
Abstract
Synaptic plasticity underlies higher brain function such as learning and memory, and the actin cytoskeleton in dendritic spines composing excitatory postsynaptic sites plays a pivotal role in synaptic plasticity. In this chapter, we review the role of drebrin in the regulation of the actin cytoskeleton during synaptic plasticity, under long-term potentiation (LTP) and long-term depression (LTD). Dendritic spines have two F-actin pools, drebrin-decorated stable F-actin (DF-actin) and drebrin-free dynamic F-actin (FF-actin). Resting dendritic spines change their shape, but are fairly constant over time at steady state because of the presence of DF-actin. Accumulation of DF-actin is inversely regulated by the intracellular Ca2+ concentration. However, LTP and LTD stimulation induce Ca2+ influx through N-methyl-d-aspartate (NMDA) receptors into the potentiated spines, resulting in drebrin exodus via myosin II ATPase activation. The potentiated spines change to excited state because of the decrease in DF-actin and thus change their shape robustly. In LTP, the Ca2+ increase via NMDA receptors soon returns to the basal level, and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) expression at the postsynaptic membrane is increased. The Ca2+ recovery and AMPAR increase coordinately induce the re-accumulation of DF-actin and change the dendritic spines from the excited state to steady state during LTP maintenance. During LTD, the prolonged intracellular Ca2+ increase inhibits the re-accumulation of DF-actin, resulting in facilitation of AMPAR endocytosis. Because of the positive feedback loop of the AMPAR decrease and drebrin re-accumulation inhibition, the dendritic spines are instable during LTD maintenance. Taken together, we propose the presence of resilient spines at steady state and plastic spines at excited state and discuss the physiological and pathological relevance of the two-state model to synaptic plasticity.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The dendritic spine shape is believed to closely correlate with higher brain functions such as learning and memory (Dailey and Smith 1996; Desmond and Levy 1983). Golgi staining of neurons shows a variety of spine shapes such as thin headless, stubby, and mushroom shapes. Because these spine shapes are believed to link to higher brain functions, the molecular base of morphological regulation was extensively studied. However, Ramon y Cajal proposed over a hundred years ago that spines dynamically move and change according to activity (Yuste 2015). In fact, learning does not link to the shapes per se, but to the shape changes. While the smaller spine is generally preferable for learning, the larger spine is not (Matsuzaki et al. 2004).
Actin is highly concentrated in dendritic spines (Matus et al. 1982). Although the average spine size within a population is constant while there is no external stimulus (Yasumatsu et al. 2008), dendritic spine shapes are not static but dynamic (Fischer et al. 1998; Maletic-Savatic et al. 1999), and the presence of dynamic and stable F-actin pools in dendritic spines was postulated to regulate dynamic spine morphology (Halpain 2000). However, how actin regulates spine morphology has not been fully elucidated.
Drebrin is a key actin-binding protein that forms the stable F-actin pool in dendritic spines. In 1999, we reported that drebrin immunostaining in neuronal somata and dendritic shafts was much weaker than in dendritic spines when cortical neurons and astrocytes were cocultured for 3 weeks, indicating that drebrin is characteristically concentrated in dendritic spines (Hayashi and Shirao 1999). However, we subsequently noticed that when we changed the culture medium from a conditioned medium to a fresh non-conditioned medium, an intense staining of cell somata and dendritic shafts with faint staining of dendritic spines was observed. This raised the hypothesis that drebrin localization changes when neurons are hyperactive in a fresh culture medium. In 2006, we found the activity-dependent translocation of drebrin between dendritic spines and the parent dendrite (Sekino et al. 2006).
In this chapter, we first review the two F-actin pools, dynamic and stable pools, in resting dendritic spines and introduce the Ca2+-dependent regulation of the drebrin-decorated F-actin stable pool. Then, we review the molecular mechanism of drebrin exodus induced in potentiated spines and the role of drebrin exodus and re-accumulation in long-term potentiation (LTP). Furthermore, we discuss the role of spine stability and instability in LTP and long-term depression (LTD) , particularly focusing on the neutral dynamic morphology of resting spines and N-methyl-D-aspartate receptor (NMDAR)-dependent morphological changes of potentiated spines. Finally, we discuss the pathological relevance of drebrin loss to Alzheimer’s disease. The role of drebrin in homeostatic synaptic plasticity is not reviewed here, but in Chaps. 8 and 10.
2 Actin Cytoskeleton in Resting Dendritic Spines
2.1 Two F-actin Pools in Resting Dendritic Spines
Actin is the main cytoskeletal element in dendritic spines that governs the spine morphology (Matus et al. 1982). In addition, dendritic spine motility is completely inhibited by actin-depolymerizing reagents such as latrunculin and cytochalasin (Fischer et al. 1998), suggesting the presence of dynamic F-actin regulating spine motility. However, spine F-actin exhibits marked resistance to cytochalasin D (Allison et al. 1998), suggesting that dendritic spine F-actin is extremely stable and plays a purely structural role. In 2000, Halpain suggested that the dynamic and stable configurations of F-actin regulate the spine shape (Halpain 2000).
Drebrin binding to F-actin results in elongation of the F-actin helical crossover (40 nm) (Sharma et al. 2012). Because drebrin has a cooperative actin-binding activity (for details, see Chap. 5), each F-actin is not covered at all or totally covered by drebrin. The resultant drebrin-decorated F-actin (DF-actin) forms a unique F-actin pool different from other normal helical crossover (36 nm) F-actin pools in dendritic spines. In addition to the structural changes, DF-actin shows a lower treadmilling and a lower depolymerization rate (Mizui et al. 2009; Mikati et al. 2013), and inhibition of actin-activated myosin II ATPase activity (Hayashi et al. 1996). Furthermore, drebrin can cross-link F-actins in a phosphorylation-dependent manner (Worth et al. 2013). Moreover, drebrin can form oligomers (see Chap. 3), which may further increase the stability of the DF-actin complex. Collectively, DF-actin likely exhibits a stable configuration in dendritic spines.
