
Abstract
In the adherens junction (AJ), cadherin and catenin proteins form a cell–cell adhesion complex that is indispensable for tissue morphogenesis and homeostasis. The complex mechanically couples neighboring cells through intercellular binding by cadherins, and actin binding and regulation by the cytoplasmic catenins. In addition, the cadherin–catenin complex participates in signaling pathways that direct cellular organization, proliferation, and motility. Some of these signaling pathways can be regulated by mechanical stimulation or posttranslational modification of the components of the AJ. In light of these findings, we discuss our current understanding of how AJ signaling and mechanical functions are regulated by phosphorylation and force, and speculate on the mechanisms underlying the coordination between these two types of modifications.
J. Tan and B.W. Benham-Pyle contributed equally with all other contributors.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Cadherin Extracellular Domain Interactions
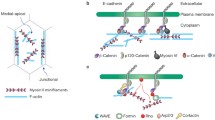
The adherens junction (AJ) contains classical cadherins, which are single-pass transmembrane proteins with five extracellular cadherin (EC) repeat domains that form a rigid curved structure stabilized by Ca++ (Shapiro and Weis 2009; Pokutta et al. 1994). Cell–cell adhesion is established through trans binding between the N-terminal EC1 domain of cadherins on opposing cells, and X-ray crystal structures have revealed two kinds of interfaces between these interacting EC1 domains (Manibog et al. 2014; Rakshit et al. 2012; Brasch et al. 2012; Harrison et al. 2010). In the first, the N-terminal β-strands of each domain exchange to form part of a β-sheet in the partner molecule (strand-swap dimer). The second interface involves association of the base of EC1 and the Ca++-binding site between it and EC2 to form an X-dimer. Kinetic and equilibrium measurements, as well as atomic force microscopy assays and steered molecular dynamics simulations, indicate that the X-dimer is an intermediate in the formation of the more stable strand-swap dimer (Manibog et al. 2014; Rakshit et al. 2012; Brasch et al. 2012; Harrison et al. 2010). The strand-swap dimer is formed by molecular interactions very similar to those in the unbound monomer, and involves the kinetically unfavorable refolding of the interacting EC1s to accommodate the partner β-strand. Thus, the X-dimer may be a low energy “encounter complex” intermediate that overcomes the kinetic barrier to the strand swap (Fig. 5.1).

Regulation of E-cadherin interactions by phosphorylation and force. Cadherin-mediated adhesion regulates the maturation of initial cell–cell recognition to loosely adherent cell clusters, to compacted groups of cells in colonies. E-cadherin is constitutively phosphorylated at S840, S846, and S847, facilitating binding to β-catenin and shuttling to the plasma membrane. E-cadherin is under constitutive tension after being incorporated into the plasma membrane and association with the actin cytoskeleton. E-cadherin trans X-dimer bonds are stabilized by force and may precede formation of stable strand-swap dimers, E-cadherin cis interactions, and the mature Adherens junction
The extracellular region of E-cadherins of the same cell can form cis interactions, which appear to contribute to the stability of cell–cell contacts. The existence of cis clusters has been inferred from crystal structures (Harrison et al. 2011), fusion constructs (Pertz et al. 1999), and chemical cross-linking (Takeda et al. 1999), but the interactions are apparently too weak to be detected in solution or in single-molecule assays, suggesting that rates of association and, thus, binding probabilities are low (Zhang et al. 2009). Combined atomic force microscopy and FRET measurements found that even though two cadherin extracellular domains do not bind in cis as single molecules, their proximity increases the probability of establishing a trans interaction (Zhang et al. 2009). Mutational disruption of E-cadherin cis interactions inferred from crystal structures prevented recruitment of endogenous E-cadherin to cell–cell junctions, indicating that cis interactions are required for AJ maturation (Harrison et al. 2011). Moreover, disruption of either trans or cis E-cadherin interactions by site-specific mutagenesis demonstrated that trans interactions in turn stabilize cis-mediated clusters of E-cadherin lacking the cytoplasmic domain, and that anchoring E-cadherin to the actin cytoskeleton guides the assembly of these clusters (Hong et al. 2013). Together, these studies indicate that trans and cis cadherin binding may cooperate during formation of cell–cell contacts.
Mechanical force may also have a role in stabilizing the cadherin adhesive interaction (Fig. 5.1). Notably, the two trans-dimer configurations have different unbinding kinetics in response to applied tension (that is, an opposing mechanical force): the X-dimer forms a catch bond, whose lifetime increases with tension, whereas the strand-swap dimer behaves as a slip bond, whose duration decreases monotonically with respect to applied tension (Rakshit et al. 2012). It is unclear if cadherin catch bonds have a significant role in vivo. Formation of E-cadherin strand-swap dimers does not seem to require tension. In vitro spectroscopy experiments indicate that most extracellular dimers can form strand-swap slip bonds after an unloaded (no tension) contact time of 3 s (Rakshit et al. 2012). Moreover, mutations that compromise the affinity of the X-dimer slow, but still permit, the formation of the strand-swap dimer (Harrison et al. 2010). The X-dimer bond is most stable at ~30 pN, a level that is unlikely to be reached by a single myosin motor (Norstrom et al. 2010) coupled to the cadherin–catenin/actin complex. It is possible that catch bond behavior enables lower levels of tension to extend the lifetime of the X-dimer bond and thereby increase the probability of transition into the more robust strand-swap dimer conformation during initial cell–cell contact formation.
Cadherins are under tension in mature cell–cell contacts. A Forster resonance energy transfer (FRET)-based tension sensor (Grashoff et al. 2010) introduced into the cytoplasmic domain of E-cadherin indicated that E-cadherin is under constitutive tension of approximately 2 pN in cultured epithelial cells (Borghi et al. 2012). Tension along E-cadherin required catenin-mediated linkage to an intact contractile actomyosin network. In another study, the same E-cadherin sensor was used to observe cadherin-specific tension during collective cell migration of border cells in the Drosophila ovary. In this context, the average tension was also ~2 pN, and was sensitive to the activity of the small Rho family GTPase Rac. Rac regulates the nucleation of branched actin filaments (Cai et al. 2014), and these may change cadherin tension by protruding into the nearby membrane and changing membrane shape. Another study also found that the morphology and contractility of the cytoskeleton influences force transmission at cadherin–catenin complexes, which experience a decrease in tension when shear force redirects intercellular tension to PECAM-1, an adhesion molecule abundant in endothelial cell–cell junctions (Conway et al. 2013).
Even though the cadherin FRET sensor has been successfully used to detect tension at cell–cell adhesions, it has a narrow dynamic range. The force versus FRET efficiency calibration curve characterized in the original vinculin FRET sensor showed that FRET indices at forces greater than 7.5 pN are indistinguishable from the background signal (Grashoff et al. 2010). Due to this limitation, the cadherin force sensor cannot be used to test if intercellular cadherin bonds in cells are ever subject to 30-pN forces, which stabilize the bonds in the X-dimer conformation. Since the inception of the vinculin tension sensor, several FRET-based genetically encoded and synthetic tension sensors have been developed (Cost et al. 2015). Unfortunately, these sensors are subject to their own unique limitations, and further techniques will need to be developed to chart a comprehensive map of forces at cell–cell junctions.
