
Abstract
Most of the length of the Y chromosome escapes recombination with the X chromosome and is strictly paternally inherited. This has profound evolutionary implications and provides (together with the matrilineal mitochondrial DNA) unique tools in human population genetics. Absence of recombination implies that a most parsimonious tree can be easily constructed from SNPs and other slowly mutating polymorphisms. The main branches of this tree, called haplogroups, have distinct geographic distributions and can be used to trace human migrations. Faster evolving polymorphisms, such as microsatellites provide readily time estimates for these migrations and other demographic events. Beyond sets of predefined polymorphism, sequencing of most of the non-recombining portion of the Y chromosome has yielded an accurate picture of the global evolution of this chromosome. Combining the Y chromosome with mitochondrial DNA has revealed sex-specific migrations, particularly in the Colonial period. Since surnames are also patrilineally inherited in many populations, the analysis of Y chromosome variation within surnames has shed light on the dynamics of surnames in populations, but has also contributed to the investigation of notorious lineages, such as the Columbus, Bourbons, and Draculs. However, it also raises the possibility of predicting a surname from an anonymous sample, which may be an important tool in forensic genetics but raises also privacy concerns for participants in genetic studies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 The Odd Chromosome and Its History
The Y chromosome is a genomic oddity: it is the squat half of the only mismatched couple in the karyotype, rather smaller than its X partner, and containing fewer genes. As we shall briefly discuss below, these features arose from its function: the presence of the Y chromosome in humans, as in eutherian mammals and other animals, steers development towards the production of a male embryo. The main masculinity switch is a gene called SRY. The human Y chromosome only recombines with the X chromosome at two small regions at the ends of the chromosome, called pseudoautosomal regions 1 and 2 (PAR1 and PAR2). The non-recombining region of the Y chromosome (NRY) covers most of its ~23 euchromatic Mb; since it does not recombine with the X chromosome, it is male specific (and thus, it is also denoted MSY) and evolves independently of the X chromosome.
Y chromosomes have arisen multiple times in evolutionary history: once in eutherian mammals, and, independently, multiple times in reptiles, amphibians, and fish (Bachtrog 2013). The sex chromosomes start their evolutionary life as an autosome pair in which a sex-determining gene appears. Any mutation that is beneficial to males but detrimental to females increases its fitness if it is inherited in tight linkage with such sex-determining gene. Thus, lack of recombination in this young Y chromosome is beneficial, and inversions seem to have been selected as a mechanism to inhibit recombination with the X chromosome. This process proceeded in stages until recombination was blocked for most of the length of the Y chromosome (Bachtrog 2013); by comparing the levels of divergence between genes in the Y chromosome and their homologues in the X chromosome, four such stages or strata have been recognized (Lahn and Page 1999). But lack of recombination comes with a price: detrimental mutations are more difficult to eliminate from the Y chromosome, and, on the contrary, beneficial mutations are harder to fixate. Recombination allows decoupling the evolution of a particular mutation from that of its immediate genomic vicinity; recombinant chromosomes lacking a particular deleterious allele will be selected for, increasing the efficiency of natural selection. However, when a deleterious mutation lands on the NRY, negative selection will affect the whole chromosome, including any weakly beneficial mutations. This has led to a drastic purge of genes on the Y chromosome (78 protein-coding genes in the MSY compared with 800 protein-coding genes in the X chromosome); most of the remaining genes have been shown to have male-related functions (Lahn and Page 1997).
The human Y chromosome is made up of five types of sequences: the pseudoautosomal regions ; a large heterochromatic block (~40 Mb); the X-degenerate region that derives from the common autosomal ancestor with the X chromosome; the more recently X-transposed region , that is unique to humans and was transposed 3–4 million years ago; and the highly repetitive ampliconic regions , which make sequencing and assembling the Y chromosome a daunting task. For a detailed discussion on the gene content of the Y chromosome, see Satta and Iwase (2015).
2 Not All Y Chromosomes Are Created Equal: Polymorphisms in the Y Chromosome
Genetic polymorphisms in the Y chromosome (as in the rest of the genome) come in different forms, each with their applications in human population genetics, as discussed below. The most commonly used types currently are UEPs (unique event polymorphisms) and STRs (short tandem repeats). Minisatellites (Bouzekri et al. 1998) and probes tested with restriction enzymes (Lucotte and Ngo 1985) are no longer favored given the technical difficulties in genotyping them and the complexity of their analysis.
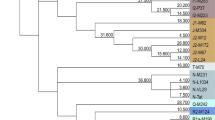
UEPs are polymorphisms created by mutations at such a slow rate that they can be assumed to have happened just once in the human lineage. They are single nucleotide polymorphisms (SNPs), short indels, and Alu insertions. Given the lack of recombination in the NRY, UEPs can be readily assorted into a maximum parsimony tree; the deepest branches of such a phylogeny (Fig. 5.1) are called haplogroups, and have particular geographical distributions (Fig. 5.2), and, thus, can be (and have been) used in exploring the geographic ancestry of male lineages. The robustness of the phylogeography of NRY UEPs allows to detect easily the few cases in which UEPs are not actually unique but have happened in different branches or have reverted to the ancestral state (see, for example Adams et al. (2006)).

