
Abstract
C-type lectin-like receptor 2 (CLEC-2) has been identified as a receptor for a platelet-activating snake venom, rhodocytin. CLEC-2 elicits powerful platelet activation signals in a manner dependent on Src, Syk kinases, and phospholipase Cγ2, similar to the collagen receptor glycoprotein (GP) VI/FcRγ-chain complex. In contrast to GPVI/FcRγ, which initiates platelet activation through tandem YxxL motifs called the immunoreceptor tyrosine-based activation motif (ITAM), CLEC-2 signals via a single YxxL motif called hemi-ITAM. An endogenous ligand of CLEC-2 has been identified as podoplanin, which is expressed on the surface of tumor cells and facilitates tumor metastasis by inducing platelet activation. CLEC-2 in platelets facilitates blood/lymphatic vessel separation by binding to podoplanin in lymphatic endothelial cells during development. In adults, platelet CLEC-2 prevents the backflow of blood into lymphatic vessels at the lymphovenous junction by forming thrombi. Moreover, platelet CLEC-2 maintains the integrity of high endothelial venules in lymph nodes by binding to podoplanin on stromal cells and in hyper-permeabilized capillaries during inflammation. In concert with GPVI, platelet CLEC-2 plays a role in thrombosis and hemostasis, although the precise mechanism remains unknown. Although CLEC-2 expression is almost specific to platelets/megakaryocytes in humans, CLEC-2 is also expressed in dendritic cells in mice, where it plays an important role in adaptive immunity. CLEC-2 may be a good target for a novel antiplatelet drug or antimetastatic drug, which could prevent arterial thrombosis and cancer metastasis, the main causes of death in developed countries. In this article, we review the signal transduction, structure, expression, and function of CLEC-2.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- CLEC-2
- Platelets
- Rhodocytin
- Podoplanin
- Lymphatic endothelial cells
- Dendritic cells
- Thrombus formation
- Tumor metastasis
- Lymphangiogenesis
- Vascular integrity
1 History
Snake venom contains a vast number of toxins that target proteins necessary for thrombosis and hemostasis. A platelet-activating snake venom, rhodocytin (also called aggretin), was purified from Calloselasma rhodostoma venom independently by the Huang (Huang et al. 1995) and Morita (Shin and Morita 1998) groups in the 1990s. Rhodocytin-induced and collagen-induced platelet aggregations are quite similar in that both agonists induce platelet aggregation with a long lag phase and are dependent on the Src family kinase, aspirin, and cytochalasin D (Suzuki-Inoue et al. 2001; Navdaev et al. 2001; Inoue et al. 1999). However, rhodocytin induces platelet aggregation independently of the collagen receptor GPVI/FcRγ-chain, a member of the immunoglobulin super family, since it can induce platelet aggregation in mice deficient in the receptor (Suzuki-Inoue et al. 2001; Navdaev et al. 2001). This indicates that there is another platelet activation receptor. Initial studies indicated that rhodocytin induces platelet aggregation by binding to integrin α2β1 and GPIb/IX/V (Suzuki-Inoue et al. 2001; Navdaev et al. 2001). However, later studies reported that rhodocytin stimulated the aggregation of platelets deficient in α2β1 or the extracellular domain of GPIb/IX/V (Bergmeier et al. 2001), indicating that there is another activation receptor for rhodocytin, although the toxin may also bind to α2β1 and/or GPIb/IX/V. Finally, by rhodocytin affinity chromatography and MS/MS-based analyses, C-type lectin-like receptor 2 (CLEC-2) was identified as a receptor for rhodocytin (Suzuki-Inoue et al. 2006). CLEC-2 was first cloned during a bioinformatic screen for C-type lectin-like receptors (Colonna et al. 2000). However, neither its function nor its ligand(s) had been elucidated for several years.
In this article, we review the signal transduction, structure, expression, and function of CLEC-2, updating our previous review (Suzuki-Inoue et al. 2011).