At the cellular scale, forces at cell–cell junctions have been inferred using traction force microscopy based on the principle of mechanical equilibrium (Maruthamuthu et al. 2011; Ng et al. 2014; Sim et al. 2015). In these experiments, cells are plated on a compliant substrate functionalized with extracellular matrix (ECM), whose deformation can be used to calculate stresses at the cell–substrate interface. Cells typically do not move substantially during the timescale of substrate deformation, so the cells are assumed to be under mechanical equilibrium in which cell–cell forces balance cell–ECM forces. Using this strategy, cell–cell junctions were found to be subject to hundreds of nN of tension. However, this tension is not confined to the AJ, as epithelial cells also form intermediate filament-bound desmosomes and an actin filament-bound tight junction. A study combining traction force microscopy and the cadherin FRET tension sensor found that average tension along cadherin molecules was constant in spite of significant changes in cell–ECM and cell–cell forces (Sim et al. 2015). Moreover, increased forces at cell–cell contacts did not result in changes of total cadherin levels at cell–cell junctions. Instead, cadherin was found to be locally enriched at the edges of the contacts as cell–cell forces increased. These findings suggest that cells may maintain molecular-level mechanical homeostasis at the AJ by modulating the localization of cadherin-based complexes.
2 Cadherin Intracellular Domain Interactions: p120-Catenin
Interactions between cadherin, catenin proteins, and the actin cytoskeleton are tightly regulated to coordinate AJ assembly and disassembly in response to external or internal cues. In epithelial tissues, the cadherin–catenin complex is composed of E-cadherin and its associated cytoplasmic catenins: p120-catenin, β-catenin, and αE-catenin. β-Catenin binds the cytoplasmic domain of E-cadherin upon synthesis in the endoplasmic reticulum. After delivery of the heterodimer to the plasma membrane, the complex is stabilized by intercellular trans E-cadherin interactions (see above) and p120-catenin binding to the cadherin juxtamembrane domain. Finally, αE-catenin mechanically integrates the cytoskeletons of adjacent cells by binding to β-catenin and linking actin filaments to the complex (Ozawa et al. 1990; Hinck et al. 1994).
p120-Catenin regulates the rate of cadherin endocytosis , and the dynamics of the actin cytoskeleton through interactions with Rho family GTPases (Fig. 5.2, left). p120-Catenin was first identified as a Src kinase substrate in a study designed to screen for genes related to transformation (Reynolds et al. 1994), but subsequent studies demonstrated that in nontransformed cells, direct binding between p120-catenin and cadherin stabilizes cadherin at the plasma membrane at the onset of strong cell–cell adhesion (Thoreson et al. 2000; Yap et al. 1998; Davis and Reynolds 2006). Moreover, internalization assays demonstrated that p120-catenin binding prevents cadherin endocytosis by blocking binding of Hakai, an E3 ligase that ubiquitylates the E-cadherin cytoplasmic domain, targeting the complex to the endocytic machinery (Hartsock and Nelson 2012; Xiao et al. 2005). Src phosphorylation of p120-catenin at Y217 and Y228 increases p120-catenin affinity for E-cadherin and RhoA GTPase (Roura et al. 1999). Similarly, Fyn/Fer kinases phosphorylate p120-catenin and increase its affinity for E-cadherin (Rosato et al. 1998). However, Src and Fer/Fyn kinases also phosphorylate β-catenin Y654 and Y142, respectively, leading to dissociation from E-cadherin and αE-catenin and subsequent deterioration of cell–cell adhesion (Roura et al. 1999; Piedra et al. 2003). Together, these results raise the question: why is the affinity of p120-catenin to cadherin increased by kinases that also destabilize cadherin’s interactions with the other catenins?

p120-Catenin-mediated regulation of actin dynamics, E-cadherin endocytosis, and phosphatase activity at the AJ. Cytosolic p120-catenin tyrosine-phosphorylated by growth factor cascades and/or Src and Fer kinases (orange) downregulates RhoA GTPase activity. Without p120-catenin binding, E-cadherin is targeted for endocytosis. Upon recruitment to the AJ at high cell densities, p120-catenin and its associated kinases can activate phosphatases (green) that counteract tyrosine-phosphorylation of β-catenin and αE-catenin, stabilizing the cadherin–catenin complex at the AJ
Phosphorylation -mediated disruption of the cadherin–catenin complex may be opposed by p120-catenin (Fig. 5.2, right). In addition to binding to E-cadherin, p120-catenin associates with several tyrosine phosphatases, including the receptor-type tyrosine phosphatases PTPμ (Zondag et al. 2000) and DEP-1 (Holsinger et al. 2002), and the cytosolic tyrosine phosphatase SHP-1 (Reynolds et al. 1994). The receptor-type tyrosine phosphatases are upregulated at high cell density (Ostman et al. 1994) and could counteract Src and Fer/Fyn phosphorylation of the cadherin–catenin complex during cell–cell junction maturation. In addition, p120-catenin recruits Fer to cell–cell adhesions, promoting activation of PTP1B, a cytosolic tyrosine phosphatase that counteracts phosphorylation of β-catenin Y142 and Y654 (El Sayegh et al. 2005; Xu et al. 2004). Thus, p120-catenin may play a critical role in maintaining the balance of kinase and phosphatase activity in the context of cell–cell adhesion.
p120-catenin also regulates actin dynamics through its interactions with Rho family GTPases (Grosheva et al. 2001; Noren et al. 2000; Anastasiadis et al. 2000). Actin dynamics regulate the architecture of the cytoskeleton, thus p120-catenin likely affects how the cadherin–catenin complex transmits mechanical stimuli. In one study, for example, overexpression of p120-catenin inhibited RhoA activity, resulting in formation of branch-like actin protrusions and destabilization of stress fibers (Reynolds et al. 1996). These findings indicate that p120-catenin, when dissociated from E-cadherin, induces a more migratory phenotype driven by branch-like actin protrusions (Noren et al. 2000; Reynolds et al. 1996). This phenotype is evident in nascent cell–cell contacts (Toret et al. 2014; Yamada and Nelson 2007), but is suppressed by RhoA activity as cell–cell contacts expand (Yamada and Nelson 2007). In addition, studies show that RhoA activity can be mechanically activated in a variety of cell types (Zhao et al. 2007a; Abiko et al. 2015) and is correlated with local levels of stress (Reffay et al. 2014). Whether p120-catenin plays a role in this pathway remains to be determined. p120-Catenin can modulate GTPase activity by acting as a guanine nucleotide dissociation inhibitor (Anastasiadis et al. 2000) or associating with guanine nucleotide exchange factors such as p190RhoGAP (Wildenberg et al. 2006) and Vav2 (Fukuyama et al. 2006). How these interactions are affected by mechanical perturbation of cell–cell contacts has not been investigated.
3 Cadherin Intracellular Interactions: β-Catenin
β-Catenin, an armadillo repeat protein (Huber et al. 1997a), binds to the cytoplasmic domain of E-cadherin distal to the juxtamembrane domain and the p120-catenin binding site. In turn, β-catenin binds the actin binding protein αE-catenin (Huber et al. 1997b). Binding of β-catenin confers structure to the cytoplasmic domain of E-cadherin, which protects cadherin from proteolysis (Huber et al. 2001) and reduces the turnover rate of the E-cadherin/β-catenin heterodimer at the plasma membrane. Calorimetry and mutagenesis studies indicate that the affinity of E-cadherin/β-catenin is increased when S840, S846, and S847 in the E-cadherin cytoplasmic domain are phosphorylated (Lickert et al. 2000; Serres et al. 2000; Choi et al. 2006; Fig. 5.1). These phosphorylation events occur constitutively (McEwen et al. 2014) and may stabilize the cadherin–catenin complex.