The presence of dynamic and stable F-actin has been detected in dendritic spines using a photoactivatable form of green fluorescent protein (PAGFP) fused to actin monomers. When PAGFP-actin molecules in F-actin are activated in dendritic spines, the fluorescence from the activated molecules decays in two phases (Honkura et al. 2008). This shows the presence of a dynamic F-actin pool with a fast treadmilling rate and a stable pool with a low treadmilling rate, because activated PAGFP-actin turns over by continuous “treadmilling.” While the dynamic F-actin is observed in the spine tip, the stable one is largely restricted to the base core of the spine head (Honkura et al. 2008).
We have shown by super resolution microscopy and electron microscopy that drebrin is located at the center of mature dendritic spines (Koganezawa et al. 2017) (Aoki et al. 2005). As mentioned above, DF-actin is more stable than drebrin-free F-actin (FF-actin). Therefore, it is considered that stable F-actin at the core of dendritic spines consists of DF-actin, and the dynamic F-actin at the periphery consists of FF-actin.
2.2 Resting Dendritic Spine Motility
At steady state, how is spine motility generated without synaptic input? When cells are cultured on a dish, motile cells form lamellipodia incessantly without any extrinsic stimulation. These lamellipodia consist of a relatively stable actin gel in the bulk and a highly dynamic one at the peripheral edge. Because dendritic spines consist of stable F-actin in the core region and dynamic F-actin at the periphery of the spine head, the underlying mechanism of spontaneous spine motility may be similar to spontaneous lamellipodium formation of motile cells.
Zimmermann and Falcke have proposed a lamellipodium formation model without any extrinsic stimulation (Zimmermann and Falcke 2014). According to this model, individual lamellipodia are formed because of random filament nucleation events amplified by autocatalytic branching and are terminated by filament bundling, severing, and capping. We have recently proposed that spine motility (periodic lamellipodium formation from the spine head) without synaptic stimulation may be similarly determined by random autocatalytic branching of FF-actin located at the periphery of the spine head (Koganezawa et al. 2017). In contrast, the stable actin structure composed of DF-actin provides a stiff substrate in the core region of the spine to push back against, to extend the plasma membrane (Fig. 11.1). Additionally, DF-actin can be stabilized by the interaction of microtubules through the interaction of drebrin with the microtubule plus-end-binding protein, EB3 (Geraldo et al. 2008).

Neutral dynamic morphology model of resting dendritic spines. Drebrin-decorated F-actin (DF-actin, 40 nm helical pitch) and drebrin-free F-actin (FF-actin, 36 nm) are localized at the core and peripheral regions of dendritic spines, respectively. Actin polymerization of FF-actin spontaneously occurs by random filament nucleation and is terminated by filament severing and capping. Despite the spontaneous spine motility, the average spine size of resting spines within the population is constant
2.3 Regulatory Mechanism Underlying DF-Actin Accumulation in Resting Dendritic Spines
Drebrin A expressed in cell soma spontaneously accumulates within dendritic spines after it is transported together with cortical F-actin to dendritic spines in the parent dendrite (Hayashi and Shirao 1999). This suggests a constitutive accumulation mechanism of DF-actin in dendritic spines. When the intracellular Ca2+ in cultured hippocampal neurons was chelated by 1,2-bis(2-aminophenoxy) ethane-N,N,N,N-tetraacetic acid acetoxymethyl (BAPTA-AM), drebrin accumulation was more than double that of the control (Fig. 11.2). Therefore, the constitutive DF-actin accumulation mechanism seems inversely regulated by the intracellular Ca2+ concentration.

The intracellular Ca2+ concentration inversely regulates drebrin-decorated F-actin in resting dendritic spines. Neurons (21 DIV) were preincubated in normal medium containing. 50 μM 1,2-bis(2-aminophenoxy) ethane-N,N,N,N-tetraacetic acid acetoxymethyl (BAPTA-AM), a membrane-permeable Ca2+ chelator, for 30 min, and stimulated with 100 μM glutamate for an additional 10 min. Images were obtained after double-labeling drebrin and F-actin. Bar graphs represent the spine/dendrite ratios (SDRs) of drebrin and actin. The F-actin images indicate that the spines kept their structure during the experiment although their shape changed. Scale bars, 5 μm. BAPTA-AM pretreatment significantly increased both the drebrin and actin SDR (n = 30 cells; p < 0.01, Scheffe’s test). In the presence of BAPTA-AM, glutamate stimulation significantly decreased both drebrin and actin SDR (n = 30 cells; p < 0.01, Scheffe’s test). Error bars represent S.E.M
Chelation of extracellular Ca2+ with ethylene glycol tetraacetic acid also increased drebrin accumulation. Similarly, blocking NMDAR by the antagonist, D-(−)-2-amino-5-phosphonopentanoic acid (APV), or L-type voltage-dependent calcium channels by nifedipine also significantly increased drebrin accumulation. However, inhibition of Ca2+ release from intracellular Ca2+ stores with thapsigargin did not affect drebrin accumulation. These findings suggest that the NMDAR and voltage-dependent calcium channels, but not intracellular Ca2+ stores, are involved in drebrin accumulation in resting dendritic spines (Mizui et al. 2014).
Ca2+ increase in the brain facilitates drebrin degradation (Arai et al. 1991). It has also been reported that overactivation of NMDAR decreased the total amount of drebrin in neurons via calpain-mediated proteolysis (Chimura et al. 2015). This raises the possibility that low Ca2+ concentration inhibits drebrin degradation, resulting in DF-actin accumulation. However, this possibility is not very likely because blocking NMDAR increased drebrin accumulation without changing the total amount of drebrin in the neuron.
We have previously reported that α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) activity regulates drebrin A accumulation during development (Takahashi et al. 2009). Fluorescence recovery after photobleaching analysis showed that the Ca2+-impermeable AMPAR activity increased the stable fraction of drebrin A in dendritic spines, consequently promoting DF-actin accumulation in dendritic spines. AMPAR may regulate drebrin accumulation by activating intracellular signaling pathways other than Ca2+ signaling (for details, see Chap. 10).