2 Mechanism of Signal Transduction
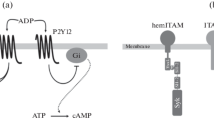
CLEC-2 has a YxxL motif in its cytoplasmic tail, which resembles the immunoreceptor tyrosine-based activation motif (ITAM; YxxL-(X)10-12-YxxL) that has two YxxL motifs. The single YxxL motif that is found in CLEC-2 and other receptors is called hemi-ITAM. ITAM is a signaling motif found in immune receptors, such as the T-cell receptor and the platelet collagen receptor GPVI/FcRγ-chain. Cross-linking of GPVI leads to tyrosine phosphorylation of ITAM in the cytoplasmic domain of the FcRγ-chain, which is constitutively associated with GPVI, by the Src family kinases, Fyn and Lyn. This leads to binding of the tandem SH2 domain of the tyrosine kinase, Syk, to the phosphorylated ITAM. Subsequent activation of Syk initiates downstream signaling events that culminate in the tyrosine phosphorylation of LAT, SLP-76, and Vav1/3 and activation of effecter enzymes including Btk, PI3-kinase, Rac/Cdc42, and phospholipaseCγ2 (PLCγ) (reviewed in Watson et al. (2005) (Fig. 6.1a)).

The signal transduction pathway mediated through the GPVI/FcRγ-chain and CLEC-2. (a) CLEC-2 is cross-linked by its endogenous ligand, podoplanin, or by an exogenous ligand (rhodocytin) and then undergoes tyrosine phosphorylation of a single YITL motif called hemi-ITAM, which leads to activation of downstream signaling. (b) GPVI is cross-linked by its endogenous ligand, collagen, and undergoes tyrosine phosphorylation on tandem YxxL motifs called ITAM, which leads to activation of downstream signaling. Lipid rafts are necessary for CLEC-2 and GPVI-mediated signal transduction. YP indicates phosphorylated tyrosine
Rhodocytin stimulates the phosphorylation of the single YxxL motif in CLEC-2, and then the tandem SH2 domains of Syk bind to the phosphorylated YxxL, and the hemi-ITAM was shown to be necessary for CLEC-2 signal transduction (Suzuki-Inoue et al. 2006; Fuller et al. 2007). CLEC-2 has only a single YxxL; however, CLEC-2 is present as a dimer in resting platelets, and the tandem SH2 domains of Syk bind to the phosphorylated YxxLs of two CLEC-2 molecules with a stoichiometry of 2:1 (Watson et al. 2009; Hughes et al. 2010a). The ITAM of the GPVI/FcRγ-chain is tyrosine phosphorylated by the Src family kinases, Fyn and Lyn, which are constitutively associated with the cytoplasmic tail of GPVI. This is followed by the binding and subsequent activation of Syk. In the case of CLEC-2, hemi-ITAM is mainly phosphorylated by Syk itself (Spalton et al. 2009). A specific Src family kinase inhibitor, PP2, also inhibits CLEC-2 tyrosine phosphorylation in human platelets, but not in murine platelets, suggesting that CLEC-2 is phosphorylated by Syk and Src family kinases in human platelets, but only by Syk in murine platelets (Suzuki-Inoue et al. 2006; Severin et al. 2011).
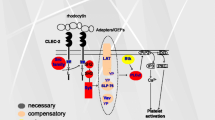
The signaling pathway of CLEC-2 downstream of Syk is almost the same as that of GPVI and includes the tyrosine phosphorylation of LAT, SLP-76, and Vav1/3 and activation of effecter enzymes including Btk and PLCγ2 (Suzuki-Inoue et al. 2006; Fuller et al. 2007). Murine platelets deficient in Syk or PLCγ2 failed to respond even to the maximal concentration of rhodocytin, suggesting that Syk and PLCγ2 are crucial for CLEC-2-mediated signal transduction. On the other hand, those deficient in the adaptor proteins, LAT or SLP-76, and the guanine nucleotide exchange factor, Vav1/3, did not respond to the low concentration, but did respond to the high concentration of rhodocytin, suggesting that these molecules are necessary, but can be compensated for during CLEC-2 signaling (Suzuki-Inoue et al. 2006). Btk, a tyrosine kinase that is necessary for PLCγ2 activation, is tyrosine phosphorylated upon rhodocytin stimulation (Suzuki-Inoue et al. 2006), but the dependence of the CLEC-2 signaling pathway on this kinase is unknown (Fig. 6.1b).