There are many posttranslational modifications that regulate the turnover of β-catenin in the cadherin–catenin complex (Fig. 5.3, top left). Phosphorylation of Y654 by Src or Abl, both cytoplasmic kinases, disrupts a hydrogen bond between the β-catenin Y654 phenolic hydroxyl group and a cadherin aspartate residue (Huber and Weis 2001), resulting in at least a fifteen-fold reduction in affinity (Roura et al. 1999; Catimel et al. 2006). Another means of perturbing this interaction is via Src-mediated phosphorylation of N-cadherin Y860, as found in endothelial cells (Qi et al. 2005). Although Src disrupts E-cadherin/β-catenin heterodimerization, the p120-catenin-associated cytoplasmic kinases Fer and Fyn (Kim and Wong 1995) disrupt β-catenin/αE-catenin interactions through tyrosine-phosphorylation of β-catenin (Rosato et al. 1998) Y142 (Piedra et al. 2003), which is located in the β-catenin/αE-catenin binding interface (Pokutta and Weis 2000).

Regulation of β-catenin localization, stability, and transcriptional activity by cell density and the balance of tyrosine kinase and phosphatase activities. Interactions of β-catenin with E-cadherin and αE-catenin are negatively regulated by phosphorylation of β-catenin by receptor and cytoplasmic tyrosine kinases EGFR, Src, Abl, Fer, and Fyn (red/orange components), which phosphorylate Y654 and Y142 residues in β-catenin. In contrast, β-catenin interactions with E-cadherin and αE-catenin are positively regulated by serine/threonine phosphorylation of E-cadherin (S840, S846, and S847) and β-catenin dephosphorylation (Y654 and Y142) by protein tyrosine phosphatases that bind p120 and β-catenin (green components). Degradation of cytoplasmic β-catenin is driven by phosphorylation by CKI and GSK3β and scaffolding by the tumor suppressors Axin and APC. The localization and phosphorylation state of β-catenin are associated with changes in cell density and affect cell–cell adhesion, cell migration, and the level of transcriptionally active β-catenin
Fer and Fyn are examples of kinases that regulate cadherin–catenin complex stability downstream of signaling pathways mediated by receptor tyrosine kinases (RTKs). One of the most studied RTKs known to regulate the cadherin–catenin complex is the epidermal growth factor (EGF) receptor. EGF receptor activation induces dissociation of cell aggregates, cell rounding, and membrane ruffling (Fujii et al. 1996). The EGF receptor can bind directly to β-catenin (Hoschuetzky et al. 1994) and phosphorylate Y654 (Hazan and Norton 1998), weakening β-catenin affinity for E-cadherin. Without the cadherin–β-catenin interaction, αE-catenin cannot link the actin cytoskeletons of neighboring cells, resulting in reduced cell–cell adhesion and transition to a migratory phenotype. There is also evidence for the intersection of Src kinase and EGFR activation pathways, as inhibition of Src kinase blocks EGF-stimulated DNA synthesis and subsequent proliferation (Bromann et al. 2004). Activation of MET tyrosine kinase, another RTK, by hepatocyte growth factor (HGF) also results in β-catenin phosphorylation and subsequent nuclear accumulation (Monga et al. 2002).
When not associated with E-cadherin, β-catenin can participate in Wnt-dependent and -independent proliferation pathways (Fig. 5.3, bottom left). These require the translocation of β-catenin to the nucleus (McCrea et al. 1991; Nelson and Nusse 2004), where it associates with TCF/LEF transcription factors and induces specific gene transcription (He et al. 1998; Korinek et al. 1997; Morin et al. 1997). The amount of cytoplasmic β-catenin and thereby its transcriptional function can be regulated by a proteasome-targeted destruction complex (Aberle et al. 1997) comprising the tumor suppressors Adenomatous Polyposis Coli (APC) (Rubinfeld et al. 1993; Su et al. 1993) and axin (Zeng et al. 1997), the serine and threonine kinases GSK-3 (Dominguez et al. 1995; He et al. 1995; Kimelman and Pierce 1996) and CK1 (Liu et al. 2002; Amit et al. 2002), protein phosphatase 2A (Seeling et al. 1999), and the E3-ligase β-TrCP (Winston et al. 1999). Axin scaffolds the phosphorylation of β-catenin S45 by CKI (Amit et al. 2002; Sakanaka 2002), and then T41, S37, and S33 by GSK3 (Liu et al. 2002; Sadot et al. 2002); phosphorylation of S33 and S37 leads to ubiquitylation by β-TrCP and destruction in the proteasome. Canonical Wnt signaling promotes cell proliferation by inhibiting the activity of the β-catenin destruction complex, and these pathways are dysfunctional in many cancers (Fodde and Brabletz 2007).
Mechanical strain activates the transcriptional function of β-catenin independently of the Wnt signaling pathway during gastrulation in Danio rerio and Drosophila melanogaster (Brunet et al. 2013; Desprat et al. 2008). During gastrulation, the blastula, a spherical sheet of cells, folds inwards to create the gastrula, a structure comprising the three germ layers that give rise to specific organs during embryonic development. Folding of the blastula requires actomyosin contractility and correlates with Src-mediated phosphorylation of β-catenin Y654. In the absence of endogenous actomyosin contractility, β-catenin phosphorylation could be rescued by exogenous compression of the blastula using magnetic beads (Brunet et al. 2013). Mechanical strain across a contact-inhibited epithelial monolayer in vitro also results in increased β-catenin nuclear signaling, and cell-cycle progression (Benham-Pyle et al. 2015). This increase in signaling requires cadherin-mediated cell–cell adhesion, as expression of a truncated E-cadherin lacking the extracellular domain blocked activation of β-catenin and cell-cycle progression following mechanical strain.
At present, it is unclear how mechanical strain is transduced to Src or β-catenin activation. Because Src phosphorylation was rescued using nonspecific magnetic compression of tissues (Desprat et al. 2008), Src may be subject to mechanical regulation independently of the cadherin–catenin complex at sites of cell–cell adhesion. Abl kinase, which affects cell–cell adhesion similarly to Src, possesses an actin binding domain (Van Etten et al. 1994), and myristoylation anchors the kinase to the plasma membrane (Hantschel et al. 2003). Interestingly, combined actin binding and myristoylation inhibit Abl activity (Hantschel et al. 2003; Woodring et al. 2001), which is lower in stable cell–cell contacts (Bays et al. 2014). These results suggest that mechanical stimuli could activate Abl at cell–cell contacts by dissociating it from the actin cytoskeleton.
Wnt-independent nuclear localization of β-catenin also depends on cell density (Dietrich et al. 2002). As cell density increases, β-catenin shifts from a nuclear pool to a junctional pool, and confluent cells stop proliferating due to contact inhibition. Cell density changes are accompanied by dramatic changes in cell morphology, and these changes may affect force generation and transmission at the AJ. Thus, it is possible that changes in mechanical strain and cell density modulate β-catenin junctional stability and transcriptional activity in similar ways (Brunet et al. 2013; Desprat et al. 2008; Benham-Pyle et al. 2015).
Phosphorylation of β-catenin and hence its transcriptional activity can be inhibited by several protein tyrosine phosphatases (PTPs) at cell–cell junctions (Fig. 5.3, right). PTPκ binds β-catenin in vitro and dephosphorylates tyrosine-phosphorylated β-catenin from cell lysates (Fuchs et al. 1996), and PTPλ similarly associates with β-catenin (Cheng et al. 1997). Several protein tyrosine phosphatases such as the cytosolic PTP-PCP2 also dephosphorylate β-catenin that had been phosphorylated downstream of growth factor signaling pathways (Yan et al. 2002). In high-density cultures, phosphatases localize to cell–cell junctions (Rijksen et al. 1993) and cadherin–catenin complexes may be directly involved in their recruitment (Piedra et al. 2003).