Intriguingly, extrinsically expressed drebrin A in immature neurons accumulated in filopodia without presynaptic terminals, forming extra-large filopodia (Mizui et al. 2005) or bifurcated abnormal filopodia (Biou et al. 2008). This suggests that AMPAR activity is necessary for accumulating DF-actin in the postsynaptic site, but not in dendritic protrusions, either dendritic filopodia or spines. Drebrin shows bundling activity of DF-actins when cyclin-dependent kinase 5 (Cdk5) phosphorylates serine 142 of drebrin (Worth et al. 2013). Additionally, the small GTPase Ras likely interacts with the C-terminal domain of drebrin, inducing changes in the actin cytoskeleton. Activation of drebrin bundling activity in dendritic spines may catch transported DF-actin along dendrites and sequester them in dendritic spines.
Taken together, DF-actin accumulation in resting dendritic spines is likely regulated by a multi-signaling pathway, such as the AMPAR-mediated intracellular signaling pathway, and by the intracellular Ca2+ concentration determined by NMDARs or L-type voltage-dependent calcium channels.
3 Activity-Dependent Changes in Drebrin Localization
3.1 Drebrin Exodus from Potentiated Synapse
When we stimulated neurons with glutamate, drebrin changed its localization from dendritic spines to the parent dendrite and cell soma (Fig. 11.3a). Interestingly, when neurons were stimulated with a low concentration of glutamate, a robust drebrin loss from dendritic spines within a partial population was observed. This weak glutamate stimulation did not induce a partial loss of drebrin from dendritic spines within a whole population. Additionally, drebrin translocation (exodus) was completely blocked by an NMDAR antagonist, indicating that the exodus specifically depends on NMDAR activity (Sekino et al. 2006). While we have shown that about 20% of spines contain no drebrin in vivo (Aoki et al. 2005), these spines may have been activated through NMDAR shortly before the analysis, and thus drebrin’s reentry was not observed.

(a) Confocal images of drebrin distribution in 21-DIV cultured cortical neurons stimulated with 100 μM glutamate (right image) and control neurons (left image). Note that the puncta immunostained with an anti-drebrin antibody are scattered as a dot-like pattern in the control neurons (left image), but not in the stimulated neurons (right image). Arrows indicate sites of neuronal somata. (b) Images were obtained from 21-DIV neurons double-labeled for drebrin and F-actin. Neurons were stimulated with glutamate in the absence and presence of APV. Scale bars in (a) and (b) are 10 μm and 3 μm, respectively
In parallel with the drebrin exodus , the F-actin content in dendritic spines decreases (Ouyang et al. 2005; Mizui et al. 2014). Does NMDAR activation specifically induce DF-actin translocation or does it generally induce both DF- and FF-actin? To answer this question, we used the neuronal culturing technique, called the Banker method (Kaech and Banker 2006; Goslin and Banker 1989), because it is difficult to discriminate the neuronal F-actin from the glial one in a cortical mixed culture.
In the Banker method, embryonic hippocampal neurons prepared from an embryonic day (ED) 18 rat brain are plated on a coverslip coated with poly-l-lysine. After attachment, the neurons are cultured on a glial sheet in a dish for 21 days in vitro (DIV). By analyzing the pure neuronal culture on the coverslip, we could determine the distribution of drebrin and F-actin in neurons without interference of the glial F-actin (Takahashi et al. 2003).
To quantify the accumulation of DF-actin and FF-actin within a single dendritic spine, we double-stained the neurons with an anti-drebrin antibody (mAb M2F6) and rhodamine-labeled phalloidin and determined the spine/dendrite ratio (SDR) of drebrin and F-actin. The accumulation levels of DF-actin can be deduced by the drebrin SDR, and the accumulation level of total F-actin (DF-actin + FF-actin) can be deduced by the actin SDR.
In mature hippocampal neurons, both drebrin SDR and actin SDR were about 1.7 (Mizui et al. 2014). After glutamate stimulation, the drebrin and actin SDRs significantly decreased to about 0.5, namely, the intensity of dendritic spines was lower than that of dendritic shafts. Conversely, this stimulation did not affect the density of spines or presynaptic terminals. When NMDARs on neurons were inhibited by the antagonist, APV, DF-actin exodus was completely blocked, but total F-actin exodus was only partially blocked (Fig. 11.3b). This suggests that DF-actin exodus is specifically regulated by NMDARs, whereas FF-actin exodus is likely regulated by another signaling pathway (Mizui et al. 2014), although we cannot exclude the possibility that FF-actin is also partly regulated by NMDARs.
We then examined whether the activation of synaptic activity is involved in drebrin exodus. When γ-aminobutyric acid (GABA)A receptors were blocked by bicuculline, drebrin exodus was induced in parallel to an increase in the spontaneous firing rate of the neuronal network (Sokal et al. 2000). When the presynaptic membrane potential was depolarized by 90 mM KCl, the exodus was also induced in parallel to glutamate release from presynaptic terminals. Thus, synaptically evoked membrane depolarization seems to activate NMDAR, resulting in drebrin exodus.
NMDAR activation is the initial step to induce LTP which is a characteristic synapse function underlying the cellular mechanism of learning and memory. LTP can be induced in cultured neuronal synapses by chemical LTP (cLTP) stimulation (200 μM glycine with 20 μM bicuculline and 1 μM strychnine) (Lu et al. 2001). We used this cLTP stimulation and succeeded to induce drebrin exodus. Interestingly, cLTP-induced drebrin exodus was transient. Drebrin exodus from dendritic spines was prominent at 5 min after the stimulation; however, after 30 min drebrin returned to a level higher than the prestimulated level. In addition, using exogenously expressed GFP-drebrin A as a tracer, we showed that the drebrin SDR began to decrease immediately after the neurons were exposed to a cLTP solution and kept declining throughout the stimulation period. When the stimulation ceased, drebrin SDR began to rise and returned to the prestimulated level within 11 min (Fig. 11.4, open circles) (Mizui et al. 2014).