3 CLEC-2 Expression
CLEC-2 was first identified from a bioinformatic screen for C-type lectin-like receptors. At that time, reverse transcriptase-PCR and Northern blot analyses indicated that CLEC-2 mRNA is expressed in the liver and several hematopoietic cell types, including monocytes, dendritic cells, NK cells, and granulocytes (Colonna et al. 2000), although platelets and megakaryocytes were not checked for expression. Later, the CLEC-2 protein expression was systematically analyzed, and the CLEC-2 protein was found to be expressed in platelets, megakaryocytic cell lines, liver sinusoidal endothelial cells (Chaipan et al. 2006), and liver Kupffer cells (Tang et al. 2010) in humans. In mice, however, it has been reported that CLEC-2 is also expressed in peripheral neutrophils (Kerrigan et al. 2009) and macrophages (Chang et al. 2010), where it mediates phagocytosis and increases the expression of proinflammatory cytokines, including tumor necrosis factor α (TNFα), as well as Kupffer cells (Tang et al. 2010). Taken together, these findings indicate that CLEC-2 is highly and relatively specifically expressed in platelets/megakaryocytes, but is also present in other types of cells at low levels, especially in mice.
4 Structure and the Mode of Ligand Binding to CLEC-2
C-type lectins can be classified as “classical” and “nonclassical” C-type lectins based on their ability to recognize carbohydrate and noncarbohydrate ligands, respectively. CLEC-2 belongs to the nonclassical C-type lectins, which contain a C-type lectin-like domain (CLTD) homologous to a carbohydrate recognition domain, but lack the consensus sequence for binding sugars and calcium (Colonna et al. 2000). In fact, Watson et al. reported that no glycosylation was observed on either of the subunits of the CLEC-2 ligand snake venom rhodocytin in the crystal structure, consistent with their bioinformatic, SDS-PAGE, and mass spectroscopy results (Watson et al. 2007). Podoplanin is a type I transmembrane sialomucin-like glycoprotein, which we found is an endogenous ligand for CLEC-2 (Suzuki-Inoue et al. 2007) (see the “Function” section for details) (Sect. 6.5). Although the association between CLEC-2 and podoplanin is dependent on the sialic acid on the O-glycans of podoplanin (Suzuki-Inoue et al. 2007), not only sialic acid but also the stereostructure of the podoplanin protein was found to be critical for the CLEC-2-binding activity of podoplanin (Kato et al. 2008). This finding is consistent with the characteristics of “nonclassical” C-type lectins.
CLEC-2 is also glycosylated and detected as 32- and 40-kDa forms in platelets, probably due to differential glycosylation. Consistent with this finding, there are two potential sites of N-glycosylation (at positions 120 and 134) (Watson et al. 2007). In addition, the double band collapses to a single band of 27 kDa, the molecular weight that is deduced from the amino acid sequence of CLEC-2 upon N-glycosidase treatment (Suzuki-Inoue et al. 2006). However, neither of the two potential N-linked glycosylation sites are supposed to play a role in at least rhodocytin binding (Watson et al. 2007). With regard to the interaction between CLEC-2 and podoplanin, the stereostructure of the CLTD in CLEC-2 is necessary for binding to podoplanin, since the extracellular domain of CLEC-2-Fc lacking only a small part of the CLTD lost the ability to bind to podoplanin (Kato et al. 2008).
5 Function
5.1 Tumor Metastasis
It has long been recognized that several kinds of tumor cells cause the aggregation of platelets, which facilitates tumor growth and metastasis (Katagiri et al. 1991; Kitagawa et al. 1989; Kato et al. 2003). Platelet aggregates surrounding tumor cells protect them from shear stress and/or NK cells (Nieswandt et al. 1999) in the blood stream and serve as a place for tumor cell nesting, and the growth factors released from activated platelets can stimulate angiogenesis and tumor growth. Podoplanin is a type I transmembrane sialomucin-like glycoprotein expressed on several kinds of tumor cells, including squamous cell carcinomas (Schacht et al. 2005; Kato et al. 2005), seminomas (Kato et al. 2004), and brain tumors (Mishima et al. 2006a, b; Kato et al. 2006), and it has been shown to induce platelet aggregation (reviewed in Tsuruo and Fujita 2008). Podoplanin expression is reportedly associated with tumor metastasis or malignant progression (Mishima et al. 2006b; Yuan et al. 2006).
