
Abstract
Dissimilatory Sulfite Reductase (dSiR) is the main redox enzyme system utilized in sulfur metabolism in both sulfur oxidizing and reducing prokaryotes. Anoxygenic phototrophic bacteria Allochromatium vinosum produces elemental sulfur during sulfur cycle which is ultimately oxidized sulfate by DSR operon. Allochromatium vinosum encodes dsrAB reverse dSiR that oxidizes thiosulfate or elemental sulfur. DsrAB is a α2β2 hetero-tetrameric complex. In our present study, we first reported the three dimensional structure of DsrAB protein complex from Allochromatium vinosum and we also predicted the protein-protein interactions between DsrA and DsrB proteins. DsrAB is a major redox enzyme complex required in both sulfur oxidation and reduction processes so this structure function relationship investigation will help in researches to predict the biochemical mechanism of sulfur-oxidation. The importance of the study lies in the fact that sulfur metabolism pathways are used in waste remediation and bio-hydrogen production. This is the most important aspect of our analysis.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
- Dsr operon
- DsrAB
- Allochromatium vinosum
- Homology modeling
- Protein-protein docking
- Active site interaction
1 Introduction
Sulfur cycle in one of the most important biochemical cycles. Sulfur has a wide range of oxidation states to perform redox reactions of sulfur anions. Sulfur oxidizing prokaryotes (SOP) catalyze a central step in the global S-cycle and are of major functional importance for a variety of natural and engineered systems, but very little is known about their structure functional aspect. Allochromatium vinosum (A. vinosum), a dominant member of purple sulfur bacteria, and having lots of significant roles in biotechnological industry [1–4] encodes dsr operon which has central role in dissimilatory sulfur metabolism. A. vinosum produces sulfur stored in periplasmic sulfur globules as an obligate intermediate during the oxidation of sulfide and thiosulfate [5, 6]. dsr operon encoding 15 genes viz. dsrABEFHCMKLJOPNRS are required for oxidation of sulfide and thiosulfate. A. vinosum carries out the complete eight-electron oxidations of sulfide and thiosulfate to sulfate [7]. Among the gene products of dsr operon, DsrAB plays a vital role in the metabolism of sulfur anions. DsrAB protein complex encodes a reverse dissimilatory sulfite reductase, the principal enzyme of sulfur oxidation, produces sulfite, which is oxidized to the final product sulfate either directly via the membrane-bound iron-sulfur molybdoprotein SoeABC or by an indirect pathway involving formation of adenosine-5-phosphosulfate (APS) catalyzed by APS reductase and ATP sulfurylase [8, 9]. DsrAB from A. vinosum is a α2β2-heterotetrameric complex known as dissimilatory sulfite reductase (dSiR). DsrAB is a cytoplasmic protein complex containing siro(heme)amide-[Fe4S4] as prosthetic group [10]. Till date no reports are available for this protein complex from A. vinosum from a structural point of view. In our present study we for the first time made three dimensional structures of DsrAB protein and investigated the molecular details of the protein–protein interactions. This study would therefore be beneficial to predict the hitherto unknown molecular details of the protein interactions in sulfur oxidation processes.
2 Material and Methods
2.1 Homology Modeling of DsrA and DsrB Proteins
The amino acid sequences of DsrA and DsrB proteins from A. vinosum were obtained from Entrez database (Accession No. YP_003443222.1 for DsrA protein and YP_003443223.1 for DsrB protein). The amino acid sequences of the DsrA and DsrB proteins were used to search Brookhaven Protein Data Bank (PBD) [11] using the software BLAST [12] for finding suitable templates for homology modeling. The X-ray crystal structure of Desulfovibrio vulgaris Dissimilatory Sulfite Reductase at 2.1 Å resolution (PDB code: 2V4J) was identified as most suitable template for homology modeling for both the DsrA and DsrB proteins. The BLAST result of DsrA showed 43 % sequence identity with the A chain of 2V4J whereas DsrB had 59 % sequence identity with the B chain of 2V4J. Homology modeling was done by MODELLER 9 v 4 [13], inbuilt in the Discovery Studio package along with loop refinement [14] as parameter. The final models of the protein were chosen from the ten models produced on the basis of the lowest value of the Modeler objective function and the least ‘discrete optimized potential energy’ (DOPE) score.