Chemical LTP and LTD stimulations induce transient and prolonged drebrin exodus. We transfected 7-DIV neurons with a GFP-drebrin A expression vector and performed time-lapse imaging at 21 DIV. Open and closed circles represent data obtained by LTP (200 μM glycine with 20 μM bicuculline and 1 μM strychnine) and LTD stimulation (100 μM glutamate with 10 μM glycine) at 30-s intervals. Error bars represent S.E.M. (n = 7 neurons)
Conversely, when drebrin exodus was induced by 100 μM glutamate stimulation for 10 min, it needed 24 h to re-accumulate in the dendritic spines (Sekino et al. 2006). Additionally, time-lapse imaging analysis showed that the drebrin SDR began to decrease immediately after chemical LTD stimulation (100 μM glutamate with 10 μM glycine) similarly to LTP stimulation. But even after the stimulation ceased, the drebrin SDR remained low and did not return to the prestimulated level within 11 min (Fig. 11.4, closed circles). These results suggest that LTD stimulation also induces drebrin exodus, but unlike LTP stimulation, the exodus is not transient.
3.2 Transient Ca2+ Influx Through NMDA Receptors Specifically Induces Drebrin Exodus
Drebrin exodus is not induced when the extracellular Ca2+ is chelated (Mizui et al. 2014), but it is induced when the intracellular Ca2+ is chelated (Fig. 11.2). In contrast, as mentioned previously, the chelation of either extracellular or intracellular Ca2+ increases drebrin content at resting dendritic spines. Additionally, blocking NMDARs inhibits drebrin exodus, but blocking either L-type voltage-dependent calcium channels or Ca2+ release from intracellular Ca2+ stores does not inhibit drebrin exodus (Mizui et al. 2014). Taken together, we concluded that the transient Ca2+ influx through NMDA receptors specifically induces the drebrin exodus, but the intracellular Ca2+ concentration of the dendritic spine at steady state does not affect drebrin exodus following the excitation.
3.3 Involvement of Myosin II ATPase in Drebrin Exodus
Myosin II ATPase activity is necessary for LTP formation (Rex et al. 2010). Because inhibition of myosin II ATPase by blebbistatin blocks drebrin exodus, myosin II ATPase activity likely plays a role in LTP formation by inducing drebrin exodus (Mizui et al. 2014).
How does myosin II ATPase activation induce drebrin exodus? Myosin II ATPase activation destabilizes the F-actin network in vitro (Haviv et al. 2008) and in vivo (Wilson et al. 2010). Murrell and Gardel have proposed an F-actin buckling model (Murrell and Gardel 2012). In disordered nonsarcomeric actomyosin networks, myosin II ATPase activation induces both tension and compression. Consequently, compressive stresses are relieved through F-actin severing. In drebrin exodus, Ca2+ influx through NMDARs activates myosin II ATPase, leading to breakup of the stable DF-actin complex via severing DF-actin in the core of dendritic spines. Consequently, tattered short DF-actins spread out evenly into the dendritic shaft.
How does transient Ca2+ influx through NMDARs activate myosin II ATPase? As well as tropomyosin, drebrin inhibits myosin II from binding to F-actin in vitro (Hayashi et al. 1996). Therefore, actin-dependent myosin II ATPase is inhibited in the core region of resting dendritic spines where DF-actin is concentrated. In the skeletal muscle, increased intracellular Ca2+ disinhibits tropomyosin-dependent myosin II ATPase inhibition. If a similar mechanism is applicable to dendritic spines, transient Ca2+ influx through NMDARs disinhibits drebrin-dependent inhibition of myosin II ATPase activity in dendritic spines (Mizui et al. 2014).
3.4 Drebrin Re-Accumulates During LTP Maintenance, but not During LTD
As mentioned above, LTP stimulation induces drebrin exodus followed by re-accumulation of DF-actin at dendritic spines (Mizui et al. 2014). Electrical LTP stimulation for 1 s induced progressive Ca2+ increases via NMDARs in dendritic spines, which returned to the basal level within 10 s (Petrozzino et al. 1995). Therefore, the Ca2+ increase via NMDARs during LTP initiation likely recovers to the basal level soon after it triggers the drebrin exodus. AMPAR levels at the postsynaptic membrane increase during LTP (Lu et al. 2001). If drebrin re-accumulation uses a DF-actin accumulation mechanism similar to that operating in resting dendritic spines, then Ca2+ recovery in conjunction with the AMPAR increase may coordinately induce the re-accumulation of DF-actin during LTP maintenance. This is consistent with previous reports that showed an increase in drebrin during LTP in vivo (Fukazawa et al. 2003) and a correlation between drebrin content and the spine head size (Kobayashi et al. 2007).
4 Role of Synaptic Stability and Instability in Synaptic Plasticity
4.1 Neutral Dynamic Morphology of Resting Dendritic Spines
Spine synapses have a tight structure–function relationship (Matsuzaki et al. 2001; Asrican et al. 2007). For example, larger spines have wider postsynaptic density (PSD) (Harris and Stevens 1989) and more AMPARs (Nusser et al. 1998; Takumi et al.1999). Additionally, the number of synaptic vesicles in the presynaptic terminal correlates with the PSD area (Harris and Stevens 1989; Knott et al.2006). Thus, the spine size closely correlates with the synaptic strength.
Although resting spines show structural changes without synaptic input, the average size of dendritic spines is constant within the population (Yasumatsu et al. 2008). According to this neutral dynamic morphology model, the size of dendritic spines is regulated by the F-actin content in dendritic spines. There are two rules we must consider to discuss this model. First, the final F-actin content is determined by the total amount of actin molecules (Korn et al.1987). Second, the actual polymerization change in the F-actin content depends on the frequency of filament nucleation events.
