
Abstract
Microbial redox reactions are mediated by a diverse set of sulfur-oxidising bacteria. These redox reactions are important to maintain the environmental sulfur balance. The sulfur oxidation reactions are performed by sulfur-oxidizing gene cluster called the sox operon comprising of genes soxEFCDYZAXBH. However, the mechanistic details of sulfur oxidation process by Hydrogenophilus thermoluteolus are yet to be determined. In this study, the three-dimensional structures of SoxY and SoxZ proteins were constructed by homology modeling. Protein-protein docking generated SoxY–Z complex. Responsible amino acid residues for the protein interactions were identified after molecular dynamics simulation of SoxY–Z complex. The best binding mode of thiosulfate with SoxY–Z complex was identified through their molecular docking. Current study thereby, provides a rational frame-work to discern molecular mechanism and biophysical characterization of sulfur-oxidation process.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others
Keywords
- Sulfur oxidation
- Homology modelling
- Sox operon
- Protein-protein interaction
- Molecular docking
- Molecular dynamics simulation
1 Introduction
Microorganisms are mainly responsible to maintain the biogeochemical cycling of sulfur [1]. Sulfur, having broad range of oxidation states, experiences varied bioinorganic reactions which reduces or oxidises sulfur. The thiosulfate, tetrathionate and sulfide are natural sources of sulfur for redox reactions. Environmentally abundant sulfur anion, thiosulfate serves a major role to maintain environmental sulfur balance. A gene cluster called sox operon is responsible for sulfur oxidation in phylogenetically diverse set of microorganisms [2–6]. Nevertheless, the biomolecular mechanism of sulfur oxidation process by sox operon is not properly understood. Most of the experiments were done in case of Paracoccus pantotrophus and Rhodovulum sulfidophilum [2–6].
It is known that two divergently transcribed transcriptional units comprising genes soxSR and soxVWXYZABCDEFGH are found in mesophilic Paracoccus pantotrophus and Rhodovulum sulfidophilum [2–6]. The proposed molecular mechanism of oxidation of thiosulfate anion in Paracoccus pantotrophus shows that thiosulfate gets coupled to carboxy terminal cysteine residue of SoxY–Z complex with the help of SoxXA. Immediately, SoxB hydrolytically cleaves the SoxY-thiosulfate adduct to release sulfate. In Paracoccus pantotrophus, Sox(CD)2 and SoxB help to recycle SoxY protein by hydrolytically releasing a sulfate from SoxY [7–9]. SoxY and SoxZ proteins are key regulators of this oxidation cycle. However, the detailed bio-molecular mechanism of sulfur oxidation process in Hydrogenophilus thermoluteolus is not yet understood. In this study, we built the computational models of SoxY and SoxZ proteins from Hydrogenophilus thermoluteolus (Hthermo). We used molecular docking to build the structure of SoxY–Z protein complex. This complex was then docked with thiosulfate. The interaction between SoxY–Z docked protein was further analyzed by molecular dynamics simulations. Molecular modelling and protein-protein docking studies reveal the strong interaction of SoxY and SoxZ proteins. As this study is the first approach towards the structural basis of involvements of SoxY and SoxZ from Hthermo in biochemical oxidation of sulfur anions, it may shed light towards investigation of complex-thiosulfate oxidation process, in moderately thermophilic Hthermo.
2 Materials and Methods
2.1 Sequence Analysis and Homology Modelling of Monomeric SoxY and SoxZ Proteins
Amino acid sequences of SoxY and SoxZ proteins of Hthermo were obtained from NCBI (Accession no: NC_ AB268546.1). They were then separately used for homology modelling by Modeller Package (version 9.10) [10].
In order to identify the suitable templates for homology modelling, BLAST against Brookhaven Protein Data Bank (PDB) [11, 12] was done using the amino acid sequences. The search results for SoxY and SoxZ picked up the X-Ray crystal structure for SoxY from Chlorobium limicola f sulfatophilum (PDB code: 2NNC A-chain) and for SoxZ from Paracoccus denitrificans (PDB code: 2OXH Z-chain) with 43 and 40 % sequence identities respectively. 2NNC and 2OXH were then used as templates for homology modelling using Modeller Package. Root Mean Squared Deviations (RMSD) for each model protein structures was calculated by superimposing the structures separately on each of the crystal templates. RMSDs were 0.269 and 0.241 Å respectively for SoxY and SoxZ proteins with their corresponding crystal templates.