2.2 Energy Minimization and Validation of the Models
The un-favorable interactions in the models of DsrA and DsrB proteins were removed by applying energy minimization technique using CHARMm [15] force field in two steps; first 500 cycles of minimizations were done using Steepest Descent (SD) algorithm and then 500 steps of Conjugate Gradient (CG) algorithm were done using Geraralized Bonn with Simple Switching (GBSW) until the structures reached the final energy derivative of 0.001 kcal /mole as employed in the simulation module of Discovery Studio 2.5. Then the stereo-chemical qualities of the three dimensional models of DsrA and DsrB proteins were checked by VERIFY3D [16] and PROCHECK [17] and Ramachandran Plots [18] were drawn in SAVes server (http://nihserver.mbi.ucla.edu/SAVES/) [19].
2.3 Docking of DsrA and DsrB Proteins
The docked complex of DsrAB was generated using ZDOCK server [20] of DS package. Angular step size for rotational sampling of ligand orientations was set to 15°. The active site residues of the DsrA and DsrB proteins were predicted before docking using the servers http://www.scfbio-iitd.res.in/dock/ActiveSite_new.jsp [21], http://dogsite.zbh.uni-hamburg.de/ [22], http://www.modelling.leeds.ac.uk/pocketfinder/ [23]. These predicted amino acid residues were used while docking the proteins in order to avoid blind docking. Top 1,000 poses were retained for evaluation using ZRANK [24] scoring function. The final docking poses were selected on the basis of the scores of the docked complexes from ZRANK.
2.4 Refinement of Docked DsrAB Protein Complex
RDOCK, an algorithm for refinement of docked complexes using the CHARMm-based minimization, was used to optimize and re-rank the docking poses to pick up near-native structures [25]. The docked poses selected from the ranking scores of ZRANK were typed with the CHARMm Polar H force field. The refined docked complex obtained from RDOCK was energy minimized using 500 cycles of Steepest Descent (SD) algorithm followed by 500 steps of Conjugate Gradient (CG) algorithm using Geraralized Bonn with Simple Switching (GBSW) as employed in the simulation module of Discovery Studio 2.5. Now the stereo-chemical qualities of the refined structures of the DsrAB complex were checked by PROCHECK and VERIFY3D as per the protocol mentioned before.
The interaction energy of stable most conformation of DsrAB protein was calculated using Discovery Studio 2.5. The interacting amino acid residues were identified and H-bonds formed were checked in DS. This study was verified by Protein Interaction Calculator (P.I.C.) [26] once again.
3 Results and Discussion
3.1 Three Dimensional Structures of DsrA and DsrB of A. vinosum
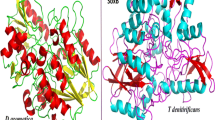
After models built in Modeller and followed by energy minimization and validation checking it was found that no residues were present in the disallowed regions of the Ramachandran Plots. The residue profile score as obtained from VERIFY3D was found to be 87.86 %; i.e., 87.86 % of residues had an averaged 3D-1D score >0.2 thereby qualifying the criterion of DsrA protein model whereas for DsrB it was 85.43 %. Modeled structures of DsrA and DsrB were then superimposed on the crystal templates without altering the coordinate systems of atomic positions in the template. The root mean squared deviations (RMSD) for the superimpositions were 0.989 Å for DsrA and 1.004 Å for DsrB on to the crystal templates indicating a good model building. The three- dimensional structures of DsrA and DsrB proteins were described in Fig. 1.

Three-dimensional model of DsrAB protein-protein complex. Figure made using Pymol. DsrA protein showing secondary structure in cyan (helix), violet (sheet), brown (loop) colour and DsrB protein showing red, yellow and green colour as helix, sheet and loop respectively
DsrA: The DsrA is a 411 amino acid residue long protein. There are a total of fifteen helices (amino acid residues 6-12, 42-52, 103-116, 137-150, 180-190, 192-196, 218-221, 238-246, 253-260, 286-290, 336-352, 359-366, 368-375, 381-383, 402-408) and six sheets (amino acid residues 89-93, 130-135, 205-209, 224-230, 301-307, 321-324).