In resting spines, the presence of the stable F-actin complex blocks the translocation of proteins into the spines (Ouyang et al. 2005). Therefore, the total amount of actin molecules is constant; consequently the final F-actin content remains constant, although the FF-actin content may be increased by random Arp2/3-dependent nucleation or decreased by severing and capping. These are the neutral dynamic morphological changes. In contrast, DF-actin forms a stable actin cytoskeleton in the core region of the spine that does not contribute to the above changes in F-actin content that directly link to spine motility. Therefore, at the risk of oversimplification, the DF-actin content in the dendritic spines inversely regulates the spine motility. Actually, overaccumulation of extrinsically expressed GFP-drebrin A in dendritic filopodia and spines stops their motility (unpublished data).
4.2 NMDA Receptor-Mediated Change of Dendritic Spines From Steady to Excited State
The concepts of spine stability and instability are fundamental to understanding the input-specific changes in synaptic strength, namely, synaptic plasticity. Spine stability is maintained by the aforementioned neutral dynamic morphology of resting dendritic spines (Fig. 11.5a). In contrast, spine instability underlying LTP and LTD requires synaptic activation of NMDARs, which leads to Ca2+ influx through the NMDAR channel and a rise in Ca2+ within the dendritic spines (Malenka and Bear 2004).

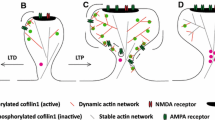
Schematic representation of the actin cytoskeleton during synaptic plasticity. (a) Dendritic spines at steady state contain dynamic F-actin (normal helical pitch F-actin) and stable F-actin (long helical pitch F-actin). (b) Once Ca2+ enters through NMDA receptors (dark brown), it activates myosin II ATPase and induces drebrin exodus. (c) During LTP, G-actin refills the spine and actin nucleation is promoted. Consequently, polymerization of dynamic F-actin is facilitated and the spine is enlarged. (d) The Ca2+ concentration returns to a normal level (light brown), and the AMPA receptor accumulates; the reconstruction of drebrin A-decorated stable F-actin is induced, contributing to the maintenance of enlarged spine morphology. (e) During LTD, the prolonged intracellular Ca2+ increase inhibits drebrin re-accumulation. Then, low drebrin facilitates AMPAR endocytosis, resulting in decreased AMPAR density at the postsynaptic membrane. Because AMPAR activity is necessary for the re-accumulation of drebrin, AMPAR decrease and drebrin re-accumulation inhibition form a positive feedback loop. Additionally, cofilin, which triggers depolymerization of F-actin, is activated. Consequently, these changes lead to spine shrinkage
Drebrin exodus is specifically triggered by Ca2+ influx through synaptic NMDARs during the LTP and LTD induction steps (Fig. 11.5b). Because of the exodus, DF-actin in potentiated dendritic spines decreases, and consequently G-actin may immediately refill the spine by diffusion from the dendritic shaft (Star et al.2002). This leads to an increase in the total actin content available for polymerization and an increase in FF-actin available for Arp2/3-dependent branching, namely, the two rules of increased F-actin described previously are satisfied (Fig. 11.5c) (Koganezawa et al. 2017). Although LTP-dependent Arp2/3 activation has not been reported, neural Wiskott–Aldrich syndrome protein (N-WASP) and the Arp2/3 complex regulate actin during the development of dendritic spines (Wegner et al. 2008). Thus, at the LTP expression step, Arp2/3-mediated actin nucleation may facilitate F-actin polymerization in dendritic spines and enlarge the spine size. Additionally, the loss of stable F-actin may induce translocation of plasticity-dependent proteins, such as actin, Ca2+/calmodulin-dependent protein kinase II, and AMPAR. Moreover, drebrin exodus may facilitate AMPAR trafficking to postsynaptic membranes by disinhibiting myosin V, which is required for AMPAR trafficking during LTP (Correia et al. 2008).
NMDAR-mediated facilitation of F-actin polymerization was observed between 3 min and 18 min after the synaptic activation and then ended (Okamoto et al. 2004). In contrast, NMDAR-mediated spine enlargement continued over 30 min. Furthermore, electrophysiological studies have shown that NMDAR-dependent LTP lasts hours, days, or even weeks and the so-called late phases or longer-lasting components of LTP require new protein synthesis and gene transcription (Abraham and Williams 2003). How can enlarged spines keep their size even after the facilitation of F-actin polymerization ends? LTP stimulation induces drebrin re-accumulation at the LTP maintenance step. As mentioned previously, drebrin accumulation in dendritic spines is inversely regulated by the intracellular Ca2+ concentration and positively by AMPAR activity. Therefore, both the Ca2+ concentration recovery to steady state and the increase in AMPAR insertion into postsynaptic membranes likely induce the drebrin re-accumulation in dendritic spines (Fig. 11.5d). Hence, the dendritic spines changes from the excited state to steady state, making it possible for the dendritic spines to keep their enlarged size with a secure increase in synaptic strength until newly synthesized proteins are delivered to these spines for the “late phases” of LTP.
Drebrin is classified into a drebrin E isoform which is widely observed in various tissues and a neuron-specific drebrin A isoform. Although both isoforms can make long helical crossover F-actin, drebrin A, but not E, is crucial for LTP formation because drebrin A-knockout mice, in which drebrin E continues to be expressed throughout life instead of drebrin A (Kojima et al. 2010), show impaired hippocampal LTP and fear learning in an age-dependent manner (Kojima et al. 2016).
4.3 Spine Instability During LTD Formation
LTD stimulation also induces a similar drebrin exodus to LTP stimulation. However, LTD stimulation downregulates Arp2/3-mediated actin nucleation (Rocca et al. 2013). Thus, because of the absence of Arp2/3-dependent F-actin branching, F-actin polymerization is not facilitated even when the total actin content available for polymerization is increased by drebrin exodus. Instead, LTD stimulation induces AMPAR internalization (Mulkey et al. 1994; Malenka and Bear 2004) and spine shrinkage (Wang et al. 2007). Drebrin exodus may increase the AMPAR internalization because endocytosis such as human immunodeficiency virus (HIV)-1 entry is suppressed by drebrin (Gordon-Alonso et al. 2013). Because of the decrease in AMPAR activity at the LTD expression stage, drebrin re-accumulation is not observed in dendritic spines (Fig. 11.5e). The absence of a stable DF-actin core makes it difficult for dynamic FF-actin to push back against and extend the plasma membrane. Additionally, cofilin, which triggers depolymerization of F-actin, is activated (Zhou et al. 2004; Cao et al. 2017). Consequently, these changes lead to spine shrinkage. This idea is consistent with our previous study demonstrating that the drebrin content correlates with the dendritic spine size (Kobayashi et al. 2007). Furthermore, according to this model, the dendritic spines during LTD maintenance are not in steady state, but in the excited state.