Suzuki-Inoue et al. noticed that the profile of podoplanin-induced platelet aggregation is similar to that of rhodocytin-induced platelet aggregation, and they identified CLEC-2 as a receptor for podoplanin (Suzuki-Inoue et al. 2007). In an experimental mouse model of metastasis, an anti-podoplanin blocking antibody significantly inhibited the number of metastatic lung nodules, consisting of tumor cells expressing podoplanin (Kato et al. 2008). Conversely, the lung metastasis of podoplanin-expressing tumors greatly inhibited in CLEC-2-deficient bone marrow chimeric mice (Shirai et al. 2013), implying that CLEC-2/podoplanin may be a promising target for antimetastatic drugs. However, tumor growth and lymphatic metastasis were not inhibited in CLEC-2-deficient chimeric mice (Shirai et al. 2013), suggesting that the CLEC-2/podoplanin interaction only plays a role in hematogenous tumor metastasis, where tumors have access to platelets in the blood flow.
5.2 Lymphatic/Blood Vessel Separation During Development
Podoplanin is expressed not only in tumor cells but also in various kinds of normal tissues, including lymphatic endothelial cells, type I alveolar cells, and kidney podocytes, after which podoplanin was named (reviewed in Tsuruo and Fujita 2008). Podoplanin is expressed in lymphatic endothelial cells, but not in vascular endothelial cells, and hence is used as a marker for lymphatic endothelial cells. Under physiological conditions, the CLEC-2 in platelets cannot interact with the podoplanin in lymphatic endothelial cells. During organ development, however, the cluster of endothelial cells in the cardinal vein is committed to the lymphatic phenotype, and these sprout to form the primary lymphatic sacs from which part of the peripheral lymphatic vasculature is generated by further centrifugal growth (reviewed in Tammela and Alitalo 2010). At this stage, the CLEC-2 in platelets can interact with the podoplanin in lymphatic endothelial cells.
Studies in CLEC-2-deficient mice revealed that CLEC-2 facilitates blood/lymphatic vessel separation during development. CLEC-2-deficient mice died at the embryonic/neonatal stages, exhibiting disorganized and blood-filled lymphatic vessels and severe edema due to abnormal blood/lymphatic vessel separation (Bertozzi et al. 2010; Suzuki-Inoue et al. 2010) (Fig. 6.2). Platelet/megakaryocyte-specific CLEC-2-deficient mice also showed blood-filled lymphatics (Finney et al. 2011; Suzuki-Inoue et al. 2010), suggesting that the CLEC-2 in platelets is required for blood/lymphatic vessel separation. Podoplanin deficiency and endothelial cell O-glycan deficiency also caused the blood/lymphatic misconnections (Fu et al. 2008), and it had been previously shown that the sialic acid present on the O-glycans of podoplanin is essential for binding to CLEC-2 (Suzuki-Inoue et al. 2007). These findings suggest that the interaction between CLEC-2 on platelets and podoplanin on lymphatic endothelial cells is important for normal lymphatic vessel development.

CLEC-2−/− and CLEC-2+/− mouse embryos. CLEC-2-deficient mouse embryos show blood-filled lymphatic vessels in the skin
Mice deficient in the signaling molecules downstream of CLEC-2, including Syk, SLP-76, and PLCγ2, showed blood/lymphatic vessel misconnection (Ichise et al. 2009; Abtahian et al. 2003). Inhibition of platelet activation by treatment of pregnant wild-type mice with acetyl salicylic acid resulted in half of the embryos exhibiting blood/lymphatic misconnection (Uhrin et al. 2010). These findings suggest that platelet activation is required for blood/lymphatic vessel separation. Platelet activation results in granule release and platelet aggregation. Platelet granules contain a vast number of angiogenic factors, growth factors, and extracellular matrix, implying that these factors could contribute to blood/lymphatic separation. Alternatively, platelet aggregates built up at the separation zone of lymph sacs and cardinal veins, which may physically help in the separation. In fact, Uhrin et al. and Bertozzi et al. reported that platelet aggregates build up at the separation zone of podoplanin-positive lymph sacs and cardinal veins in wild-type embryos, but not in podoplanin-deficient or SLP-76-deficient embryos (Uhrin et al. 2010; Bertozzi et al. 2010). However, mice deficient in integrin αIIbβ3, which is necessary for platelet aggregation, but not for granule release, do not show the non-separation phenotype (Bertozzi et al. 2010), suggesting a role for granule release. In line with this notion, supernatants from activated platelets and their main content, TGFβ, inhibited the migration and proliferation of lymphatic endothelial cells (Osada et al. 2012). However, patients who lack sufficient platelet density or a-granules also do not exhibit defective blood/lymphatic vessel separation (Michelson 2013). Further studies are therefore necessary to address these mechanisms.