4 Cadherin–Catenin Intracellular Interactions: αE-Catenin
αE-catenin, which binds to cadherin through β-catenin, anchors the AJ to the actin cytoskeleton directly or indirectly through different actin-binding partners. The amino terminus of αE-catenin comprises a β-catenin binding domain and, in the mammalian homologue, an overlapping homodimerization domain (Pokutta and Weis 2000). The N-terminus is followed by a modulation domain that binds several actin-binding proteins including vinculin (Hazan et al. 1997; Choi et al. 2012), l-afadin (Pokutta et al. 2002), formin-1 (Kobielak et al. 2004), and α-actinin (Knudsen et al. 1995); the C-terminal domain also binds ZO-1 (Itoh et al. 1997) and EPLIN (Abe and Takeichi 2008). Thus, the cadherin–catenin complex can bind the actin cytoskeleton and regulate its nucleation (Kobielak et al. 2004; Tang and Brieher 2012) and morphology (Abe and Takeichi 2008) through multiple actin binding partners. The C-terminal domain of αE-catenin binds directly to actin filaments (Pokutta et al. 2002; Rimm et al. 1995; Fig. 5.4, left).

Regulation of cytosolic and junctional αE-catenin. Cytosolic αE-catenin can be dephosphorylated by Shp2 phosphatase (green), and can also form homodimers that have a higher affinity for actin filaments and inhibit Arp2/3-mediated branching. Junctional αE-catenin is subject to phosphorylation by CKI/II (orange), and acto-myosin generated tension which increases the actin binding affinity of the cadherin–catenin complex by modulating transitions between weakly and strongly bound catch bond states. Under tension, αE-catenin acquires an open conformational state associated with vinculin recruitment (dark purple), and possibly other actin binding proteins (see domain organization)
αE-Catenin contains a bone fide actin-binding domain and a long-standing hypothesis in the field is that the cadherin–catenin complex binds to actin filaments directly. However, a simple actin pelleting assay was unable to reconstitute this interaction in vitro (Yamada et al. 2005) because binding to β-catenin decreases the actin binding affinity of αE-catenin by >20-fold (Drees et al. 2005; Miller et al. 2013). These findings were puzzling inasmuch as other experiments demonstrated that actin binding is necessary for cell–cell adhesion (Imamura et al. 1999) and that adhesion can be induced by E-cadherin-αE-catenin chimeras (Nagafuchi et al. 1994; Pacquelet and Rørth 2005).
Because E-cadherin is under constitutive tension in cells (see above), an optical trap was used to reconstitute a direct cadherin–catenin/actin interaction by applying tension to the αE-catenin/F-actin bond (Buckley et al. 2014). This work supported a two-state catch bond model in which increasing tension shifts the cadherin–catenin/actin bond from a weakly bound state to a strongly bound state (Fig. 5.4, right). However, the molecular basis for cadherin–catenin/actin catch bond states is unclear due to a lack of detailed structural information. Crystal structures of nearly full-length dimeric αE-catenin have been published (Rangarajan and Izard 2012; Rangarajan and Izard 2013), but because β-catenin–bound αE-catenin behaves differently from the dimer in in vitro biochemical assays (Drees et al. 2005; Miller et al. 2013), the available structures may not provide a strong basis for understanding actin binding by the complex. It is also possible that the kinetic states in the two-state cadherin–catenin/actin catch bond model are associated with the conformation of actin filaments, which change upon cooperative binding of αE-catenin (Hansen et al. 2013).
Mechanical tension may regulate the affinity of αE-catenin for several of its binding partners. In cell culture models, an antibody that recognizes the vinculin binding domain of αE-catenin localizes to cell–cell junctions as long as actomyosin is contractile (Yonemura et al. 2010). Although full-length αE-catenin does not bind full-length vinculin in solution, the vinculin head domain readily binds part of the modulation domain of αE-catenin, and the affinity decreases as flanking domains of αE-catenin are included (Choi et al. 2012). Significantly, stretching of αE-catenin using magnetic tweezers promotes vinculin head domain binding (Yao et al. 2014), but whether this force-mediated structural change is sufficient to recruit full-length vinculin to the cadherin–catenin complex at the AJ is unclear. Pulling on cadherin-coated magnetic beads attached to cells recruits full-length vinculin to cadherin-mediated attachment sites (le Duc et al. 2010), and this recruitment requires Src/Abl phosphorylation of vinculin Y822 in the head domain (Bays et al. 2014). Abl phosphorylates the vinculin head domain in vitro, but it may not phosphorylate full-length vinculin due to autoinhibitory interactions between the actin-binding domain and the rest of the molecule. Together, these data indicate that the actin-binding activity of vinculin at cell–cell junctions may be coactivated by force-induced conformational changes of αE-catenin and phosphorylation by Abl. It is possible that phosphorylation regulates αE-catenin interactions as well; for example, the linker that connects the αE-catenin modulation and actin binding domains is constitutively phosphorylated by CKI and CKII (Fig. 5.4, right). However, these particular modifications do not seem to affect binding of actin (Drees et al. 2005) or vinculin and other actin binding partners (Drees et al. 2005; Escobar et al. 2015).
αE-catenin may also mediate crosstalk between the cadherin–catenin complex and other adhesion complexes at cell–cell junctions. A prominent example is the nectin family of Ig superfamily adhesion proteins (Takai et al. 2008), which can affect the spatial localization of cadherin–catenin complexes during assembly by recruiting them to nascent cell–cell junctions. The recruitment may occur through afadin, an actin-binding protein that can bind directly to nectins (Takai et al. 2008) and the cadherin–catenin complex through αE-catenin (Pokutta et al. 2002). Ponsin and vinculin may also mediate interactions between afadin and the cadherin–catenin complex (Tachibana et al. 2000; Mandai et al. 1999). However, ponsin does not bind afadin and vinculin simultaneously in vitro (Mandai et al. 1999), but vinculin coimmunoprecipitates with ponsin when αE-catenin is present (Peng et al. 2012), suggesting it may be necessary to reconstitute a ponsin/afadin/vinculin complex.