In fact, the preferential sites for morphological changes are small spines (Matsuzaki et al. 2004) that express a small number of AMPARs (Matsuzaki et al. 2001). Conversely, large spines express AMPARs abundantly and are stable for months in vivo (Trachtenberg et al. 2002).
5 Pathological Relevance of Drebrin Loss to Alzheimer’s Disease
Alzheimer’s disease (AD) is a neurodegenerative disorder accompanied by severe progressive memory and cognitive impairment. Drebrin decrease is a hallmark of the AD brain (Harigaya et. al 1996; Kojima and Shirao 2007; Ma et al. 2015), as well as amyloid plaques (senile plaques) and neurofibrillary tangles (Selkoe and Hardy 2016). Drebrin is markedly decreased in dendritic spines before significant presynaptic changes appear.
As drebrin loss confers a loss of drebrin-decorated stable F-actin, the balance between the stability and instability of spines may be disrupted in AD. According to the drebrin exodus model, dendritic spines without drebrin accumulation should be constitutively instable because of the lack of stable F-actin. Therefore, although LTP stimulation can enlarge the dendritic spine in the AD brain, it is likely to soon be restored to the previous size, resulting in failure of LTP formation. This AD model has been reviewed in detail by Shirao et al. (2017).
6 Conclusion
In this chapter, we discussed the role of the actin cytoskeleton in dendritic spines, particularly focusing on synaptic plasticity, and concluded that synaptic plasticity begins with drebrin exodus, by which dendritic spines change from steady state to excited state. Here, we proposed a “two-state model of synaptic plasticity” (Fig. 11.6).

Two-state model of synaptic plasticity. (a) LTP formation. Dendritic spines without tetanic stimulation are at steady state with high drebrin. These spines can spontaneously change their size by random actin nucleation even at steady state, but they are resilient and seldom change into the excited state. Once LTP stimulation induces Ca2+ influx, drebrin is decreased because of the drebrin exodus. Consequently, dendritic spines change into the excited state and become plastic. Shortly after LTP stimulation, the Ca2+ concentration returns to normal, and F-actin polymerization is facilitated by the high actin nucleation activity, resulting in the enlargement of spine size and the increase in AMPAR density. Normal Ca2+ and increased AMPAR coordinately increase drebrin, and then the spines return to steady state. (b) LTD formation. Once LTD stimulation induces Ca2+ influx, drebrin is decreased because of the drebrin exodus. After LTD stimulation, the Ca2+ concentration remains elevated, and the AMPAR density decreases because of the facilitated endocytosis of AMPAR, inhibiting the re-accumulation of drebrin. Even after the Ca2+ concentration returns to normal, the low drebrin content confers high instability to dendritic spines. The diameter of the circles shows the dendritic spine head size. The AMPAR density on the postsynaptic membrane is shown by a red line. The Ca2+ and drebrin content in the spine head are shown by black and blue lines, respectively
The resting spine at steady state has a high drebrin content. According to the aforementioned neutral dynamic morphology model, dendritic spines at steady state are resilient, namely, the average size of dendritic spines does not change at steady state because of the presence of stable F-actin, although they spontaneously increase their size by random nucleation and decrease it by severing and capping.
In the initial process of either LTP or LTD, the Ca2+ influx through NMDARs triggers myosin-driven drebrin exodus, resulting in drebrin decrease. Thus, potentiated dendritic spines change from steady state to excited state, where dynamic F-actin predominates and F-actin begins to polymerize by activation of actin nucleation.
Because of the increase in dynamic F-actin, potentiated spines become plastic and exhibit LTP or LTD as a result of two different processes. During LTP, the increased Ca2+ soon returns to the basal level, and AMPAR density and the spine size increase. The increase in AMPAR density in conjunction with Ca2+ recovery coordinately accumulates drebrin in dendritic spines, leading the spine to steady state. Thus, the resilient spines keep their enlarged size with a secure increase in synaptic strength during LTP maintenance.
In contrast during LTD, the intracellular Ca2+ increase is prolonged, which inhibits drebrin re-accumulation. Then, low drebrin facilitates AMPAR endocytosis, resulting in decreased AMPAR density at the postsynaptic membrane. Because AMPAR activity is necessary for the re-accumulation of drebrin, AMPAR decrease and drebrin re-accumulation inhibition form a positive feedback loop, keeping high plasticity of the dendritic spines. This is consistent with the observation that small spines are preferable for synaptic plasticity.
According to this model, the resilient spines in Alzheimer’s disease likely show high spine instability because of low drebrin. This may cause defects in LTP maintenance in Alzheimer’s disease. Thus, it is important to examine the pathological relevance of spine instability to various types of dementia.