Schematic tree of the Y chromosome haplogroups. Branch lengths are proportional to the number of UEPs in the 2013 ISOGG tree. Dotted lines indicate the length of the deepest branch in each haplogroup (http://www.isogg.org/tree/ISOGG_YDNATreeTrunk.html)

Haplogroup frequencies in selected world populations
The initial search for diversity in the human Y chromosome was rather disappointing (Jobling and Tyler-Smith 1995). Two large restriction fragment length polymorphism (RFLP) studies screened, on average, 833 bp (Jakubiczka et al. 1989) and 2215 bp (Malaspina et al. 1990) in each of 22 Y chromosomes and found between them only three polymorphisms. Seielstad et al. (1994) sequenced 1.4 kb of DNA in 12–16 chromosomes of diverse geographical origins and a single polymorphic nucleotide substitution was found, and Dorit et al. (1995) did not find any variation in a 729 bp intron in the ZFY gene in 38 males.
The pace of discovery accelerated when denaturing high-performance liquid chromatography (DHPLC) was used to screen for polymorphisms in the Y chromosome. Just 2 years after Jobling and Tyler-Smith (1995) published a summary of the nine UEPs known at the time, Underhill et al. (1997) discovered 19 new polymorphisms. Three years later, the same group had increased the total number of known NRY UEPs to 166, arranged in a phylogeography of 116 haplogroups that is quite similar to the current one (Semino et al. 2000). In parallel, Hammer et al. (2001) used single-stranded conformation polymorphism (SSCP) to detect 43 new variants, which they subsequently genotyped in >2500 men from 50 world populations. At that point, an effort was made to merge the two new large phylogenies and create a new nomenclature for Y chromosome haplogroups (see below) that was modeled on that used for mitochondrial DNA (Calafell et al. 2002; The Y Chromosome Consortium 2002); it was subsequently updated in 2008 (Karafet et al. 2008), and in 2013 (Van Geystelen et al. 2013). In parallel, the International Society for Genetic Genealogy maintains a non-peer-reviewed, continuously updated tree (http://www.isogg.org/tree/ISOGG_YDNATreeTrunk.html).
The diversity, phylogeny, and evolution of many haplogroups were studied in a series of monographic papers that deepened the general knowledge about those haplogroups: A (Cruciani et al. 2011), C (Zhong et al. 2010), E-P2 (Trombetta et al. 2011), E-M215 (Cruciani et al. 2004), E-M78 (Cruciani et al. 2006), G (Rootsi et al. 2012), I (Rootsi et al. 2004), J (Semino et al. 2004), N (Rootsi et al. 2007), R1a (Underhill et al. 2010), and R1b-M269 (Myres et al. 2010).
The next leap in the number of known polymorphisms in the Y chromosome happened with the advent of next-generation sequencing technologies, and projects such as the 1000 Genomes Project (http://www.1000genomes.org/).In a pilot project, a 1.8X coverage sequencing of 77 Y chromosomes yielded 2840 high-confidence Y-SNPs, 74% of them new (Durbin et al. 2010). Additionally, Rocca et al. (2012) mined the 1000 Genomes Project data set and established the main branches of the phylogeny of haplogroup R1b-M269 in Western Europe. Subsequently, Y chromosome specific projects have analyzed extensive NRY sequences in population samples. Thus, Wei et al. (2013) focused on ~9 Mb of sequence in 36 worldwide individuals, which allowed them to produce a calibrated phylogeny for the Y chromosome, with dates for its root and some of the main groups. Francalacci et al. (2013) produced low-pass (1.2X) sequences for the same ~9 Mb region in 1204 Sardinian males; they used the Sardinian-specific clades and the known age of the colonization of Sardinia to calibrate the molecular clock and produce a date for the Y chromosome phylogeny (~180,000–200,000 years ago) that is commensurate with the age of the mitochondrial DNA tree. A similar result was reached in a back-to-back paper (Poznik et al. 2013), in which they analyzed a slightly larger sequence (~10 Mb) at a much deeper coverage (~300X) in a smaller sample (69 African, Asian, and Native American males).
Unlike UEPs, STRs mutate at a fast rate (10−3–10−4 per meiosis), and, given their stepwise mutations, new mutants are often alleles that already existed in the population; thus, reliable phylogenies cannot be build solely on STRs. However, STR alleles do carry information on the haplogroup background they are found on (and actually, STR variability is partitioned by haplogroups to a greater extent than by populations (Bosch et al. 1999)), to the point that main haplogroups can be reasonable predicted from the STR haplotypes they carry (Athey 2005; Schlecht et al. 2008), but see Larmuseau et al. (2014) for a counterexample. The fast mutation rate in STRs implies that they can be used to time relatively recent events in human evolution, at a prehistoric (Arredi et al. 2004; Alonso et al. 2005; Berniell-Lee et al. 2009; Balaresque et al. 2010; Batini et al. 2011) or historic time scale (Bosch et al. 2003; McEvoy et al. 2006; Adams et al. 2008), or to date specific patrilineal lineages, linked to historic figures or to surnames (Skorecki et al. 1997; Zerjal et al. 2003; McEvoy and Bradley 2006; Hammer et al. 2009; King and Jobling 2009a; Martinez-Cruz et al. 2012; Martínez-González et al. 2012).