5.3 Maintenance of the Vascular Integrity in Adults
5.3.1 Prevention of the Backflow of Blood into Lymphatic Vessels at the Lymphovenous (LV) Junction
The vascular networks connect at the LV junction, where lymph drains into blood. An LV valve (LVV) prevents the backflow of blood into lymphatic vessels. Hess et al. reported that the loss of CLEC-2 resulted in backfilling of the lymphatic network with blood from the thoracic duct in both neonatal and mature mice, even when the LVVs were intact (Hess et al. 2014). Fibrin-containing platelet thrombi were observed at the LVV and in the terminal thoracic duct in wild-type mice, but not in CLEC-2-deficient mice. An analysis of mice lacking LVVs or lymphatic valves revealed that platelet-mediated thrombus formation limits LV backflow even under conditions of impaired valve function. This indicates that hemostasis via CLEC-2 functions with the LVV to safeguard the lymphatic vascular network throughout life.
5.3.2 Integrity of High Endothelial Venules in Lymph Nodes
Circulating lymphocytes continuously enter lymph nodes for immune surveillance through specialized blood vessels named high endothelial venules (HEV). This process increases markedly during immune responses. Herzog et al. revealed the mechanism by which HEVs permit lymphocyte transmigration while maintaining the vascular integrity (Herzog et al. 2013). The podoplanin expressed on fibroblastic reticular cells, which surround HEVs, stimulates platelets by binding to its receptor, CLEC-2. Sphingosine-1-phosphate released from activated platelets promotes the expression of VE-cadherin on HEVs, which is essential for overall vascular integrity. Mice deficient in CLEC-2, podoplanin, or sphingosine-1-phosphate exhibited spontaneous bleeding in mucosal lymph nodes and bleeding in the draining peripheral lymph nodes after immunization. A role for platelet activation via CLEC-2 in the maintenance of HEV integrity during immune responses was added to the list of the roles platelets play beyond clotting.
5.3.3 Integrity of Vessels During Inflammation
It is known that platelets play a critical role in maintaining the vascular integrity, especially during inflammation, when inflammatory cytokines enhance vascular permeability. For this reason, petechial hemorrhage was observed upon the induction of severe thrombocytopenia even without traumatic injury. However, how platelets maintain the vascular integrity during inflammation is unclear. Boulaftali et al. proved that ITAM signaling, but not G-protein-coupled receptor (GPCR) signaling, is critical for the prevention of inflammation-induced hemorrhage (Boulaftali et al. 2013). Inflammation-induced hemorrhage in thrombocytopenic mice was rescued by the transfusion of wild-type platelets or thrombin receptor-deficient platelets, but not by the transfusion of platelets deficient in CLEC-2, GPVI, or SLP-76. These results indicate that controlling the vascular integrity is a major function of immune-type receptors in platelets. On the other hand, signaling from GPCR is not necessary for controlling the vascular integrity, although it is important for hemostasis. Upon platelet leakage from vessels, GPVI signaling is likely activated at sites of inflammation by collagen and/or laminin, the two physiological ligands for GPVI found in the vessel wall (Ozaki et al. 2009) (Fig. 6.3b). How platelets are activated through CLEC-2 at the site of inflammation is difficult to explain, but Boulaftali et al. speculated that podoplanin on the surface of infiltrating macrophages, or an as yet unidentified ligand expressed in tissues around vessels or in the vessel wall, stimulates platelets via CLEC-2.