Recent work has shown that αE-catenin also regulates the Hippo pathway protein YAP1, implicating the AJ in another cell proliferation pathway. Initially discovered in Drosophila, the Hippo pathway is a serine/threonine kinase cascade comprising Hippo (Harvey et al. 2003), Warts (Xu et al. 1995), Salvador (Pantalacci et al. 2003), and Mats (Lai et al. 2005). To control organ size during development, the Hippo–Salvador complex activates the Warts–Mats complex, which phosphorylates Drosophila YAP1, deactivates YAP1 transcriptional activity, and excludes it from the nucleus (Dong et al. 2007; Oh and Irvine 2008; Zhao et al. 2007b). Recent studies indicate that αE-catenin acts as a suppressor of the transcriptional activity of YAP1 (Schlegelmilch et al. 2011; Silvis et al. 2011). This function of αE-catenin is cell-density dependent and requires an interaction with the scaffolding protein 14-3-3 to sequester YAP1 at the AJ and in the cytosol. Expression of a truncated E-cadherin lacking the extracellular domain disrupts YAP1 sequestration in the cytoplasm (Benham-Pyle et al. 2015), suggesting that trans interactions between E-cadherin and mechanical coupling between cells may be required for sequestration of YAP1 in the cytoplasm or interaction with the cadherin–catenin complex. As does β-catenin, YAP1 becomes localized to the nucleus and transcriptionally active upon mechanical strain of contact-inhibited epithelial cells, but the molecular mechanism of activation is unknown (Benham-Pyle et al. 2015). Because YAP1 activation is sensitive to the morphology and contractile state of the actin cytoskeleton (Dupont et al. 2011; Wada et al. 2011), it is possible that YAP1 is mechanically activated through interactions with cytosolic αE-catenin. Cytosolic αE-catenin forms homodimers that bind and bundle actin filaments in the absence of tension and inhibit Arp2/3-mediated actin polymerization (Drees et al. 2005; Benjamin et al. 2010). Finally, both YAP1 and αE-catenin have been linked to the β-catenin destruction complex (Brunet et al. 2013), indicating that YAP1 phosphorylation independent of the Hippo pathway may disrupt Yap1 interactions with the 14-3-3 scaffold.
5 Moving Forward
Several themes emerge from the large body of work seeking to understand how cadherin and catenin proteins are regulated by phosphorylation and mechanical force. Cell biology, biochemistry, and genetic data indicate that a balance of cell density-dependent phosphorylation and dephosphorylation events regulates cadherin-mediated adhesion. At low cell densities, high tyrosine kinase activity, some of which is downstream of growth factor signaling pathways, upregulates the motility and proliferation machinery necessary to develop a dense multicellular organization. As cell density increases, tyrosine phosphatase activity increases, perhaps to the point of counteracting kinase activity, resulting in the stabilization of the cadherin–catenin complex at cell–cell junctions while turning off motility and proliferation signals. Interestingly, many receptor protein tyrosine phosphatases possess extracellular domains similar to those found in cell adhesion molecules (Stoker 2005), and thus these phosphatases may be recruited and activated by cadherin-mediated adhesion via mechanisms similar to those reconstituted on lipid bilayers (Hui and Vale 2014; Greene et al. 2014; Lin et al. 2014).
Recent biophysical and bioengineering methods have uncovered evidence that cadherin and catenin biology is regulated mechanically. A salient finding is that cadherin and αE-catenin form catch bonds between trans-interacting E-cadherin extracellular domains (Manibog et al. 2014) and F-actin (Buckley et al. 2014), respectively. However, additional experiments are needed to determine whether E-cadherin and αE-catenin catch bonds contribute to signaling in a cellular environment. The rate of tension loaded in force spectroscopy experiments is much faster than that generated by molecular motors associated with the cytoskeleton in the cytoplasm (Finer et al. 1994). If this rate is too low, then bonds dissociate before experiencing levels of tension that slow down unbinding (Dudko et al. 2008). Thus, it is not clear if cadherin and αE-catenin “feel” sufficient force in vivo to display catch bond behavior. To date, the best evidence of a catch bond operating in physiological conditions comes from studies of neutrophils detaching from selectin-binding surfaces under shear flow (Schmidtke and Diamond 2000; Yago et al. 2004). Gathering additional evidence for this type of cadherin/cadherin or αE-catenin/F-actin bond in vivo will likely require a combination of FRET-based force measurements and single-molecule tracking.
It seems increasingly likely that mechanical force not only alters the structure and molecular composition of the AJ, but also contributes to signaling from the AJ to regulate growth, invasion, and cell division. Mechanical strain across contact-inhibited epithelial monolayers induces cell-cycle entry and DNA synthesis, which require trans interactions between neighboring cells (Benham-Pyle et al. 2015). Density-dependent mechanical properties regulate the exclusion of transcription factors (YAP1, β-catenin) from the nucleus. Moreover, mechanical perturbations of the AJ can result in numerous phosphorylation events, triggering remodeling and release of previously sequestered signaling molecules (Brunet et al. 2013; Desprat et al. 2008; Benham-Pyle et al. 2015). It remains unknown how mechanical force at the AJ triggers increased kinase activity or release of sequestered transcription factors, and this will be an important topic for future work.
The morphology of the actin cytoskeleton regulates how force is generated and transmitted at the AJ. Actin networks can adopt distinct architectures: a highly branched network that is nucleated downstream of Rac1 and Cdc42, and an unbranched contractile network downstream of RhoA (Ridley 2006). These types of networks have different mechanical properties. Branched networks can tolerate compressive forces better than linear networks because network-level forces dissipate at the nodes connecting actin branches, and the high spatial density of these nodes generates short branches that buckle at larger compressive forces (Pujol et al. 2012). Thus, a branched network is better suited for generating protrusive forces, such as those found at the leading edge of migrating cells. At the AJ, these protrusive forces may move the plasma membrane locally and associated cadherin–catenin complexes. In addition, these complexes could experience an increase in tension if they are anchored to actin filament bundles that do not move with respect to the branching network. In contrast to branched networks, contractile networks are comprised of actin filaments bundled by myosin motors or other actin bundling proteins such as α-actinin and cytosolic αE-catenin. In these networks, motors generate contractile forces, and the bundled filaments transmit tension without undergoing much deformation (strain) given the Young’s modulus of individual actin filaments (~50 pN/nm; Kojima et al. 1994). Due to this mechanical resilience, a contractile actin network can efficiently induce mechanical strain on associated protein scaffolds (Claessens et al. 2006). In turn, the strain on these components can manifest as changes in conformation and dissociation rates. This myosin-dependent process drives morphogenetic changes such as planar cell intercalation, where AJs perpendicular to the axis of elongation disassemble to give rise to aligned AJs (Bertet et al. 2004).
The combination of phosphorylation and mechanical studies of the cadherin–catenin complex generate a model in which a stable E-cadherin/β-catenin/α-catenin complex is buttressed on either end by force-dependent interactions with E-cadherin molecules on neighboring cells and F-actin in the cytoplasm. The stability of E-cadherin/β-catenin/α-catenin interactions can then be tightly regulated by kinases and phosphatases to quickly dissociate the complex when needed, for example, in response to tissue wounding or other morphogenetic signals. It is likely that the combination of mechanical and biochemical modifications facilitates switches between different functions of cadherin and catenin proteins. As such, it will be important to address how mechanical forces contribute to phosphatase and kinase activities at the AJ, and how these modifications then contribute to the regulation of cell migration and growth.