3 Climbing Down the Y Tree
As mentioned above, the UEPs provide a sound scaffold to produce a phylogenetic tree for the Y chromosome. The main branches are called haplogroups, but, in turn, given the tree structure of the phylogeny, they are subdivided in subhaplogroups, which in turn are further subdivided. The nomenclature of haplogroups (The Y Chromosome Consortium 2002) starts with a capital Roman letter (A, B,..., T); subhaplogroups are indicated with numbers (A1, A2,...), the next division uses a lowercase letter (A1a, A1b), and, subsequently, numbers and lowercase letters alternate (see, for instance, E1b1b1b2a1 or R1b1a2a1a2a1a1a1). Given how unwieldy this nomenclature can become, haplogroups are often designated with a deep main branch (e.g., E or R1b), plus the most derived UEP that subhaplogroup carries; then, the latter examples become E-M34 and R1b-M153, respectively. In this nomenclature, a final star indicates a paragroup. A Y chromosome falls in a paragroup if it belongs in a haplogroup for which further UEPs have been described but that it is ancestral for those (for instance, E-M78 contains E-V12, E-V13, E-V22, and E-V65; if a Y chromosome is derived for M78 but ancestral for V12, V13, V22, and V65, then it belongs to paragroup E-M78*).
In this section, we will describe the structure, geographical distribution, and history of the main branches of the Y chromosome phylogeny. Further details may be found in the chapters in this book devoted to specific geographic regions. When first described, the deepest branch in the Y tree was labeled A. It is found at moderate frequencies in populations of Eastern and Southern Africa, and at lower frequencies in Central and Northern Africa. Cruciani et al. (2011) discovered new polymorphisms and found that clade A1b split first from the root; it has been subsequently renamed A0. But its position close to the last common ancestor did not last for long: Mendez et al. (2013) discovered that an African-American man carried the ancestral states at all the UEPs known that far; after sequencing a 300-Kb fragment of his Y chromosome, the authors found a new branch of the tree, the closest to the root, which they called A00. It is also found at a very low frequency in Cameroon.
Haplogroup B is also found only in African populations or in populations of African descent. It reaches moderate frequencies in West and Central Africa, and some of its branches are particularly prevalent in Pygmies (Batini et al. 2011).
The next major division of the Y chromosome tree brings together haplogroups D and E, which share one of the first UEPs to be discovered, namely the eponymous YAP (Y Alu Polymorphism). However, the geographical distribution of both haplogroups is quite different. Haplogroup D is found mainly in Japan and Tibet, while different branches of haplogroup E are the most frequent haplogroups in almost all African populations, and reach the Middle East and Southern Europe. E-M81 is prevalent in North Africa and has been used to trace the medieval North African migrations into Iberia and Sicily (Bosch et al. 2001; Adams et al. 2008); similarly, E-V38 may track Bantu migration from central to southern Africa.
Haplogroup C has a vast geographical distribution, from Southern Europe, across Eurasia, and into native North Americans and Polynesians. Several branches of this haplogroup reach their highest frequencies in Central Asia, Japan, New Guinea, and Australia. Within haplogroup C, Zerjal et al. (2003) identified a lineage carried by 8% of men in a large region of Asia, and that, given its age and origin, could represent the descendants of Genghis Khan.
Next to branch is haplogroup F; it contains a few minor branches dispersed over Eurasia, but it also harbors all other haplogroups from G to T. In fact, most men outside of Africa carry some branch of haplogroup F. It has been recently discovered that the first such haplogroup to branch off F is haplogroup G (Poznik et al. 2013). It reaches its peak frequency in the Caucasus, but it is found, probably because of the Neolithic expansion, from Western Europe to South Asia. Haplogroup H, until recently thought to be a sister clade to haplogroup G, is prevalent in South Asia, and its presence in European Roma (Gypsy) men bears testimony to their Indian ancestry.
Two Y chromosome haplogroups that are indeed sister clades are I and J. Haplogroup I is found mostly in Europe, with branches that are among the most frequent among men in Scandinavia, the Balkans, and Sardinia. Haplogroup J peaks in the Middle East, and is found throughout southern Europe, North Africa, and South Asia. The famous “Cohen haplotype” belongs to haplogroup J (Skorecki et al. 1997; Hammer et al. 2009).
The rest of Y chromosome haplogroups is grouped under superhaplogroup K, which, like haplogroup F, has a few minor branches with a sparse distribution found in Oceania and Australia, and only at low frequency in South Asia and the Malay Archipelago. The first clade to branch off K consists of the sister haplogroups L and T: the former is prevalent in South Asia, the Middle East, and Central Italy, while the latter has a sparse distribution from the Middle East to SW Europe.
Another linked pair of haplogroups is constituted by N and O, which represent much of the male lineages in East Asia; N has a more northerly distribution, reaching Northeast Europe to the west, while O is more prevalent to the south, reaching Polynesia. Haplogroup M is also found in New Guinea, extending to Melanesia and Polynesia. A similar, but more restricted distribution, is that of haplogroup S, which is most prevalent in the highlands of New Guinea.
We are reaching the final superhaplogroup: P, which, unlike F and K, has no minor branches. The paragroup P* is present at low to moderate frequencies among South Asian populations. P has two main branches, namely Q and R. Haplogroup Q is found sparsely in Europe and the Middle East, at moderate frequencies in northern Asia, but, with the occasional exception of haplogroup C, is the sole lineage found in native Americans. Finally, haplogroup R has three main branches: R1a, R1b, and R2. R1a is found at high frequencies from Central, Eastern, and Northern Europe, to Central and South Asia. R1b is most prevalent in Western Europe, with peaks >75% in Iberia and Ireland; finally, R2 is much less frequent and mostly restricted to the Middle East.
