2.5, 1.25 mg/mL in distilled water) and mix thoroughly with a further with 900 μL of distilled water. For negative control add 100 μL of the larval suspension to 1400 μL of distilled water and for the positive ivermectin controls add 100 μL of the larval suspension to 1390 μL of distilled water and 10 μL of 1000 μg/mL ivermectin solution.
-
2.
Incubate tubes horizontally at 25°C for 2 h.
-
3.
Add 10 μL of FITC labelled E. coli and incubate tubes horizontally for a further 18 h at 25°C.
-
4.
Microcentrifuge tubes at 3000g for 20 s to “pellet” worms, remove 750 μL of supernatant.
-
5.
Examine larvae from the pellet under inverted fluorescence microscope fitted with blue filter.
-
6.
Determine the number of feeding/fed and non-feeding/unfed larvae at each test concentration (Fig. 3.5).
-
7.
Calculate mean numbers of eggs and larvae at each concentration and the percentage hatched using the formula:
$\begin{array}{l} {\rm{Percentage\ feeding}} =\\ {\rm{(Number\ of\ fed\ larvae) 100/(Number\ of\ fed\ and\ unfed\ larvae)}}\end{array}$


First stage larvae that have fed on FITC labelled E. coli (a) and those that have been unable to feed (b) as seen under a fluorescence microscope
Plot the percentage feeding at each concentration on a graph and calculate the LF50 value (concentration of compound at which 50% of the larvae fail to feed).
3.3.5 Larval Exsheathment Assay (LEA)
This test uses infective third stage larvae in a two-stage process aimed at examining the effect of the test plant product on larval exsheathment induced by a diluted sodium hypochlorite solution. This is an adaptation of the rapid exsheathment process described previously [3]. The aim is to obtain a progressive exsheathment of the larvae, making microscopic observation of the process feasible. Examples of ensheathed and exsheathed larvae at × 100 magnification are provided in Figs. 3.6 and 3.7 respectively. For control, a 100% exsheathment after 60–70 min is sought. Negative controls of PBS and extracts of rye grass (or some other forage that is known to be non-bioactive) are incorporated into the assay.


Ensheathed third stage larva (× 100 magnification)


Exsheathed third stage larva (× 100)
Because the rate of exsheathment varies with the parasite species and/or strains, it is important in a preliminary step, to undertake a preliminary test on control larvae, to determine the best dilution to apply for the exsheathment fluid in order to obtain a gradual exsheathment process within a 60-min time interval. For this, the step 2 of the procedure described herein is applied with a range of concentrations of exsheathment fluid, usually from 1 in 100 to 1 in 500.
3.3.5.1 Materials
-
Infective larvae are harvested from monospecific cultures in the manner described previously
-
PBS (0.1 M phosphate containing 0.05 M NaCl, pH 7.2)
-
2% w/v sodium hypochlorite solution
-
16.5% w/v sodium chloride solution
-
24-well multiwell plates
3.3.5.2 Procedure
-
1.
Pre-incubate 1500 ensheathed L3 larvae in PBS at pH 7.2 with the test product (600 μg/mL) or rye grass extract (600 μg/mL) or PBS (pH 7.2) at 22°C for 3 h.
-
2.
Examine the larvae and if no exsheathment has occurred then move to the second stage of the procedure.
-
3.
Wash the larvae centrifugally 3 times using PBS (pH 7.2) and reduce volume so that 100 μL contains 100 ensheathed L3 larvae. Dispense the larvae (100 μL) into the wells using 6 replicates per plant product. Also, use larvae that have been exposed to PBS alone or rye grass extract (600 μg/mL) to provide negative controls
-
4.
Dilute the 2% w/v hypochlorite solution and the 16.5% w/v sodium chloride solutions 1 in 300 with PBS (pH 7.2) add 1900 μL of this mixture to each well that contains 100 ensheathed larvae in 100 μL.
-
5.
Observe the rate of exsheathment by examining material from the wells at × 200 magnification at 10, 20, 30, 40, 50 and 60 min after adding the exsheathment fluid.
-
6.
Calculate the mean exsheathment rate for the test substances and analyse dif-ferences using an appropriate statistical technique such as GLM.
3.3.6 Larval Development Assay (LDA)
This test uses fresh eggs like the egg hatch assay, but is carried out over a longer period. It measures the ability of the parasite to hatch and develop to the infective third stage larva in the presence of the test substance [2, 5, 9, 16, 18]. In this assay, it is possible to test for activity of plant products against eggs and developing larva. Dilute 1000 μg/mL stock anthelmintic solution with distilled water to produce test concentrations. Pure anthelmintics such as thiabendazole (TBZ), ivermectin and levamisole may be used in this assay to provide positive controls. Final concentrations in the control wells should be 2.5 μg/mL for TBZ and 20 μg/mL for Levamisole and Ivermectin. Since the technique involves a 7-day incubation period, which increases the risk of bacterial or fungal “overgrowths”, it is necessary to incorporate antibiotics and antifungals in the assay. Although the technique has clear advantages, the long incubation period has meant that fewer laboratories have used the assay as the primary screen for activity in vitro.