Modelled proteins were then energy minimized in two steps. Steepest decent technique was followed by conjugate gradient technique to minimize the overall modelled structures using the Discovery studio software until the structures reached the final RMS gradient of 0.0001 kcal/mole. All energy minimizations were done using CHARMM force field by fixing the backbones of the proteins [13].
2.2 Validation of Models
PROSA web server was used to calculate the Z-score of the predicted homology models. The predicted models were well inside the range of typical native structures [14]. The residue profiles of the three-dimensional models were further checked by VERIFY3D [15]. PRO-CHECK [16] analysis was performed to assess the stereo-chemical qualities of the models. Ramachandran plots [17] were drawn. No residues were found to be present in disallowed regions of individual Ramachandran plots.
2.3 Protein-Protein Molecular Docking Simulations
In order to study interactions between SoxY–Z protein complex, SoxY and SoxZ proteins were docked using the software Cluspro 2.0 [18]. The resulting complex structure was energy minimized by fixing backbones of the proteins in the complex by steepest descent technique using CHARMM force field [13].
2.4 Molecular Dynamics Simulations
In order to study the interactions between the proteins, molecular dynamics simulations (MDs) were done using the Gromacs suite, version 4.6.5 [19]. The SoxY–Z protein complex was dissolve in a SPC216 water-solvated periodic environment. Water molecules were added using the truncated octahedron box extending 3 Å from each atom. Neutralization of molecular systems was done with counter ions as required. For the purpose of this study, all MDs were performed using the NVT followed by NPT in a canonical environment at 300 K and a step size equal to 2 fs for a total of 100 ps simulation time. Final MD run was done using step size 2 fs for a total of 70 ns simulation time at 300 K. Change in back bone RMSDs were plotted against time to check the progress of the simulation run.
2.5 Calculation of Protein-Protein Interaction
To calculate the interactions between the SoxY, SoxZ, Protein Interaction Calculator (PIC) web server was used. Protein complex (SoxY–Z) structure was provided to P.I.C web server to calculate various kinds of interactions; such as disulfide bonds, hydrophobic interactions, ionic interactions, hydrogen bonds, aromatic-sulfur interactions, aromatic-aromatic interactions and cation-π interactions. PIC web server recognizes a single protein or protein in complex for the examination in the interaction [20].
2.6 Protein-Ligand Molecular Docking Interactions
The complex docked structure of SoxY and SoxZ protein was again docked with thiosulfate anion by AutoDock package (version 4.2) [21, 22]. It accepts PDBQT molecular structure file format as input and output [22]. Only the structures of molecules to be docked and specification of the search space including the binding site was required. Interactions between SoxY–Z complex and thiosulfate anion were observed and examined by PyMOL.
3 Results
3.1 Description of the Structure of SoxY
The modelled structure of SoxY is a 123 amino acid residue long protein. The predicted structure is very similar to the structure of Sulfur Carrier Protein SoxY from Chlorobium Limicola F Thiosulfatophilum (PDB code: 2NNC A chain). The protein is made up of α-helix, β-strands and random coil region. N-terminal position was made up of α-helical region (amino acid residues 2-11). Majority part of the structure is made up of β strands spanned by coil regions (mainly amino acid residues 26-28, 32-35, 43-52, 58-63, 72-76, 84-91, 95-103 and 106-114 are the β strand regions). The structure is presented in Fig. 1.

Model structure of SoxY protein from Htherm with distinct α-helix (cyan), β-structure (purple) and random coil (tan)
3.2 Description of the Structure of SoxZ
The model of SoxZ protein consists of 102 amino acid residues. The predicted structure is very similar to the structure of SoxZ protein from Paracoccus denitrificans (PDB Code: 2OXH_Z). It is made up of seven β-strands (amino acid residues 7-10, 15-21, 45-51, 54-61, 70-76, 81-89 and 92-98) interspersed with coil region. Both N and C terminal ends of the protein are composed of β-strand regions. The structure is presented in Fig. 2.

Model structure of SoxZ protein from Hthermo showing distinct β-structure (purple shade) and random coil (tan shade)
3.3 Interactions of SoxY with SoxZ
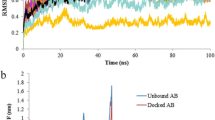
In order to find the interactions between the SoxY and SoxZ, the three-dimensional structures of the proteins were docked by software tool Cluspro 2.0. SoxY and SoxZ interact strongly with each other mainly via H-bonding between both the main and the side chains of the SoxY–Z complex as observed from P.I.C. The SoxY–Z complex is also stabilized by the hydrophobic and ionic interactions. Tables 1 and 2 describe the residue level interactions in SoxY–Z protein complex. The complex structure is presented in Fig. 3. The interactions were analyzed after performing molecular dynamics simulation. The progress of the simulation run was measured by plotting the backbone RMDS against time in picoseconds as in Fig. 4.