4 La Donna è Mobile … Ma Non Troppo
The Y chromosome diversity tracks the demographic history of men, and, in particular, their migration patterns and rates, and their effective population size. The matrilineal counterpart of the Y chromosome is mitochondrial DNA (mtDNA; see Chap. 6 in this book): although present both in males and females, it is inherited from the mother (with some extremely rare exceptions). The comparison of the diversity and phylogeography of NRY and the mtDNA has been used to learn about the specificities of the demographic history of each sex.
Back in 1987, one of the first genetic pieces of evidence to be found for the Out of Africa model of human evolution came from the mtDNA phylogeography. The mtDNA sequences (analyzed by restriction mapping) were found to coalesce to a common ancestor that may have lived in Africa 200,000 years ago (Cann et al. 1987). This ancestor was promptly dubbed “mitochondrial Eve,” and it should be clear that she was not the only woman living at that time, but rather just the one to whom all current humans can trace their maternal ancestry. “Y chromosome Adam” took a little longer to find, but it came with a surprise: he lived only 59,000 years ago (Thomson et al. 2000), a much more recent timeframe than that of his female counterpart. A difference in effective population sizes between the sexes can help resolve this discrepancy. Effective population size, the theoretical figure that governs the intensity of genetic drift, may be lower in men than in women, because the variance in reproductive success may be higher in men. That would be particularly the case in polygynous societies, where a few men can afford many wives (and children), and others may be left childless. However, this may be a recent phenomenon in human history (certainly unheard of in hunter-gatherer societies), and it is unclear for how long male effective population size needs to be reduced to produce such a large discrepancy. The time to the most recent common ancestor (TMRCA) for mtDNA and the NRY seem to have converged recently to a figure of 120,000–200,000 years ago (Soares et al. 2009; Poznik et al. 2013), so differences in effective population sizes between men and women, albeit important in other aspects (see below), need no longer be invoked to resolve any gap between the TMRCAs.
Y chromosome variants tend to be more localized geographically than those of mtDNA and the autosomes. The fraction of variation within human populations for Y chromosome SNPs (as opposed to variation between populations) was estimated globally at 35.5% (Seielstad et al. 1998), versus 80–85% for the autosomes and mtDNA (Barbujani et al. 1997; Romualdi et al. 2002). Note that Jorde et al. (2000) did not find this difference, since they were comparing NRY STRs to mitochondrial and autosomal SNPs; the mutation pattern in STRs, in which the same alleles are produced numerous times, runs counter to the demographic differentiation processes. A higher female than male migration rate (via patrilocality, the tendency for a wife to move into her husband’s natal household) explains most of this discrepancy; polygyny may also contribute (Dupanloup et al. 2003). Luca Cavalli-Sforza, the distinguished population geneticist who was the senior author of Seielstad et al. (1998) summarized this pattern in his native Italian: la donna è mobile. At a local scale, Pérez-Lezaun et al. (1999) found that the patterns of mtDNA and NRY differentiation between low- and high-altitude communities in central Asia suggested a strong bottleneck in the initial colonization of the high-altitude habitat, but that subsequent female migration from the lowlands had replenished mtDNA, but not NRY, diversity. Similarly, comparison of the mtDNA and NRY allowed to conclude that social mobility among castes in India is easier for women than for men (Wooding et al. 2004). And African Pygmy women are more likely than men to marry Bantu farmers (Verdu et al. 2009).
However, at a continental scale, the comparison of NRY and the mtDNA tips the scales in favor of male mobility. For instance, many urban mestizo populations throughout Latin America are the result of triple admixture from European, African, and Native American ancestries, although the proportions differ by sex. The contributions of Native American women and European men are larger than those of Native American men and European women (Torroni et al. 1994; Bosch et al. 2003; Mendizabal et al. 2008; Corach et al. 2010; Núñez et al. 2010; Guerra et al. 2011); among the first European colonizers of the Americas, men were much more frequent than women. Obviously, that sexual asymmetry stemmed also from the power inequality, manifest also in the fact that European admixture into African Americans is much larger for the NRY than for mtDNA (Stefflova et al. 2009; Lao et al. 2010; Battaggia et al. 2012; Torres et al. 2012). Similar situations have happened in other continents and historic times, such as in the fifteenth century rediscovery of the Canary Islands (Flores et al. 2003; Gonzalez et al. 2003), the French colonization of the Réunion Island (Berniell-Lee et al. 2008), the incredible journey of the Southeast Asian seafarers into Madagascar (Hurles et al. 2005; Tofanelli et al. 2009), or the dual colonization of Iceland with Scandinavian men and Irish women (Helgason et al. 2000a; Helgason et al. 2000b).
5 Male Lines
The Y chromosome is carried by men, who inherit it from their fathers. Thus, the NRY behaves as a marker for paternal lines of descent, as surnames do in many cultures. It is expected, then, that all the descendants of a founder of a surname carry copies of the same Y chromosome, modified only by mutation. In the usual time depth of surnames (about 700 years in most western European countries, although they are more recent in Japan and more ancient in China), most mutations that can be detected within the Y chromosomes of carriers of the same surname will be in STRs; given the ascertainment scheme used to find the SNPs usually typed in the NRY, the descendants of the founder of a surname are very unlikely to carry detectable SNP variability in their Y chromosomes (obviously, this is not the case if the whole NRY is sequenced; we are referring specifically to the panels of SNPs that are normally used to classify Y chromosomes into haplogroups). Conversely, STR and SNP variation can be used to sort the Y chromosomes into groups of descendants from different surname founders. After the medieval founding of surnames, Y chromosomes may have introgressed into a surname by at least three different mechanisms: false paternity, adoption, and the anomalous inheritance of the maternal surname. It may be particularly difficult to distinguish a founder’s Y chromosome from a Y chromosome that introgressed into a surname early in its history (King and Jobling 2009b).