DsrB: DsrB is a 356 amino acid residue long protein. The modeled three diemnsional structure consists of thirteen helices (amino acid residues 21-25, 64-78, 97-109, 140-156, 195-201, 204-210, 231-233, 245-247, 290-306, 313-320, 322-329, 335-337, and 342-347) and eleven sheets (amino acid residues 31-34, 40-45, 48-56, 83-85, 88-94, 165-169, 181-186, 216-220, 225-229, 256-261, 276-279) joined by loop regions.
3.2 Calculation of Interactions Between DsrAB Protein Complex
The interacting amino acids from DsrAB protein complexes of A. vinosum calculated by Discovery Studio 2.5 are listed in Table 1.
The total interaction energy of DsrAB complex was found −827.71532 kcal/mol and electrostatic and VDW interaction energies of major amino acid residues were shown in Fig. 2.

Electrostatic and VDW interaction energy of some major amino acid residues
Several H-bonds were formed between DsrA and DsrB protein complex (Fig. 3a–c).



a Hbond interaction showing in Black dotted line between His30, Lys86, Asp136 of DsrA (A chain in red colour and residues are in violet) and Glu9, Gly11, Cys12, Tyr18 of DsrB (B chain in green colour and residues are in orange) protein. b Hbond interaction showing in Black dotted line between Asp145, Glu 146, Asp 149 of DsrA (A chain in red colour and residues are in violet) and Arg60, Gln 128, Gly 176 of DsrB (B chain in green colour and residues are in orange) protein. c Hbond interaction showing in Black dotted line between Arg 185, Asn 189, Asp 334 of DsrA (A chain in red colour and residues are in violet) and Glu 203 and Arg204 of DsrB (B chain in green colour and residues are in orange) protein
Some hydrophobic and ionic interactions were also present within DsrAB complex as shown in Table 2.
The DsrAB protein complex is bound by strong interactions. These interactions help to bind the proteins together and allow them to be involved in sulfur oxidation processes.
4 Conclusion
DsrAB protein complex is one of the central players in sulfur oxidation process. In the present work we have analyzed the three dimensional structures of the proteins by homology modeling techniques. By molecular docking techniques we analyzed the mode of binding between the proteins. This is the first report to predict the biochemical mechanism of binding of these proteins. This study would therefore be essential to predict the hitherto unknown molecular biochemistry of sulfur oxidation process.
References
Sasikala, C., Ramana, C.V.: Biotechnological potentials of anoxygenic phototrophic bacteria. 1. Production of single-cell protein, vitamins, ubiquinones, hormones, and enzymes and use in waste treatment. Adv. Appl. Microbiol. 41, 173–226 (1995)
Sasikala, C., Ramana, C.V.: Biotechnological potentials of anoxygenic phototrophic bacteria. 2. Bio-polyesters, biopesticide, biofuel, and biofertilizer. Adv. Appl. Microbiol. 41, 227–278 (1995)
Liebergesell, M., Steinbüchel, A.: New knowledge about the pha-locus and P (3HB) granule-associated proteins in Chromatium vinosum. Biotechnol. Lett. 18, 719–724 (1996)
Sasikala, K., Ramana, C.V., Rao, P.R., Kovács, K.L.: Anoxygenic photosynthetic bacteria: physiology and advances in hydrogen production technology. Adv. Appl. Microbiol. 38, 211–295 (1993)
Grein, F.: Biochemical, biophysical and functional analysis of the DsrMKJOP transmembrane complex from Allochromatium vinosum. PhD thesis. Rhenish Friedrich Wilhelm University, Bonn (2010)
Pott, A.S.: Dahl. C.: Sirohaem-sulfite reductase and other proteins encoded in the dsr locus of Chromatium vinosum are involved in the oxidation of intracellular sulfur. Microbiology 144, 1881–1894 (1998)
Dahl, C., Engels, S., Pott-Sperling, A.S., Schulte, A., Sander, J., Lübbe, Y., Oliver Deuster, O., Brune. D.C.: Novel genes of the dsr gene cluster and evidence for close interaction of Dsr Proteins during sulfur oxidation in the phototrophic sulfur bacterium Allochromatium vinosum. J. Bacteriol. 187, 1392–1404 (2005)