3.3.6.1 Materials
-
Fresh nematode egg suspension
-
Phosphate buffered saline (PBS) solution
-
5 mg/mL Amphotericin B in PBS
-
30,000 U/mL Nystatin in distilled water
-
Streptopen solution (10,000 μg streptomycin 10,000 units penicillin per mL PBS)
-
Earls balanced salt solution (EBSS)
-
1 g/90 mL yeast extract in 0.85% (w/v) saline solution
-
Helminthological iodine (250 g Potassium iodide and 50 g Iodine in 500 mL water)
-
Sealed plastic humidity chamber
3.3.6.2 Procedure
-
1.
Harvest eggs from faeces as described above. Incubate eggs overnight in the antifungal and antibiotic solution made up with PBS and 2% Nystatin, Amphotericin and Streptopen solutions. Following the overnight incubation, centrifuge in a microcentrifuge at 18,000g for 2 min, then remove supernatant and resuspend the eggs in sterile distilled water. Repeat the washing step using sterile distilled water and centrifugation three times.
-
2.
Count ten aliquots to determine the concentration of eggs. Adjust volume with sterile distilled water to give a concentration of 100-eggs/60 μL.
-
3.
Add 20 μL of yeast extract suspension, 20 μL of lyophilised E. coli and 60 μL of egg suspension to each well of microtitre plate. Add 20 μL streptopen, 20 μL amphotericine B and 10 μL nystatin to each well. Fill each of the outer wells of the microtitre plate with 200 μL distilled water and seal using the microtitre plate lid.
-
4.
Incubate at 22°C for 24 h and then add plant product or anthelmintic to duplicate wells. The control wells should incorporate DMSO at the same concentration that it is present in the positive control (anthelmintic) test wells.
-
5.
Re-cover the microtitre plate with a lid and seal its edges with PVC tape. Incubate in a 100% relative humidity chamber at 22°C for 7 days.
-
6.
Add 10 μL of helminthological iodine to each well and count the numbers of L3 larvae at × 40 using an inverted microscope. The mean larval development for each drug concentration is calculated using the standard formula:
-
7.
Larval development = (Number of live L3/total number added to wells with anthelmintic)]/[(Number of live L3/total number added to control tubes)
-
8.
Plot the percentage larval development at each concentration on a graph and calculate the LC50 value (concentration at which 50% of the parasites fail to develop) using an appropriate statistical method such as probit analysis.
3.3.7 Adult Motility Assay (AMA)
This test uses freshly collected adult worms that are maintained in a culture medium or in the medium containing the test plant product for several days [13, 14]. Activity of the worms is scored over the period by counting the numbers of active worms at regular times throughout the assay. Paralysing anthelmintics such as ivermectin or levamisole can be used to provide positive control material for the assay. Negative controls (i.e. worms in PBS) are also included.
3.3.7.1 Materials
-
Freshly collected adult worms
-
Plant product dilutions (suggested range 75–1200 μg/mL in distilled water). Phosphate buffered saline (PBS)-antibiotic solution (with 4% w/v penicillin and 4% streptomycin)
-
2% w/v Levamisole solution for positive control
-
24 (for Haemonchus) or 48 (for Teladorsagia and Trichostrongylus) multiwell
-
Plates
-
Inverted microscope or stereomicroscope.
3.3.7.2 Procedure
-
1.
Add 1 mL of the plant product in the PBS antibiotic solution to each of three wells.
-
2.
For the 3 negative control wells, use 1 mL of the PBS/antibiotic solution.
-
3.
For the 3 positive control wells, add 1 mL of a 1% w/v levamisole solution made in the PBS-antibiotic solution.
-
4.
Count adult worms into each of the wells for large worms such as Haemonchus and then use about 2–3 worms per well and increase the numbers for smaller worms (e.g. about 4–6 for adult Teladorsagia and adult Trichostrongylus). If too many worms are used in the assay, it becomes difficult to make accurate observations.
-
5.
Observe and score the activity of the adult worms after 6, 24, 48 or 72 h of incubation at 37°C, 100% relative humidity in a 5% CO2/air mixture. In general, the survival of control Haemonchus is less (48 h) than for Trichostrongylus or Teladorsagia (72 h).
-
6.
Change the incubation medium every 24 h. Medium with the plant concentration can be prepared at the start of the assay and stored at 4°C but the solution have to be placed at 37°C before the changes.
-
7.
For each time point, calculate a motility index based on the numbers of immobile worms divided by the total number of worms in the well.
-
8.
Analyse the survival curve, taking into account the different concentrations, using a suitable non-parametric test such as the stratified Cox regression test run using suitable software.