Sykes and Irven (2000) pioneered the study of Y chromosome diversity in a surname, namely Sykes, which opened the floodgates for genetic genealogy; thousands of men, particularly in the USA, have found their kin in some men carrying their same surname, with the help of companies such as Family Tree DNA (King and Jobling 2009b). Of a broader scientific interest is the analysis of an entire surname system, such as that of England (King and Jobling 2009a) or Ireland (McEvoy and Bradley 2006). In the English case, King and Jobling (2009a) found that surname frequency is driven by polyphyletism; that is, the more frequent surnames are common because they were founded numerous times: Smith is an obvious example, but the patronym surnames (those that derive from first names, such as Jones, Davies or Williams) are both likely to have been founded multiple times and to be quite frequent in the UK. King and Jobling also estimated the rate of Y chromosome introgression into a surname at about 2% per generation. The Irish case (McEvoy and Bradley 2006) was strikingly different: some frequent surnames, such as O’Sullivan and Ryan, were founded by a single ancestor, and came to be frequent in Ireland by differences in fecundity engendered by the social power and prestige associated with the name and its bearers in the past.
6 A Royal Mystery in Three Acts
Some particular male lineages have been investigated by their historical connections. Thus, it has been established that Thomas Jefferson fathered one of his slaves’ sons (King et al. 2007). The Italian Colombo and Catalan Colom surnames have been investigated to decide whether it would be genetically possible to validate an alternate, Catalan hypothesis for Columbus’ origin (Martínez-González et al. 2012). It has also been established that the current bearers of the Basarab surname are not the descendants of the most famous bearer of that surname, count Vlad Dracul (Martinez-Cruz et al. 2012). And then, a bizarre story unfolded in three acts. Enter first an antique gourd that emerged and was brought to the attention of a group of geneticists. It was pyrographed with portraits of French revolutionaries and with an inscription that stated that the gourd contained a handkerchief that had been dipped in the blood of Louis XVI of France immediately after he was guillotined. Indeed, the gourd contained a black residue with human DNA consistent with a blue-eyed male. mtDNA and NRY profiles were also obtained (Lalueza-Fox et al. 2011). In the second act, a severed, mummified head that appeared in the Parisian attic of a tax collector was anatomically shown to be compatible with Henri IV’s missing head. A partial NRY STR profile could be retrieved (Charlier et al. 2013), and showed one mismatch with his seven-generation descendant, Louis XVI. The mismatch could have been caused by a mutation in these seven generations. But, in the (final?) third act, three living Bourbons enter the stage. They share a common ancestor that is intermediate between Henri IV and Louis XVI; they are also shown (Larmuseau et al. 2013) to share the same Y chromosome haplotype, which is different either from that of the severed head and that in the handkerchief. These results can be explained by forgery, contamination, or false paternity in various possible combinations.
7 Hacking the Y Chromosome
The fact that the NRY and the surname are inherited together opens the door to intriguing possibilities, such as the prediction of the surname from the observation of a particular NRY haplotype, which would be a welcome tool in crime investigation. Given the high rate of Y chromosome introgression into a surname, many false positives can be expected. However, Gitschier (2009) succeeded in inferring the surnames of 20 of the 30 Utah males in the CEU sample, by accessing their genotypes from the Hapmap project and their surnames from the http://www.ysearch.org/, where both STR haplotypes and surnames are stored. It can be argued that this was possible due to both the relative closed nature of the Latter-Day Saint community in Utah and to their devotion to genealogy. But Gymrek et al. (2013) systematized this search and estimated that the probability of assigning a correct surname to the STR haplotype of a European-American male is ~12%, and, that, if information on year of birth and US state is known, then the list of potential matches averages only 12 men. They demonstrated this approach by recovering Craig Venter’s name from the NRY STRs in his publicly available, identified genome (they failed in two other public genomes). It should be noted that this exercise was conducted by using publicly available databases and with minimal informatic sophistication.