References
Abe K, Takeichi M (2008) EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc Natl Acad Sci U S A 105:13–19
Aberle H, Bauer A, Stappert J, Kispert A, Kemler R (1997) beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J 16:3797–3804
Abiko H et al (2015) Rho guanine nucleotide exchange factors involved in cyclic-stretch-induced reorientation of vascular endothelial cells. J Cell Sci 128:1683–1695
Amit S et al (2002) Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev 16:1066–1076
Anastasiadis PZ et al (2000) Inhibition of RhoA by p120 catenin. Nat Cell Biol 2:637–644
Bays JL et al (2014) Vinculin phosphorylation differentially regulates mechanotransduction at cell-cell and cell-matrix adhesions. J Cell Biol 205:251–263
Benham-Pyle BW, Pruitt BL, Nelson WJ (2015) Cell adhesion. Mechanical strain induces E-cadherin-dependent Yap1 and β-catenin activation to drive cell cycle entry. Science 348:1024–1027
Benjamin JM et al (2010) AlphaE-catenin regulates actin dynamics independently of cadherin-mediated cell-cell adhesion. J Cell Biol 189:339–352
Bertet C, Sulak L, Lecuit T (2004) Myosin-dependent junction remodelling controls planar cell intercalation and axis elongation. Nature 429:667–671
Borghi N et al (2012) E-cadherin is under constitutive actomyosin-generated tension that is increased at cell-cell contacts upon externally applied stretch. Proc Natl Acad Sci U S A 109:12568–12573
Brasch J, Harrison OJ, Honig B, Shapiro L (2012) Thinking outside the cell: how cadherins drive adhesion. Trends Cell Biol 22:299–310
Bromann PA, Korkaya H, Courtneidge SA (2004) The interplay between Src family kinases and receptor tyrosine kinases. Oncogene 23:7957–7968
Brunet T et al (2013) Evolutionary conservation of early mesoderm specification by mechanotransduction in Bilateria. Nat Commun 4:2821
Buckley CD et al (2014) Cell adhesion. The minimal cadherin-catenin complex binds to actin filaments under force. Science 346:1254211
Cai D et al (2014) Mechanical feedback through E-cadherin promotes direction sensing during collective cell migration. Cell 157:1146–1159
Catimel B et al (2006) In situ phosphorylation of immobilized receptors on biosensor surfaces: application to E-cadherin/beta-catenin interactions. Anal Biochem 357:277–288
Cheng J et al (1997) A novel protein-tyrosine phosphatase related to the homotypically adhering kappa and mu receptors. J Biol Chem 272:7264–7277
Choi HJ, Huber AH, Weis WI (2006) Thermodynamics of beta-catenin-ligand interactions: the roles of the N- and C-terminal tails in modulating binding affinity. J Biol Chem 281:1027–1038
Choi HJ et al (2012) alphaE-catenin is an autoinhibited molecule that coactivates vinculin. Proc Natl Acad Sci U S A 109:8576–8581
Claessens MM, Bathe M, Frey E, Bausch AR (2006) Actin-binding proteins sensitively mediate F-actin bundle stiffness. Nat Mater 5:748–753
Conway DE et al (2013) Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr Biol 23:1024–1030
Cost AL, Ringer P, Chrostek-Grashoff A, Grashoff C (2015) How to measure molecular forces in cells: a guide to evaluating genetically-encoded fret-based tension sensors. Cell Mol Bioeng 8:96–105
Davis MA, Reynolds AB (2006) Blocked acinar development, E-cadherin reduction, and intraepithelial neoplasia upon ablation of p120-catenin in the mouse salivary gland. Dev Cell 10:21–31
Desprat N, Supatto W, Pouille PA, Beaurepaire E, Farge E (2008) Tissue deformation modulates twist expression to determine anterior midgut differentiation in Drosophila embryos. Dev Cell 15:470–477
Dietrich C, Scherwat J, Faust D, Oesch F (2002) Subcellular localization of beta-catenin is regulated by cell density. Biochem Biophys Res Commun 292:195–199
Dominguez I, Itoh K, Sokol SY (1995) Role of glycogen synthase kinase 3 beta as a negative regulator of dorsoventral axis formation in Xenopus embryos. Proc Natl Acad Sci U S A 92:8498–8502
Dong J et al (2007) Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130:1120–1133
Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI (2005) Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell 123:903–915
Dudko OK, Hummer G, Szabo A (2008) Theory, analysis, and interpretation of single-molecule force spectroscopy experiments. Proc Natl Acad Sci U S A 105:15755–15760
Dupont S et al (2011) Role of YAP/TAZ in mechanotransduction. Nature 474:179–183
El Sayegh TY et al (2005) Phosphorylation of N-cadherin-associated cortactin by Fer kinase regulates N-cadherin mobility and intercellular adhesion strength. Mol Biol Cell 16:5514–5527
Escobar DJ et al (2015) α-Catenin phosphorylation promotes intercellular adhesion through a dual-kinase mechanism. J Cell Sci 128:1150–1165
Finer JT, Simmons RM, Spudich JA (1994) Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature 368:113–119
Fodde R, Brabletz T (2007) Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol 19:150–158
Fuchs M, Müller T, Lerch MM, Ullrich A (1996) Association of human protein-tyrosine phosphatase kappa with members of the armadillo family. J Biol Chem 271:16712–16719
Fujii K, Furukawa F, Matsuyoshi N (1996) Ligand activation of overexpressed epidermal growth factor receptor results in colony dissociation and disturbed E-cadherin function in HSC-1 human cutaneous squamous carcinoma cells. Exp Cell Res 223:50–62
Fukuyama T, Ogita H, Kawakatsu T, Inagaki M, Takai Y (2006) Activation of Rac by cadherin through the c-Src-Rap1-phosphatidylinositol 3-kinase-Vav2 pathway. Oncogene 25:8–19
Grashoff C et al (2010) Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature 466:263–266
Greene AC et al (2014) Spatial organization of EphA2 at the cell-cell interface modulates trans-endocytosis of ephrinA1. Biophys J 106:2196–2205
Grosheva I, Shtutman M, Elbaum M, Bershadsky AD (2001) p120 catenin affects cell motility via modulation of activity of Rho-family GTPases: a link between cell-cell contact formation and regulation of cell locomotion. J Cell Sci 114:695–707
Hansen SD et al (2013) alphaE-catenin actin-binding domain alters actin filament conformation and regulates binding of nucleation and disassembly factors. Mol Biol Cell 24:3710–3720
Hantschel O et al (2003) A myristoyl/phosphotyrosine switch regulates c-Abl. Cell 112:845–857
Harrison OJ et al (2010) Two-step adhesive binding by classical cadherins. Nat Struct Mol Biol 17:348–357
Harrison OJ et al (2011) The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure 19:244–256
Hartsock A, Nelson WJ (2012) Competitive regulation of E-cadherin juxtamembrane domain degradation by p120-catenin binding and Hakai-mediated ubiquitination. PLoS One 7:e37476
Harvey KF, Pfleger CM, Hariharan IK (2003) The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 114:457–467
Hazan RB, Norton L (1998) The epidermal growth factor receptor modulates the interaction of E-cadherin with the actin cytoskeleton. J Biol Chem 273:9078–9084
Hazan RB, Kang L, Roe S, Borgen PI, Rimm DL (1997) Vinculin is associated with the E-cadherin adhesion complex. J Biol Chem 272:32448–32453
He X, Saint-Jeannet JP, Woodgett JR, Varmus HE, Dawid IB (1995) Glycogen synthase kinase-3 and dorsoventral patterning in Xenopus embryos. Nature 374:617–622
He TC et al (1998) Identification of c-MYC as a target of the APC pathway. Science 281:1509–1512