Dahl, C., Franz, B., Hensen, D., Kesselheim, A., Zigann, R.: Sulfite oxidation in the purple sulfur bacterium Allochromatium vinosum: Identification of SoeABC as a major player and relevance of SoxYZ in the process. Microbiology 159, 2626–2638 (2013)
Sanchez, O., Ferrera, I., Dahl, C., Mas, J.: In vivo role of APS reductase in the purple sulfur bacterium Allochromatium vinosum. Arch. Microbiol. 176, 301–305 (2001)
Lübbe, Y.J., Youn, H., Timkovich, R., Dahl, C.: Siro (haem) amide in Allochromatium vinosum and relevance of DsrL and DsrN, a homolog of cobyrinic acid a, c-diamide synthase, for sulphur oxidation. FEMS Microbiol. Lett. 261, 194–202 (2006)
Berman, H.M.: The Protein Data Bank: a historical perspective. Acta Crystallogr. A 64, 88–95 (2008)
Altschul, S.F., Gish,W., Miller, W., Myers, E.W., Lipman, D.J.: Basic local alignment search tool. J. Mol. Biol. 215(3), 403–410 (1990)
Sali, A., Pottertone, L., Yuan, F., van Vlijmen, H., Karplus, M.: Evaluation of comparative protein modeling by MODELLER. Proteins 23, 318–326 (1995)
Fiser, A., Kinh Gian Do, R., Sali, A.: Modeling of loops in protein structures. Protein Sci. 9, 1753–1773 (2000)
Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., Karplus, M.: CHARMM: a program for macromolecular energy minimization and dynamics calculations. J. Comp. Chem. 4, 187–217 (1983)
Lüthy, R., Bowie, J.U., Eisenberg, D.: Assessment of protein models with three-dimensional profiles. Nature 356(6364), 83–5 (1992)
Laskowski, R.A., McArthur, M.W., Moss, D.S., Thornton, J.M.: PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 (1993)
Ramachandran, G.N., Ramakrishnan, C., Sasisekharan, V.: Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 7, 95–99 (1963)
Structural Analysis and Verification Server, http://nihserver.mbi.ucla.edu/SAVES/
Chen, R., Weng, Z.: Docking unbound proteins using shape complementarity, desolvation, and electrostatics. Proteins 47, 281–294 (2002)
Supercomputing Facility for Bioinformatics & Computational Biology, IIT Delhi, Active Site Prediction, http://www.scfbio-iitd.res.in/dock/ActiveSite_new.jsp
University Hamburg, Centre of Bioinformatics, DoGSiteScorer: Active Site Prediction and Analysis Server, http://dogsite.zbh.uni-hamburg.de/
Pocket-Finder, http://www.modelling.leeds.ac.uk/pocketfinder/
Pierce, B., Weng, Z.: ZRANK: reranking protein docking predictions with an optimized energy function. Proteins 67, 1078–1086 (2007)
Li, L., Chen, R., Weng, Z.: RDOCK: refinement of rigid-body protein docking predictions. Proteins 53, 693–707 (2003)
Tina, K.G., Bhadra, R., Srinivasan, N.: PIC: Protein interaction calculator. Nucleic Acid Res. 35, Web server issue W473–W476 (2007)
Acknowledgment
Ms. Semanti Ghosh is thankful to the University of Kalyani, West Bengal, India and University Grant Commission, India for the financial support. We would like to thank the Bioinformatics Infrastructure Facility and also the DST-PURSE program 2012–2015 going on in the Department of Biochemistry and Biophysics, University of Kalyani for their support.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer India
About this paper
Cite this paper
Ghosh, S., Bagchi, A. (2015). Intermolecular Interaction Study of Dissimilatory Sulfite Reductase (DsrAB) from Sulfur Oxidizing Proteobacteria Allchromatium vinosum . In: Mandal, J., Satapathy, S., Kumar Sanyal, M., Sarkar, P., Mukhopadhyay, A. (eds) Information Systems Design and Intelligent Applications. Advances in Intelligent Systems and Computing, vol 340. Springer, New Delhi. https://doi.org/10.1007/978-81-322-2247-7_3
Download citation
DOI: https://doi.org/10.1007/978-81-322-2247-7_3
Published:
Publisher Name: Springer, New Delhi
Print ISBN: 978-81-322-2246-0
Online ISBN: 978-81-322-2247-7
eBook Packages: EngineeringEngineering (R0)