References
Abraham WC, Williams JM (2003) Properties and mechanisms of LTP maintenance. Neuroscientist 9:463–474
Allison DW, Gelfand VI, Spector I, Craig AM (1998) Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci 18:2423–2436
Aoki C, Sekino Y, Hanamura K, Fujisawa S, Mahadomrongkul V, Ren Y, Shirao T (2005) Drebrin A is a postsynaptic protein that localizes in vivo to the submembranous surface of dendritic sites forming excitatory synapses. J Comp Neurol 483:383–402
Arai H, Sato K, Uto A, Yasumoto Y (1991) Effect of Transient cerebral ischemia in mongolian gerbils on synaptic vesicle protein (SVP-38) and developmentally regulated brain protein (drebrin). Neurosci Res Commun 9:143–150
Asrican B, Lisman J, Otmakhov N (2007) Synaptic strength of individual spines correlates with bound Ca2+-calmodulin-dependent kinase II. J Neurosci 27:14007–14011
Biou V, Brinkhaus H, Malenka RC, Matus A (2008) Interactions between drebrin and Ras regulate dendritic spine plasticity. Eur J Neurosci 27:2847–2859
Cao F, Zhou Z, Pan X, Leung C, Xie W, Collingridge G, Jia Z (2017) Developmental regulation of hippocampal long-term depression by cofilin-mediated actin reorganization. Neuropharmacology 112:66–75
Chimura T, Launey T, Yoshida N (2015) Calpain-mediated degradation of drebrin by excitotoxicity in vitro and in vivo. PLoS One 10:e0125119
Correia SS, Bassani S, Brown TC, Lise MF, Backos DS, El-Husseini A, Passafaro M, Esteban JA (2008) Motor protein-dependent transport of AMPA receptors into spines during long-term potentiation. Nat Neurosci 11:457–466
Dailey ME, Smith SJ (1996) The dynamics of dendritic structure in developing hippocampal slices. J Neurosci 16:2983–2994
Desmond NL, Levy WB (1983) Synaptic correlates of associative potentiation/depression: an ultrastructural study in the hippocampus. Brain Res 265:21–30
Fischer M, Kaech S, Knutti D, Matus A (1998) Rapid actin-based plasticity in dendritic spines. Neuron 20:847–854
Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K (2003) Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron 38:447–460
Geraldo S, Khanzada UK, Parsons M, Chilton JK, Gordon-Weeks PR (2008) Targeting of the F-actin-binding protein drebrin by the microtubule plus-tip protein EB3 is required for neuritogenesis. Nat Cell Biol 10:1181–1189
Gordon-Alonso M, Rocha-Perugini V, Alvarez S, Ursa A, Izquierdo-Useros N, Martinez-Picado J, Munoz-Fernandez MA, Sanchez-Madrid F (2013) Actin-binding protein drebrin regulates HIV-1-triggered actin polymerization and viral infection. J Biol Chem 288:28382–28397
Goslin K, Banker G (1989) Experimental observations on the development of polarity by hippocampal neurons in culture. J Cell Biol 108(4):1507–1516
Halpain S (2000) Actin and the agile spine: how and why do dendritic spines dance? Trends Neurosci 23:141–146
Harigaya Y, Shoji M, Shirao T, Hirai S (1996) Disappearance of actin-binding protein, drebrin, from hippocampal synapses in Alzheimer’s disease. J Neurosci Res 43(1):87–92
Harris KM, Stevens JK (1989) Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci 9:2982–2997
Haviv L, Gillo D, Backouche F, Bernheim-Groswasser A (2008) A cytoskeletal demolition worker: myosin II acts as an actin depolymerization agent. J Mol Biol 375:325–330
Hayashi K, Shirao T (1999) Change in the shape of dendritic spines caused by overexpression of drebrin in cultured cortical neurons. J Neurosci 19:3918–3925
Hayashi K, Ishikawa R, Ye LH, He XL, Takata K, Kohama K, Shirao T (1996) Modulatory role of drebrin on the cytoskeleton within dendritic spines in the rat cerebral cortex. J Neurosci 16:7161–7170
Honkura N, Matsuzaki M, Noguchi J, Ellis-Davies GC, Kasai H (2008) The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron 57:719–729
Kaech S, Banker G (2006) Culturing hippocampal neurons. Nat Protoc 1:2406–2415
Knott GW, Holtmaat A, Wilbrecht L, Welker E, Svoboda K (2006) Spine growth precedes synapse formation in the adult neocortex in vivo. Nat Neurosci 9(9):1117–1124. Epub 2006 Aug 6
Kobayashi C, Aoki C, Kojima N, Yamazaki H, Shirao T (2007) Drebrin a content correlates with spine head size in the adult mouse cerebral cortex. J Comp Neurol 503:618–626
Koganezawa N, Hanamura K, Sekino Y, Shirao T (2017) The role of drebrin in dendritic spines. Mol Cell Neurosci. doi:10.1016/j.mcn.2017.01.004
Kojima N, Hanamura K, Yamazaki H, Ikeda T, Itohara S, Shirao T (2010) Genetic disruption of the alternative splicing of drebrin gene impairs context-dependent fear learning in adulthood. Neuroscience 165:138–150
Kojima N, Shirao T (2007) Synaptic dysfunction and disruption of postsynaptic drebrin-actin complex: a study of neurological disorders accompanied by cognitive deficits. Neurosci Res 58(1):1–5. Epub 2007 Feb 11
Korn ED, Carlier MF, Pantaloni D (1987) Actin polymerization and ATP hydrolysis. Science 238(4827):638–644
Kojima N, Yasuda H, Hanamura K, Ishizuka Y, Sekino Y, Shirao T (2016) Drebrin A regulates hippocampal LTP and hippocampus-dependent fear learning in adult mice. Neuroscience 324:218–226
Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT (2001) Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29:243–254
Ma Q, Ruan YY, Xu H, Shi XM, Wang ZX, Hu YL (2015) Safflower yellow reduces lipid peroxidation, neuropathology, tau phosphorylation and ameliorates amyloid β-induced impairment of learning and memory in rats. Biomed Pharmacother 76:153–164. doi: 10.1016/j.biopha.2015.10.004. Epub 2015 Nov 19
Malenka RC, Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44:5–21
Maletic-Savatic M, Malinow R, Svoboda K (1999) Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science 283:1923–1927
Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429:761–766
Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H (2001) Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci 4:1086–1092