References
Adams SM, King TE, Bosch E, Jobling MA (2006) The case of the unreliable SNP: recurrent back-mutation of Y-chromosomal marker P25 through gene conversion. Forensic Sci Int 159:14–20
Adams SM, Bosch E, Balaresque PL et al (2008) The genetic legacy of religious diversity and intolerance: paternal lineages of Christians, Jews, and Muslims in the Iberian Peninsula. Am J Hum Genet 83:725–736
Alonso S, Flores C, Cabrera V et al (2005) The place of the Basques in the European Y-chromosome diversity landscape. Eur J Hum Genet 13:1293–1302
Arredi B, Poloni ES, Paracchini S et al (2004) A predominantly neolithic origin for Y-chromosomal DNA variation in North Africa. Am J Hum Genet 75:338–345
Athey TW (2005) Haplogroup prediction from Y-STR values using an allele- frequency approach. J Genet Geneal 1:1–7
Bachtrog D (2013) Y-chromosome evolution: emerging insights into processes of Y-chromosome degeneration. Nat Rev Genet 14:113–124
Balaresque P, Bowden GR, Adams SM et al (2010) A predominantly neolithic origin for European paternal lineages. PLoS Biol 8:e1000285
Barbujani G, Magagni A, Minch E, Cavalli-Sforza LL (1997) An apportionment of human DNA diversity. Proc Natl Acad Sci USA 94:4516–4519
Batini C, Ferri G, Destro-Bisol G et al (2011) Signatures of the pre-agricultural peopling processes in sub-Saharan Africa as revealed by the phylogeography of early Y chromosome lineages. Mol Biol Evol 28:2603–2613
Battaggia C, Anagnostou P, Boschi I et al (2012) Detecting sex-biased gene flow in African-Americans through the analysis of intra- and inter-population variation at mitochondrial DNA and Y- chromosome microsatellites. Balk J Med Genet 15:7–14
Berniell-Lee G, Plaza S, Bosch E et al (2008) Admixture and sexual bias in the population settlement of La Reunion Island (Indian Ocean). Am J Phys Anthropol 136:100–107
Berniell-Lee G, Calafell F, Bosch E et al (2009) Genetic and demographic implications of the bantu expansion: insights from human paternal lineages. Mol Biol Evol 26:1581–1589
Bosch E, Calafell F, Santos FR et al (1999) Variation in short tandem repeats is deeply structured by genetic background on the human Y chromosome. Am J Hum Genet 65:1623–1638
Bosch E, Calafell F, Comas D et al (2001) High-resolution analysis of human Y-chromosome variation shows a sharp discontinuity and limited gene flow between northwestern Africa and the Iberian Peninsula. Am J Hum Genet 68:1019–1029
Bosch E, Calafell F, Rosser ZH et al (2003) High level of male-biased Scandinavian admixture in Greenlandic Inuit shown by Y-chromosomal analysis. Hum Genet 112:353–363
Bouzekri N, Taylor PG, Hammer MF, Jobling MA (1998) Hypervariable digital DNA codes for human paternal lineages: MVR-PCR at the Y-specific minisatellite, MSY1 (DYF155S1). Hum Mol Genet 7:643–653
Calafell F, Comas D, Bertranpetit J (2002) Why names. Genome Res 12:219–221
Cann R, Stoneking M, Wilson AC (1987) Mitochondrial DNA and human evolution. Nature 325:31–36
Charlier P, Olalde I, Sole N et al (2013) Genetic comparison of the head of Henri IV and the presumptive blood from Louis XVI (both kings of France). Forensic Sci Int 226:38–40. https://doi.org/10.1016/j.forsciint.2012.11.018
Corach D, Lao O, Bobillo C et al (2010) Inferring continental ancestry of argentineans from autosomal, Y-chromosomal and mitochondrial DNA. Ann Hum Genet 74:65–76
Cruciani F, La Fratta R, Santolamazza P et al (2004) Phylogeographic analysis of haplogroup E3b (E-M215) y chromosomes reveals multiple migratory events within and out of Africa. Am J Hum Genet 74:1014–1022
Cruciani F, La Fratta R, Torroni A et al (2006) Molecular dissection of the Y chromosome haplogroup E-M78 (E3b1a): a posteriori evaluation of a microsatellite-network-based approach through six new biallelic markers. Hum Mutat 27:831–832
Cruciani F, Trombetta B, Massaia A et al (2011) A revised root for the human y chromosomal phylogenetic tree: the origin of patrilineal diversity in Africa. Am J Hum Genet 88:814–818
Dorit R, Akashi H, Gilbert W (1995) Absence of polymorphism at the ZFY locus on the human Y chromosome. Science 268:1183–1185
Dupanloup I, Pereira L, Bertorelle G et al (2003) A recent shift from polygyny to monogamy in humans is suggested by the analysis of worldwide Y-chromosome diversity. J Mol Evol 57:85–97
Durbin RM, Abecasis GR, Altshuler DL et al (2010) A map of human genome variation from population-scale sequencing. Nature 467:1061–1073
Flores C, Maca-Meyer N, Perez JA et al (2003) A predominant European ancestry of paternal lineages from canary islanders. Ann Hum Genet 67:138–152
Francalacci P, Morelli L, Angius A et al (2013) Low-pass DNA sequencing of 1200 Sardinians reconstructs European Y-chromosome phylogeny. Science 341:565–569
Gitschier J (2009) Inferential genotyping of Y chromosomes in latter-day saints founders and comparison to Utah samples in the HapMap project. Am J Hum Genet 84:251–258
Gonzalez AM, Brehm A, Perez JA et al (2003) Mitochondrial DNA affinities at the Atlantic fringe of Europe. Am J Phys Anthropol 120:391–404