The suggested physiological and pathological roles of platelet CLEC-2. (a) Podoplanin expressed in tumor cells facilitates tumor metastasis by inducing platelet activation through CLEC-2. Platelets adhering to tumor cells protect them from shear stress and/or NK cells, provide tumor cells with a scaffold for extravasation, and release growth factors or angiogenic factors to facilitate tumor growth. (b) Podoplanin expressed in lymphatic endothelial cells facilitates blood/lymphatic separation in the developmental stage. (c) CLEC-2 stabilizes thrombus formation under a normal blood flow, at least partly through homophilic interactions. (d) CLEC-2 maintains the vascular integrity. The vascular integrity under inflammatory conditions is maintained by signaling mediated through CLEC-2 and GPVI. High endothelial venule integrity is maintained by the interaction between the CLEC-2 on platelets and the podoplanin on fibroblastic reticular cells. The integrity of the lymphovenous junction is maintained by thrombus formation induced by the interaction between the CLEC-2 on platelets and the podoplanin on lymphatic endothelial cells
5.4 Thrombosis and Hemostasis
Studies to investigate the role of CLEC-2 in thrombosis and hemostasis demonstrated that CLEC-2-null mice exhibit mortality at the embryonic/neonatal stages, while irradiated chimeric animals were rescued by transplantation of a CLEC-2−/− fetal liver (Suzuki-Inoue et al. 2010). Antibody-induced CLEC-2-deficient mice were also utilized for this purpose. The injection of an anti-CLEC-2 antibody into mice leads to a specific loss of CLEC-2 in circulating platelets for several days, although the precise mechanism underlying this observation remains unknown. CLEC-2-deficient platelets resulting from both methods displayed normal adhesion under flow conditions, but the subsequent thrombus formation was severely impaired in vitro, although the in vitro platelet aggregation induced by agonists other than rhodocytin was normal (May et al. 2009; Suzuki-Inoue et al. 2010). FeCl3-induced thrombus formation in the artery was also inhibited in CLEC-2-deficient chimeras (May et al. 2009). These mice also showed mildly increased or mild but not significantly increased tail bleeding (Suzuki-Inoue et al. 2010; May et al. 2009; Tang et al. 2010). These findings suggest that CLEC-2 is involved in thrombus stabilization in vitro and in vivo.
In contrast to these reports, Hughes et al. reported that CLEC-2 is neither required for platelet aggregate stability in the presence of arteriolar shear nor for normal hemostasis (Hughes et al. 2010b). The discrepancies between studies may be because the role of CLEC-2 in thrombosis and hemostasis is relatively minor and depends on the experimental conditions. Simultaneous deletion of GPVI and CLEC-2 from mice, however, caused a severe bleeding tendency and profound impairment of arterial thrombus formation (Bender et al. 2013). These findings suggest that CLEC-2 plays a definite role in thrombosis and hemostasis, although the deletion of CLEC-2 alone results in a relatively minor phenotype. The mechanism by which CLEC-2 and GPVI play a complementary role in thrombosis and hemostasis is a topic of interest for future investigation.
5.5 A Role for CLEC-2 on the Surface of Dendritic Cells in Immunity
CLEC-2 is also expressed on the surface of dendritic cells in mice, but not in humans. Recently, accumulating evidence has suggested that the CLEC-2 in dendritic cells plays an important role in adaptive immunity.