Matus A, Ackermann M, Pehling G, Byers HR, Fujiwara K (1982) High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sci U S A 79:7590–7594
Mikati MA, Grintsevich EE, Reisler E (2013) Drebrin-induced stabilization of actin filaments. J Biol Chem 288:19926–19938
Mizui T, Takahashi H, Sekino Y, Shirao T (2005) Overexpression of drebrin A in immature neurons induces the accumulation of F-actin and PSD-95 into dendritic filopodia, and the formation of large abnormal protrusions. Mol Cell Neurosci 30:149–157
Mizui T, Kojima N, Yamazaki H, Katayama M, Hanamura K, Shirao T (2009) Drebrin E is involved in the regulation of axonal growth through actin-myosin interactions. J Neurochem 109:611–622
Mizui T, Sekino Y, Yamazaki H, Ishizuka Y, Takahashi H, Kojima N, Kojima M, Shirao T (2014) Myosin II ATPase activity mediates the long-term potentiation-induced exodus of stable F-actin bound by drebrin A from dendritic spines. PLoS One 9:e85367
Mulkey RM, Endo S, Shenolikar S, Malenka RC (1994) Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature 369:486–488
Murrell MP, Gardel ML (2012) F-actin buckling coordinates contractility and severing in a biomimetic actomyosin cortex. Proc Natl Acad Sci U S A 109:20820–20825
Nusser Z, Lujan R, Laube G, Roberts JD, Molnar E, Somogyi P (1998) Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron 21(3):545–559
Okamoto K, Nagai T, Miyawaki A, Hayashi Y (2004) Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci 7:1104–1112
Ouyang Y, Wong M, Capani F, Rensing N, Lee CS, Liu Q, Neusch C, Martone ME, Wu JY, Yamada K, Ellisman MH, Choi DW (2005) Transient decrease in F-actin may be necessary for translocation of proteins into dendritic spines. Eur J Neurosci 22:2995–3005
Petrozzino JJ, Pozzo Miller LD, Connor JA (1995) Micromolar Ca2+ transients in dendritic spines of hippocampal pyramidal neurons in brain slice. Neuron 14:1223–1231
Rex CS, Gavin CF, Rubio MD, Kramar EA, Chen LY, Jia Y, Huganir RL, Muzyczka N, Gall CM, Miller CA, Lynch G, Rumbaugh G (2010) Myosin IIb regulates actin dynamics during synaptic plasticity and memory formation. Neuron 67:603–617
Rocca DL, Amici M, Antoniou A, Blanco Suarez E, Halemani N, Murk K, McGarvey J, Jaafari N, Mellor JR, Collingridge GL, Hanley JG (2013) The small GTPase Arf1 modulates Arp2/3-mediated actin polymerization via PICK1 to regulate synaptic plasticity. Neuron 79:293–307
Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8(6):595–608. doi: 10.15252/emmm.201606210
Sekino Y, Tanaka S, Hanamura K, Yamazaki H, Sasagawa Y, Xue Y, Hayashi K, Shirao T (2006) Activation of N-methyl-D-aspartate receptor induces a shift of drebrin distribution: disappearance from dendritic spines and appearance in dendritic shafts. Mol Cell Neurosci 31:493–504
Sharma S, Grintsevich EE, Hsueh C, Reisler E, Gimzewski JK (2012) Molecular cooperativity of drebrin1-300 binding and structural remodeling of F-actin. Biophys J 103:275–283
Shirao T, Hanamura K, Koganezawa N, Ishizuka Y, Yamazaki H, Sekino Y (2017) The role of drebrin in neurons. J Neurochem. doi:10.1111/jnc.13988
Sokal DM, Mason R, Parker TL (2000) Multi-neuronal recordings reveal a differential effect of thapsigargin on bicuculline- or gabazine-induced epileptiform excitability in rat hippocampal neuronal networks. Neuropharmacology 39:2408–2417
Star EN, Kwiatkowski DJ, Murthy VN (2002) Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci 5(3):239–246
Takahashi H, Yamazaki H, Hanamura K, Sekino Y, Shirao T (2009) Activity of the AMPA receptor regulates drebrin stabilization in dendritic spine morphogenesis. J Cell Sci 122:1211–1219
Takahashi H, Sekino Y, Tanaka S, Mizui T, Kishi S, Shirao T (2003) Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine mor phogenesis. J Neurosci 23:6586–6595
Takumi Y, Ramírez-León V, Laake P, Rinvik E, Ottersen OP (1999) Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat Neurosci 2(7):618–624
Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K (2002) Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420:788–794
Wang XB, Yang Y, Zhou Q (2007) Independent expression of synaptic and morphological plasticity associated with long-term depression. J Neurosci 27:12419–12429
Wegner AM, Nebhan CA, Hu L, Majumdar D, Meier KM, Weaver AM, Webb DJ (2008) N-wasp and the arp2/3 complex are critical regulators of actin in the development of dendritic spines and synapses. J Biol Chem 283:15912–15920
Wilson CA, Tsuchida MA, Allen GM, Barnhart EL, Applegate KT, Yam PT, Ji L, Keren K, Danuser G, Theriot JA (2010) Myosin II contributes to cell-scale actin network treadmilling through network disassembly. Nature 465:373–377
Worth DC, Daly CN, Geraldo S, Oozeer F, Gordon-Weeks PR (2013) Drebrin contains a cryptic F-actin-bundling activity regulated by Cdk5 phosphorylation. J Cell Biol 202:793–806
Yasumatsu N, Matsuzaki M, Miyazaki T, Noguchi J, Kasai H (2008) Principles of long-term dynamics of dendritic spines. J Neurosci 28:13592–13608
Yuste R (2015) The discovery of dendritic spines by Cajal. Front Neuroanat 9:18
Zhou Q, Homma KJ, Poo MM (2004) Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44:749–757
Zimmermann J, Falcke M (2014) Formation of transient lamellipodia. PLoS One 9:e87638
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Japan KK
About this chapter
Cite this chapter
Sekino, Y., Koganezawa, N., Mizui, T., Shirao, T. (2017). Role of Drebrin in Synaptic Plasticity. In: Shirao, T., Sekino, Y. (eds) Drebrin. Advances in Experimental Medicine and Biology, vol 1006. Springer, Tokyo. https://doi.org/10.1007/978-4-431-56550-5_11
Download citation
DOI: https://doi.org/10.1007/978-4-431-56550-5_11
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-56548-2
Online ISBN: 978-4-431-56550-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)