Guerra DC, Perez CF, Izaguirre MH et al (2011) Gender differences in ancestral contribution and admixture in Venezuelan populations. Hum Biol 83:345–361
Gymrek M, McGuire AL, Golan D et al (2013) Identifying personal genomes by surname inference. Science 339:321–324
Hammer MF, Karafet TM, Redd AJ et al (2001) Hierarchical patterns of global human Y-chromosome. Diversity 18:1189–1203
Hammer MF, Behar DM, Karafet TM et al (2009) Extended Y chromosome haplotypes resolve multiple and unique lineages of the Jewish priesthood. Hum Genet 126:707–717
Helgason A, Sigureth ardottir S, Gulcher JR et al (2000a) mtDNA and the origin of the Icelanders: deciphering signals of recent population history. Am J Hum Genet 66:999–1016
Helgason A, Sigureth ardottir S, Nicholson J et al (2000b) Estimating Scandinavian and Gaelic ancestry in the male settlers of Iceland. Am J Hum Genet 67:697–717
Hurles ME, Sykes BC, Jobling MA, Forster P (2005) The dual origin of the Malagasy in island Southeast Asia and East Africa: evidence from maternal and paternal lineages. Am J Hum Genet 76:894–901
Jakubiczka S, Arnemann J, Cooke HJ et al (1989) A search for restriction fragment length polymorphism on the human Y chromosome. Hum Genet 84:86–88
Jobling MA, Tyler-Smith C (1995) Fathers and sons: the Y chromosome and. Hum Evol 11:449–456
Jorde LB, Watkins WS, Bamshad MJ et al (2000) The distribution of human genetic diversity: a comparison of mitochondrial, autosomal, and Y-chromosome data. Am J Hum Genet 66:979–988
Karafet TM, Mendez FL, Meilerman MB et al (2008) New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res 18:830–838
King TE, Jobling MA (2009a) Founders, drift, and infidelity: the relationship between Y chromosome diversity and patrilineal surnames. Mol Biol Evol 26:1093–1102
King TE, Jobling MA (2009b) What’s in a name? Y chromosomes, surnames and the genetic genealogy revolution. Trends Genet 25:351–360
King TE, Bowden GR, Balaresque PL et al (2007) Thomas Jefferson’s Y chromosome belongs to a rare European lineage. Am J Phys Anthropol 132:584–589
Lahn BT, Page DC (1997) Functional coherence of the human Y chromosome. Science 278:675–680
Lahn BT, Page DC (1999) Four evolutionary strata on the human X chromosome. Science 286:964–967
Lalueza-Fox C, Gigli E, Bini C et al (2011) Genetic analysis of the presumptive blood from Louis XVI, King of France. Forensic Sci Int Genet 5:459–463. https://doi.org/10.1016/j.fsigen.2010.09.007
Lao O, Vallone PM, Coble MD et al (2010) Evaluating self-declared ancestry of U.S. Americans with autosomal, Y-chromosomal and mitochondrial DNA. Hum Mutat 31:1875–1893
Larmuseau MH, Delorme P, Germain P et al (2013) Genetic genealogy reveals true Y haplogroup of House of Bourbon contradicting recent identification of the presumed remains of two French Kings. Eur J Hum Genet 22(5):681–687
Larmuseau MHD, Vanderheyden N, Van Geystelen A et al (2014) Recent radiation within Y-chromosomal Haplogroup R-M269 resulted in high Y-STR haplotype resemblance. Ann Hum Genet 78:92–103. https://doi.org/10.1111/ahg.12050
Lucotte G, Ngo NY (1985) p49F, a highly polymorphic probe that detects TaqI RFLPs on the human Y chromosome. Nucleic Acids Res 13:8285
Malaspina P, Persichetti F, Novelletto A et al (1990) The human Y chromosome shows a low level of DNA polymorphism. Ann Hum Genet 54:297–305
Martinez-Cruz B, Ioana M, Calafell F et al (2012) Y-chromosome analysis in individuals bearing the Basarab name of the first dynasty of Wallachian kings. PLoS One 7:e41803. https://doi.org/10.1371/journal.pone.0041803
Martínez-González LJ, Martínez-Espín E, Álvarez JC et al (2012) Surname and Y chromosome in southern Europe: a case study with Colom/Colombo. Eur J Hum Genet 20:211–216. https://doi.org/10.1038/ejhg.2011.162
McEvoy B, Bradley DG (2006) Y-chromosomes and the extent of patrilineal ancestry in Irish surnames. Hum Genet 119:212–219
McEvoy B, Brady C, Moore LT, Bradley DG (2006) The scale and nature of Viking settlement in Ireland from Y-chromosome admixture analysis. Eur J Hum Genet 14:1288–1294
Mendez FL, Krahn T, Schrack B et al (2013) An African American paternal lineage adds an extremely ancient root to the human Y chromosome phylogenetic tree. Am J Hum Genet 92:454–459
Mendizabal I, Sandoval K, Berniell-Lee G et al (2008) Genetic origin, admixture, and asymmetry in maternal and paternal human lineages in Cuba. BMC Evol Biol 8:213
Myres NM, Rootsi S, Lin AA et al (2010) A major Y-chromosome haplogroup R1b Holocene era founder effect in Central and Western Europe. Eur J Hum Genet 19:95–101
Núñez C, Baeta M, Sosa C et al (2010) Reconstructing the population history of Nicaragua by means of mtDNA, Y-chromosome STRs, and autosomal STR markers. Am J Phys Anthropol 143:591–600
Pérez-Lezaun A, Calafell F, Comas D et al (1999) Sex-specific migration patterns in central Asian populations revealed by the analysis of Y-chromosome STRs and mtDNA. Am J Hum Genet 65:208–219
Poznik GD, Henn BM, Yee MC et al (2013) Sequencing Y chromosomes resolves discrepancy in time to common ancestor of males versus females. Science 341:562–565