Hinck L, Näthke IS, Papkoff J, Nelson WJ (1994) Dynamics of cadherin/catenin complex formation: novel protein interactions and pathways of complex assembly. J Cell Biol 125:1327–1340
Holsinger LJ, Ward K, Duffield B, Zachwieja J, Jallal B (2002) The transmembrane receptor protein tyrosine phosphatase DEP1 interacts with p120(ctn). Oncogene 21:7067–7076
Hong S, Troyanovsky RB, Troyanovsky SM (2013) Binding to F-actin guides cadherin cluster assembly, stability, and movement. J Cell Biol 201:131–143
Hoschuetzky H, Aberle H, Kemler R (1994) Beta-catenin mediates the interaction of the cadherin-catenin complex with epidermal growth factor receptor. J Cell Biol 127:1375–1380
Huber AH, Weis WI (2001) The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell 105:391–402
Huber AH, Nelson WJ, Weis WI (1997a) Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell 90:871–882
Huber O, Krohn M, Kemler R (1997b) A specific domain in alpha-catenin mediates binding to beta-catenin or plakoglobin. J Cell Sci 110(Pt 15):1759–1765
Huber AH, Stewart DB, Laurents DV, Nelson WJ, Weis WI (2001) The cadherin cytoplasmic domain is unstructured in the absence of beta-catenin. A possible mechanism for regulating cadherin turnover. J Biol Chem 276:12301–12309
Hui E, Vale RD (2014) In vitro membrane reconstitution of the T-cell receptor proximal signaling network. Nat Struct Mol Biol 21:133–142
Imamura Y, Itoh M, Maeno Y, Tsukita S, Nagafuchi A (1999) Functional domains of alpha-catenin required for the strong state of cadherin-based cell adhesion. J Cell Biol 144:1311–1322
Itoh M, Nagafuchi A, Moroi S, Tsukita S (1997) Involvement of ZO-1 in cadherin-based cell adhesion through its direct binding to alpha catenin and actin filaments. J Cell Biol 138:181–192
Kim L, Wong TW (1995) The cytoplasmic tyrosine kinase FER is associated with the catenin-like substrate pp 120 and is activated by growth factors. Mol Cell Biol 15:4553–4561
Kimelman D, Pierce SB (1996) Regulation of dorsal-ventral axis formation in Xenopus by intercellular and intracellular signalling. Biochem Soc Symp 62:13–23
Knudsen KA, Soler AP, Johnson KR, Wheelock MJ (1995) Interaction of alpha-actinin with the cadherin/catenin cell-cell adhesion complex via alpha-catenin. J Cell Biol 130:67–77
Kobielak A, Pasolli HA, Fuchs E (2004) Mammalian formin-1 participates in adherens junctions and polymerization of linear actin cables. Nat Cell Biol 6:21–30
Kojima H, Ishijima A, Yanagida T (1994) Direct measurement of stiffness of single actin filaments with and without tropomyosin by in vitro nanomanipulation. Proc Natl Acad Sci U S A 91:12962–12966
Korinek V et al (1997) Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 275:1784–1787
Lai ZC et al (2005) Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell 120:675–685
le Duc Q et al (2010) Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J Cell Biol 189:1107–1115
Lickert H, Bauer A, Kemler R, Stappert J (2000) Casein kinase II phosphorylation of E-cadherin increases E-cadherin/beta-catenin interaction and strengthens cell-cell adhesion. J Biol Chem 275:5090–5095
Lin WC et al (2014) H-Ras forms dimers on membrane surfaces via a protein-protein interface. Proc Natl Acad Sci U S A 111:2996–3001
Liu C et al (2002) Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108:837–847
Mandai K et al (1999) Ponsin/SH3P12: an l-afadin- and vinculin-binding protein localized at cell-cell and cell-matrix adherens junctions. J Cell Biol 144:1001–1017
Manibog K, Li H, Rakshit S, Sivasankar S (2014) Resolving the molecular mechanism of cadherin catch bond formation. Nat Commun 5:3941
Maruthamuthu V, Sabass B, Schwarz US, Gardel ML (2011) Cell-ECM traction force modulates endogenous tension at cell-cell contacts. Proc Natl Acad Sci U S A 108:4708–4713
McCrea PD, Turck CW, Gumbiner B (1991) A homolog of the armadillo protein in Drosophila (plakoglobin) associated with E-cadherin. Science 254:1359–1361
McEwen AE, Maher MT, Mo R, Gottardi CJ (2014) E-cadherin phosphorylation occurs during its biosynthesis to promote its cell surface stability and adhesion. Mol Biol Cell 25:2365–2374
Miller PW et al (2013) Danio rerio alphaE-catenin is a monomeric F-actin binding protein with distinct properties from Mus musculus alphaE-catenin. J Biol Chem 288:22324–22332
Monga SP et al (2002) Hepatocyte growth factor induces Wnt-independent nuclear translocation of beta-catenin after Met-beta-catenin dissociation in hepatocytes. Cancer Res 62:2064–2071
Morin PJ et al (1997) Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275:1787–1790
Nagafuchi A, Ishihara S, Tsukita S (1994) The roles of catenins in the cadherin-mediated cell adhesion: functional analysis of E-cadherin-alpha catenin fusion molecules. J Cell Biol 127:235–245
Nelson WJ, Nusse R (2004) Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303:1483–1487
Ng MR, Besser A, Brugge JS, Danuser G (2014) Mapping the dynamics of force transduction at cell-cell junctions of epithelial clusters. Elife 3:e03282
Noren NK, Liu BP, Burridge K, Kreft B (2000) p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J Cell Biol 150:567–580
Norstrom MF, Smithback PA, Rock RS (2010) Unconventional processive mechanics of non-muscle myosin IIB. J Biol Chem 285:26326–26334
Oh H, Irvine KD (2008) In vivo regulation of Yorkie phosphorylation and localization. Development 135:1081–1088
Ostman A, Yang Q, Tonks NK (1994) Expression of DEP-1, a receptor-like protein-tyrosine-phosphatase, is enhanced with increasing cell density. Proc Natl Acad Sci U S A 91:9680–9684
Ozawa M, Ringwald M, Kemler R (1990) Uvomorulin-catenin complex formation is regulated by a specific domain in the cytoplasmic region of the cell adhesion molecule. Proc Natl Acad Sci U S A 87:4246–4250
Pacquelet A, Rørth P (2005) Regulatory mechanisms required for DE-cadherin function in cell migration and other types of adhesion. J Cell Biol 170:803–812
Pantalacci S, Tapon N, Léopold P (2003) The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat Cell Biol 5:921–927
Peng X, Maiers JL, Choudhury D, Craig SW, DeMali KA (2012) α-Catenin uses a novel mechanism to activate vinculin. J Biol Chem 287:7728–7737
Pertz O et al (1999) A new crystal structure, Ca2+ dependence and mutational analysis reveal molecular details of E-cadherin homoassociation. EMBO J 18:1738–1747
Piedra J et al (2003) p120 Catenin-associated Fer and Fyn tyrosine kinases regulate beta-catenin Tyr-142 phosphorylation and beta-catenin-alpha-catenin interaction. Mol Cell Biol 23:2287–2297
Pokutta S, Weis WI (2000) Structure of the dimerization and beta-catenin-binding region of alpha-catenin. Mol Cell 5:533–543
Pokutta S, Herrenknecht K, Kemler R, Engel J (1994) Conformational changes of the recombinant extracellular domain of E-cadherin upon calcium binding. Eur J Biochem 223:1019–1026
Pokutta S, Drees F, Takai Y, Nelson WJ, Weis WI (2002) Biochemical and structural definition of the l-afadin- and actin-binding sites of alpha-catenin. J Biol Chem 277:18868–18874
Pujol T, du Roure O, Fermigier M, Heuvingh J (2012) Impact of branching on the elasticity of actin networks. Proc Natl Acad Sci U S A 109:10364–10369
Qi J, Chen N, Wang J, Siu CH (2005) Transendothelial migration of melanoma cells involves N-cadherin-mediated adhesion and activation of the beta-catenin signaling pathway. Mol Biol Cell 16:4386–4397