References
Alvarez-Sanchez, M.A., J.P. Garcia, D. Bartley, F. Jackson, and F.A. Rojo-Vazquez. 2005. The larval feeding inhibition assay for the diagnosis of nematode anthelmintic resistance. Exp. Parasitol. 110:56–61.
Amarante, A.F.T., W.E. Pomroy, W.A.G Charleston, D.M. Leathwick, and M.T.T. Tornero. 1997. Evaluation of a larval development assay for the detection of anthelmintic resistance in Ostertagia circumcincta. Int. J. Parasitol. 27:305–311.
Bahuaud, D., C.M.O. De Montellano, , S. Chauveau, F. Prevot, F. Torres-Acosta, , I. Fouraste, and H. Hoste. 2006. Effects of four tanniferous plant extracts on the in vitro exsheathment of third-stage larvae of parasitic nematodes. Parasitology. 132:545–554.
Barrau, E., N. Fabre, I. Fouraste, and H. Hoste. 2005. Effect of bioactive compounds from Sainfoin (Onobrychis viciifolia Scop.) on the in vitro larval migration of Haemonchus contortus: role of tannins and flavonol glycosides. Parasitology. 131:531–538.
Coles, G.C. and K.G. Simpkin. 1977. Resistance of nematode eggs to the ovicidal activity of benzimidazoles. Res. Vet. Sci. 22:386–387.
Coles, G.C., C. Bauer, F.H.M. Borgsteede, S. Geerts, T.R. Klei, M.A. Taylor and P.J. Waller. 1992. World-Association-For-The-Advancement-Of-Veterinary-Parasitology (WAAVP): methods for the detection of anthelmintic resistance In nematodes of veterinary importance. Vet. Parasitol. 44:35–44.
Geary, T.G., S.M. Sims, E.M. Thomas, L. Vanover, J.P. Davis, C.A. Winterrowd, R.D. Klein, N.F.H. Ho, and D.P. Thompson. 1993. Haemonchus-contortus – ivermectin-induced paralysis of the pharynx. Exp. Parasitol. 77:88–96.
Gill, J.H., J.M. Redwin, J.A. van Wyk, and E. Lacey. 1991. Detection of resistance to ivermectin in Haemonchus-contortus. Int. J. Parasitol. 21:771–776.
Hubert, J. and D. Kerboeuf. 1992. A microlarval development assay for the detection of anthelmintic resistance in sheep nematodes. Vet. Rec. 130:442–446.
Hunt, K.R. and M.A. Taylor. 1989. Use of the egg hatch assay on sheep faecal samples for the detection of benzimidazole resistant nematodes. Vet. Rec. 125:153–154.
Le Jambre, L.F., W.H. Southcott, and K.M. Dash. 1976. Resistance of selected lines of Haemonchus contortus to thiabendazole, morantel tartrate and levamisole. Int. J. Parasitol. 6:217–222.
Ministry of Agriculture, Fisheries and Food. 1986. Manual of Veterinary Parasitological Laboratory Techniques, Her Majesty’s Stationery Office, London. Edition 3.
O’Grady, J. and A.C. Kotze. 2004. Haemonchus contortus: in vitro drug screening assays with the adult life stage. Exp. Parasitol. 106 (3–4):164–172.
Paolini, V., I. Fouraste, and H. Hoste. 2004. In vitro effects of three woody plant and sainfoin on third-stage larvae and adult worms of three gastrointestinal nematodes. Parasitology. 129:69–77.
Rabel, B., R. McGregor, and P.G.C. Douch. 1994. Improved bioassay for estimation of inhibitory effects of ovine gastrointestinal mucus and anthelmintics on nematode larval migration. Int. J. Parasitol. 24:671–676
Taylor, M.A. 1990. A larval development test for the detection of anthelmintic resistance in nematodes of sheep. Res. Vet. Sci. 49:198–202.
Van Wyk, J.A., J. Cabaret, and L.M. Michael. 2004. Morphological identification of nematode larvae of small ruminants and cattle simplified. Vet. Parasitol. 119:277–306.
Varady, M. and J. Corba. 1999. Comparison of six in vitro tests in determining benzimidazole and levamisole resistance in Haemonchus contortus and Ostertagia circumcincta of sheep. Vet. Parasitol. 80:239–249.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 International Atomic Energy Agency
About this chapter
Cite this chapter
Jackson, F., Hoste, H. (2010). In Vitro Methods for the Primary Screening of Plant Products for Direct Activity against Ruminant Gastrointestinal Nematodes. In: Vercoe, P., Makkar, H., Schlink, A. (eds) In vitro screening of plant resources for extra-nutritional attributes in ruminants: nuclear and related methodologies. Springer, Dordrecht. https://doi.org/10.1007/978-90-481-3297-3_3
Download citation
DOI: https://doi.org/10.1007/978-90-481-3297-3_3
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-90-481-3296-6
Online ISBN: 978-90-481-3297-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)