Rocca RA, Magoon G, Reynolds DF et al (2012) Discovery of Western European R1b1a2 Y chromosome variants in 1000 genomes project data: an online community approach. PLoS One 7:e41634
Romualdi C, Balding D, Nasidze IS et al (2002) Patterns of human diversity, within and among continents, inferred from biallelic DNA polymorphisms. Genome Res 12:602–612
Rootsi S, Magri C, Kivisild T et al (2004) Phylogeography of Y-chromosome haplogroup I reveals distinct domains of prehistoric gene flow in Europe. Am J Hum Genet 75:128–137
Rootsi S, Zhivotovsky LA, Baldovic M et al (2007) A counter-clockwise northern route of the Y-chromosome haplogroup N from Southeast Asia towards Europe. Eur J Hum Genet 15:204–211
Rootsi S, Myres NM, Lin AA et al (2012) Distinguishing the co-ancestries of haplogroup G Y-chromosomes in the populations of Europe and the Caucasus. Eur J Hum Genet 20:1275–1282
Satta Y, Iwase M (2015) Genes on X and Y chromosomes. In: Evolution of the human genome I. Evolutionary studies. Springer, Tokyo
Schlecht J, Kaplan ME, Barnard K et al (2008) Machine-learning approaches for classifying haplogroup from Y chromosome STR data. PLoS Comput Biol 4:e1000093
Seielstad MT, Hebert JM, Lin AA et al (1994) Construction of human Y-chromosomal haplotypes using a new polymorphic A to G transition. Hum Mol Genet 3:2159–2161
Seielstad MT, Minch E, Cavalli-Sforza LL (1998) Genetic evidence for a higher female migration rate in humans. Nat Genet 20:278–280
Semino O, Passarino G, Oefner PJ et al (2000) The genetic legacy of Paleolithic Homo sapiens sapiens in extant Europeans: a Y chromosome perpective. Science 290:1155–1159
Semino O, Magri C, Benuzzi G et al (2004) Origin, diffusion, and differentiation of Y-chromosome haplogroups E and J: inferences on the neolithization of Europe and later migratory events in the Mediterranean area. Am J Hum Genet 74:1023–1034
Skorecki K, Selig S, Blazer S et al (1997) Y chromosomes of Jewish priests. Nature 385:32
Soares P, Ermini L, Thomson N et al (2009) Correcting for purifying selection: an improved human mitochondrial molecular clock. Am J Hum Genet 84:740–759
Stefflova K, Dulik MC, Pai AA et al (2009) Evaluation of group genetic ancestry of populations from Philadelphia and Dakar in the context of sex-biased admixture in the Americas. PLoS One 4:e7842
Sykes B, Irven C (2000) Surnames and the Y chromosome. Am J Hum Genet 66:1417–1419
The Y Chromosome Consortium (2002) A nomenclature system for the tree of human Y-chromosomal binary haplogroups. Genome Res 12:339–348
Thomson R, Pritchard JK, Shen P et al (2000) Recent common ancestry of human Y chromosomes: evidence from DNA sequence data. Proc Natl Acad Sci U S A 97:7360–7365
Tofanelli S, Bertoncini S, Castri L et al (2009) On the origins and admixture of Malagasy: new evidence from high-resolution analyses of paternal and maternal lineages. Mol Biol Evol 26:2109–2124
Torres JB, Doura MB, Keita SO, Kittles RA (2012) Y chromosome lineages in men of west African descent. PLoS One 7:e29687
Torroni A, Chen YS, Semino O et al (1994) mtDNA and Y-chromosome polymorphisms in four native American populations from southern Mexico. Am J Hum Genet 54:303–318
Trombetta B, Cruciani F, Sellitto D, Scozzari R (2011) A new topology of the human Y chromosome haplogroup E1b1 (E-P2) revealed through the use of newly characterized binary polymorphisms. PLoS One 6:e16073
Underhill PA, Jin L, Lin AA et al (1997) Detection of numerous Y chromosome biallelic polymorphisms by denaturing high-performance liquid chromatography. Genome Res 7:996–1005
Underhill PA, Myres NM, Rootsi S et al (2010) Separating the post-glacial coancestry of European and Asian Y chromosomes within haplogroup R1a. Eur J Hum Genet 18:479–484
Van Geystelen A, Decorte R, Larmuseau MH (2013) Updating the Y-chromosomal phylogenetic tree for forensic applications based on whole genome SNPs. Forensic Sci Int Genet 7:573–580
Verdu P, Austerlitz F, Estoup A et al (2009) Origins and genetic diversity of pygmy hunter-gatherers from Western Central Africa. Curr Biol 19:312–318
Wei W, Ayub Q, Chen Y et al (2013) A calibrated human Y-chromosomal phylogeny based on resequencing. Genome Res 23:388–395
Wooding S, Ostler C, Prasad BV et al (2004) Directional migration in the Hindu castes: inferences from mitochondrial, autosomal and Y-chromosomal data. Hum Genet 115:221–229
Zerjal T, Xue Y, Bertorelle G et al (2003) The genetic legacy of the mongols. Am J Hum Genet 72:717–721
Zhong H, Shi H, Qi XB et al (2010) Global distribution of Y-chromosome haplogroup C reveals the prehistoric migration routes of African exodus and early settlement in East Asia. J Hum Genet 55:428–435
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Japan KK, part of Springer Nature
About this chapter

Cite this chapter
Calafell, F., Comas, D. (2021). The Y Chromosome. In: Saitou, N. (eds) Evolution of the Human Genome II. Evolutionary Studies. Springer, Tokyo. https://doi.org/10.1007/978-4-431-56904-6_5
Download citation
DOI: https://doi.org/10.1007/978-4-431-56904-6_5
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-56902-2
Online ISBN: 978-4-431-56904-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)