Rakshit S, Zhang Y, Manibog K, Shafraz O, Sivasankar S (2012) Ideal, catch, and slip bonds in cadherin adhesion. Proc Natl Acad Sci U S A 109:18815–18820
Rangarajan ES, Izard T (2012) The cytoskeletal protein alpha-catenin unfurls upon binding to vinculin. J Biol Chem 287:18492–18499
Rangarajan ES, Izard T (2013) Dimer asymmetry defines alpha-catenin interactions. Nat Struct Mol Biol 20:188–193
Reffay M et al (2014) Interplay of RhoA and mechanical forces in collective cell migration driven by leader cells. Nat Cell Biol 16:217–223
Reynolds AB et al (1994) Identification of a new catenin: the tyrosine kinase substrate p120cas associates with E-cadherin complexes. Mol Cell Biol 14:8333–8342
Reynolds AB, Daniel JM, Mo YY, Wu J, Zhang Z (1996) The novel catenin p120cas binds classical cadherins and induces an unusual morphological phenotype in NIH3T3 fibroblasts. Exp Cell Res 225:328–337
Ridley AJ (2006) Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol 16:522–529
Rijksen G, Völler MC, van Zoelen EJ (1993) The role of protein tyrosine phosphatases in density-dependent growth control of normal rat kidney cells. FEBS Lett 322:83–87
Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS (1995) Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci U S A 92:8813–8817
Rosato R, Veltmaat JM, Groffen J, Heisterkamp N (1998) Involvement of the tyrosine kinase fer in cell adhesion. Mol Cell Biol 18:5762–5770
Roura S, Miravet S, Piedra J, García de Herreros A, Duñach M (1999) Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J Biol Chem 274:36734–36740
Rubinfeld B et al (1993) Association of the APC gene product with beta-catenin. Science 262:1731–1734
Sadot E et al (2002) Regulation of S33/S37 phosphorylated beta-catenin in normal and transformed cells. J Cell Sci 115:2771–2780
Sakanaka C (2002) Phosphorylation and regulation of beta-catenin by casein kinase I epsilon. J Biochem 132:697–703
Schlegelmilch K et al (2011) Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell 144:782–795
Schmidtke DW, Diamond SL (2000) Direct observation of membrane tethers formed during neutrophil attachment to platelets or P-selectin under physiological flow. J Cell Biol 149:719–730
Seeling JM et al (1999) Regulation of beta-catenin signaling by the B56 subunit of protein phosphatase 2A. Science 283:2089–2091
Serres M et al (2000) The disruption of adherens junctions is associated with a decrease of E-cadherin phosphorylation by protein kinase CK2. Exp Cell Res 257:255–264
Shapiro L, Weis WI (2009) Structure and biochemistry of cadherins and catenins. Cold Spring Harb Perspect Biol 1:a003053
Silvis MR et al (2011) α-catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Sci Signal 4:ra33
Sim JY et al (2015) Apatial distribution of cell-cell and cell-ecm adhesions regulates force balance while maintaining E-cadherin molecular tension in cell pairs. Mol Biol Cell 26(13):2456–2465
Stoker AW (2005) Protein tyrosine phosphatases and signalling. J Endocrinol 185:19–33
Su LK, Vogelstein B, Kinzler KW (1993) Association of the APC tumor suppressor protein with catenins. Science 262:1734–1737
Tachibana K et al (2000) Two cell adhesion molecules, nectin and cadherin, interact through their cytoplasmic domain-associated proteins. J Cell Biol 150:1161–1175
Takai Y, Miyoshi J, Ikeda W, Ogita H (2008) Nectins and nectin-like molecules: roles in contact inhibition of cell movement and proliferation. Nat Rev Mol Cell Biol 9:603–615
Takeda H, Shimoyama Y, Nagafuchi A, Hirohashi S (1999) E-cadherin functions as a cis-dimer at the cell-cell adhesive interface in vivo. Nat Struct Biol 6:310–312
Tang VW, Brieher WM (2012) α-Actinin-4/FSGS1 is required for Arp2/3-dependent actin assembly at the adherens junction. J Cell Biol 196:115–130
Thoreson MA et al (2000) Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J Cell Biol 148:189–202
Toret CP, Collins C, Nelson WJ (2014) An Elmo-Dock complex locally controls Rho GTPases and actin remodeling during cadherin-mediated adhesion. J Cell Biol 207:577–587
Van Etten RA et al (1994) The COOH terminus of the c-Abl tyrosine kinase contains distinct F- and G-actin binding domains with bundling activity. J Cell Biol 124:325–340
Wada K, Itoga K, Okano T, Yonemura S, Sasaki H (2011) Hippo pathway regulation by cell morphology and stress fibers. Development 138:3907–3914
Wildenberg GA et al (2006) p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell 127:1027–1039
Winston JT et al (1999) The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev 13:270–283
Woodring PJ, Hunter T, Wang JY (2001) Inhibition of c-Abl tyrosine kinase activity by filamentous actin. J Biol Chem 276:27104–27110
Xiao K et al (2005) p120-Catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol Biol Cell 16:5141–5151
Xu T, Wang W, Zhang S, Stewart RA, Yu W (1995) Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development 121:1053–1063
Xu G et al (2004) Continuous association of cadherin with beta-catenin requires the non-receptor tyrosine-kinase Fer. J Cell Sci 117:3207–3219
Yago T et al (2004) Catch bonds govern adhesion through L-selectin at threshold shear. J Cell Biol 166:913–923
Yamada S, Nelson WJ (2007) Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell-cell adhesion. J Cell Biol 178:517–527
Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ (2005) Deconstructing the cadherin-catenin-actin complex. Cell 123:889–901
Yan HX et al (2002) Physical and functional interaction between receptor-like protein tyrosine phosphatase PCP-2 and beta-catenin. Biochemistry 41:15854–15860
Yao M et al (2014) Force-dependent conformational switch of alpha-catenin controls vinculin binding. Nat Commun 5:4525
Yap AS, Niessen CM, Gumbiner BM (1998) The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J Cell Biol 141:779–789
Yonemura S, Wada Y, Watanabe T, Nagafuchi A, Shibata M (2010) alpha-Catenin as a tension transducer that induces adherens junction development. Nat Cell Biol 12:533–542
Zeng L et al (1997) The mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell 90:181–192
Zhang Y, Sivasankar S, Nelson WJ, Chu S (2009) Resolving cadherin interactions and binding cooperativity at the single-molecule level. Proc Natl Acad Sci U S A 106:109–114
Zhao XH et al (2007a) Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J Cell Sci 120:1801–1809
Zhao B et al (2007b) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21:2747–2761
Zondag GC, Reynolds AB, Moolenaar WH (2000) Receptor protein-tyrosine phosphatase RPTPmu binds to and dephosphorylates the catenin p120(ctn). J Biol Chem 275:11264–11269
Acknowledgments
This work was supported by Predoctoral Fellowships from the NSF (JT, BB-P), a Stanford Bio-X Pre-doctoral Fellowship (JT), NSF EFRI Award (1136790) to WJN and WIW, and NIH GM35527 (WJN).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this chapter
Cite this chapter
Tan, J., Benham-Pyle, B.W., Weis, W.I., Nelson, W.J. (2016). Regulation of Cadherin–Catenin Biology by Mechanical Force and Phosphorylation. In: Suzuki, S., Hirano, S. (eds) The Cadherin Superfamily. Springer, Tokyo. https://doi.org/10.1007/978-4-431-56033-3_5
Download citation
DOI: https://doi.org/10.1007/978-4-431-56033-3_5
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-56031-9
Online ISBN: 978-4-431-56033-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)