Represents docked SoxY–Z complex with distinct α-helix (cyan), β-structure (purple) and random coil (tan)

Represents the plotting of backbone RMSD versus time in picoseconds
3.4 Interactions of SoxY–Z-Thiosulfate Complex
Thiosulphate anion was docked using AutoDock package (version 4.2). The plugin of PyMOL helped to identify that thiosulfate anion mainly binds to Gly120, Cys121, Gly122 and Lys90 of SoxY–Z complex via intermolecular H-bonds. Furthermore, Lys90 provides stability to the complex structure. Interestingly, SoxY of Hthermo has a sulfur anion binding signature GGCGG sequence at carboxy terminal region of the protein containing a free thiol group in the Cysteine residue 121 which binds the sulfur anion (thiosulfate). Figure 5 represents the binding.

Describes binding of Thiosuphate to Gly120, Cys121, Gly122 and Lys90 of SoxY–Z complex via intermolecular H-bonds (green dots)
4 Discussion
In this study, an attempt was made to analyze the structural basis of the involvements of SoxY, SoxZ, their mutual binding and role in sulfur oxidation. For this purpose, three-dimensional structures of SoxY and SoxZ have been built and analysed using comparative modelling technique. Protein-protein docking studies predicted putative binding orientations of SoxY and SoxZ proteins. P.I.C webserver identified the potential residues for the protein-protein interactions. The interaction scheme revealed the presence of H-bonding in between both main and side chains of SoxY–Z complex. Structural analysis revealed that main chain-side chain H-bonding interactions in the complex of Hthermo were formed among Asn69-Ala41, Thr87-Pro4 and Val74-Asn68 of SoxY and SoxZ proteins respectively as shown in Table 1. It also unveiled that three side chain-side chain H-bonding interactions in the complex of Hthermo were formed among Asn7 of SoxY with Ser35 of SoxZ protein as shown in Table 2. On the basis of in silico docking studies, possible molecular basis of binding of thiosulfate ion with SoxY–Z complex has been described. From the interaction pattern it is predicted that intermolecular H-bonding is mainly involved in SoxY–Z and thiosulfate interaction. Thiosulfate ion mainly interacts with Gly120, Cys121, Gly122 and Lys90 of SoxY–Z complex via intermolecular H-bonds. Furthermore, Lys90 aids in stability of the complex. This is so far the first report to analyze the molecular details of the binding interactions between the proteins and thiosulfate in Hthermo. So this report may help us to understand the three-dimensional structures of SoxY and SoxZ as well as to elucidate the structural basis of the molecular functions of these proteins. In a nutshell, present study provides a rational framework for designing experiments to determine the contribution of the various amino acid residues in SoxY and SoxZ proteins and to predict the molecular basis of the interactions both, among themselves as well as with various sulfur anions.
5 Conclusion
This is the first report to predict the molecular details of the interactions between SoxY–Z protein complex with thiosulfate. This study would therefore be beneficial to understand the molecular basis of the interactions between these proteins in sulphur oxidation process. There were reports on alpha proteobacteria but no reports were available for betaproteobacteria. From that point of view this work would provide a new dimension to the sulphur oxidation process.
Abbreviations
- Hydrogenophilus thermoluteolus :
-
Hthermo
- Protein interaction calculator:
-
P.I.C
References
Friedrich, C.G., Bardischewsky, F., Rother, D., Quentmeier, A., Fischer, J.: Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 8, 253–259 (2005)
Freidrich, C.G.: Physiology and genetics of sulfur- oxidizing bacteria. Adv. Microb. Physiol. 39, 235–289 (1998). doi:10.1016/S0065-2911(08)60018-1
Appia-Ayme, C., et al.: Cytochrome complex essential for photosynthetic oxidation of both thiosulfate and sulfide in Rhodovulum sulfidophilum. J. Bacteriol. 183, 6107–6118 (2001)
Bagchi, A., Ghosh, T.C.: Structural insight into the interactions of SoxV, SoxW and SoxS in the process of transport of reductants during sulfur oxidation by the novel global sulfur oxidation reaction cycle. Biophys. Chem. 119, 7–13 (2006)
Bagchi, A., et al.: Homology modeling of a tran-scriptional regulator SoxR of the lithotrophic sulfur oxidation (Sox) operon in α-proteobacteria. J. Biomol. Struct. Dyn. 22, 571–578 (2005)
Bagchi, A.; and Roy, P.; Structural insight into SoxC and SoxD interaction and their role in electron transport process in the novel global sulfur cycle in Paracoccus pantotrophus. Biochem. Biophys. Res. Commun. 331:1107–1103 (2005)
Bagchi, A.: Structural modeling of SoxF protein from Chlorobium tepidum: an approach to understand the molecular basis of thiosulfate oxidation. Biochem. Biophys. Res. Commun. 414, 409–411 (2011)
Bagchi, A., Ghosh, T.C.: Structural analyses of the interactions of SoxY and SoxZ from thermo-neutrophilic Hydrogenobacter thermophiles. J. Biophys. Chem. 2(4), 408–413 (2011)
Bagchi, A.: Structural insight into the mode ofinteractions of SoxL from Allochromatium vinosum in the global sulfur oxidation cycle. Mol. Biol. Rep. 39(12), 10243–10248 (2012)
Sali, A., Potterton, L., Yuan, F., Van Vlijmen, H., Karplus, M.: Evaluation of comparative protein modeling by MODELLER. Proteins 23(3), 318–326 (1995)
Berman, M.H., et al.: The protein data bank. Nucleic Acids Res 28, 235–242 (2000)
Altschul, S.F., et al.: Basic local alignment search tool. J. Mol. Biol. 25, 403–410 (1990)
Brooks, B.R., Bruccoleri, R.E., Olafson, B.D. States, D.J., Swaminathan, S., Karplus, M.: CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 4,187–217 (1983)
Sippl, M.J.: Recognition of errors in three-dimen- sional structures in proteins. Proteins 17, 355–362 (1993)
Eisenberg, D., et al.: VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 277, 396–404 (1997)
Laskowski, R.A., et al.: PROCHECK: a program to check the stereochemistry of protein structures. J. Appl. Crystallogr. 26, 283–291 (1993)
Ramachandran, G.N., Sashisekharan, V.: Con- formation of polypeptides and proteins. Adv. Protein Chem. 23, 283–438 (1968)
Comeau, S.R., et al.: ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 20, 45–50 (2004)
Hess, B., Kutzner, C., Van Der Spoel, D., Lindahl, E.: GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theor. Comput. 4(3), 435–447 (2008)
Tina, K.G., Bhadra, R., Srinivasan, N.: PIC: protein interactions calculator. Nucleic Acids Res. 35, W473–W476 (2007)
Morris, G.M., Huey, R., Lindstrom, W., Sanner, M.F., Belew, R.K., Goodsell, D.S., Olson, A.J.: Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J. Comput. Chem. 30, 2785–2791 (2009)
Cosconati, S., Forli, S., Perryman, A.L., Harris, R., Goodsell, D.S., Olson, A.J.: Virtual screening with AutoDock: theory and practice. Expert Opin. Drug Discovery 5, 597–607 (2010)
Acknowledgment
The authors would like to thank the DST-PURSE programme 2012–2015 going on in the Department of Biochemistry and Biophysics, University of Kalyani for providing different instrumental and infrastructural support. Authors are also thankful to the DBT sponsored Bioinformatics Infrastructure Facility in the Department of Biochemistry and Biophysics, University of Kalyani for the necessary support.
Conflict of Interest
None
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer India
About this paper
Cite this paper
Ray, S., Bagchi, A. (2015). An In-Silico Structural Analysis of the Interactions of SoxY and SoxZ from Moderately Thermophilic Betaproteobacterium, Hydrogenophilus thermoluteolus in the Global Sulfur Oxidation Cycle. In: Mandal, J., Satapathy, S., Kumar Sanyal, M., Sarkar, P., Mukhopadhyay, A. (eds) Information Systems Design and Intelligent Applications. Advances in Intelligent Systems and Computing, vol 340. Springer, New Delhi. https://doi.org/10.1007/978-81-322-2247-7_1
Download citation
DOI: https://doi.org/10.1007/978-81-322-2247-7_1
Published:
Publisher Name: Springer, New Delhi
Print ISBN: 978-81-322-2246-0
Online ISBN: 978-81-322-2247-7
eBook Packages: EngineeringEngineering (R0)