To initiate adaptive immunity, dendritic cells move from parenchymal tissues to lymphoid organs by migrating along stromal scaffolds consisting of podoplanin-positive fibroblastic reticular cells (FRCs). Acton et al. reported that CLEC-2 deficiency in DCs impaired their entry into the lymphatics and their trafficking to and within lymph nodes, thereby reducing T-cell priming (Acton et al. 2012). CLEC-2 engagement by podoplanin induces the spreading and migration of dendritic cells along stromal surfaces. CLEC-2 activation triggered cell spreading via downregulation of RhoA activity and Rac1 activation. In turn, CLEC-2 in dendritic cells controls the fibroblastic reticular network tension and lymph node expansion by generating signals downstream of podoplanin in FRCs (Acton et al. 2014; Astarita et al. 2015). Immunogenic challenge induces the infiltration and division of lymphocytes, which markedly increases the lymph node cellularity, leading to lymph node expansion. In the resting state, podoplanin signaling in stromal FRCs maintains the physical elasticity of the lymph nodes by inducing actomyosin contractility in FRCs via the activation of RhoA/C. Upon immunogenic challenge, the engagement of CLEC-2 in mature dendritic cells causes podoplanin clustering and rapidly uncouples podoplanin from RhoA/C activation, relaxing the actomyosin cytoskeleton and permitting FRC stretching. This process allows increased lymph node cellularity and provides places for antigen presentation by dendritic cells to lymphocytes. Thus, the CLEC-2/podoplanin axis induces bidirectional signaling between dendritic cells and FRCs, leading to increased motility of dendritic cells and FRC extension, both of which are necessary for efficient T-cell priming.
Benezech et al. showed that a constitutive lack of CLEC-2 expression leads to defective lymphatic cell proliferation, resulting in impaired development of the lymphatic vascular structures and involution of the LN anlagen in the embryo (Benezech et al. 2014). In contrast, the deletion of CLEC-2 on the megakaryocyte/platelet lineage still allows the formation of lymph nodes, although they are blood-filled lymph nodes, as previously reported (Herzog et al. 2013). Thus, the interaction between the CLEC-2 on dendritic cells and podoplanin on stromal cells may regulate lymph node development.
6 Concluding Remarks
The discovery of the novel platelet activation receptor, CLEC-2, revealed that platelets have various roles within and beyond clotting. Platelets regulate tumor metastasis, blood/lymphatic vessel separation, and the integrity of vessels, including HEVs in lymph nodes and the hyper-permeabilized capillaries that occur during inflammation through the interaction between CLEC-2 and its endogenous ligand, podoplanin. In concert with GPVI, the CLEC-2 in platelets plays a role in thrombosis and hemostasis, although the precise mechanism remains unknown. The roles of CLEC-2 in platelets are summarized in Fig. 6.3. CLEC-2 in dendritic cells plays an important role in adaptive immunity in mice. There is growing evidence that CLEC-2 has other endogenous and exogenous ligands, the discovery of which would also provide additional advances in the field of platelet biology. With regard to the clinical aspects of the research on CLEC-2, it could be a good target for an anti-hematogenous metastasis drug or as an antiplatelet drug. There is still a long way to go before there could be a practical use of these drugs, but research on CLEC-2 may lead to a better understanding of cancer and arterial thrombosis, which are the main causes of death in developed countries.
References
Abtahian F, Guerriero A, Sebzda E, Lu MM, Zhou R, Mocsai A, Myers EE, Huang B, Jackson DG, Ferrari VA, Tybulewicz V, Lowell CA, Lepore JJ, Koretzky GA, Kahn ML (2003) Regulation of blood and lymphatic vascular separation by signaling proteins SLP-76 and Syk. Science 299(5604):247–251
Acton SE, Astarita JL, Malhotra D, Lukacs-Kornek V, Franz B, Hess PR, Jakus Z, Kuligowski M, Fletcher AL, Elpek KG, Bellemare-Pelletier A, Sceats L, Reynoso ED, Gonzalez SF, Graham DB, Chang J, Peters A, Woodruff M, Kim YA, Swat W, Morita T, Kuchroo V, Carroll MC, Kahn ML, Wucherpfennig KW, Turley SJ (2012) Podoplanin-rich stromal networks induce dendritic cell motility via activation of the C-type lectin receptor CLEC-2. Immunity 37(2):276–289
Acton SE, Farrugia AJ, Astarita JL, Mourao-Sa D, Jenkins RP, Nye E, Hooper S, van Blijswijk J, Rogers NC, Snelgrove KJ, Rosewell I, Moita LF, Stamp G, Turley SJ, Sahai E, Reis e Sousa C (2014) Dendritic cells control fibroblastic reticular network tension and lymph node expansion. Nature 514(7523):498–502
Astarita JL, Cremasco V, Fu J, Darnell MC, Peck JR, Nieves-Bonilla JM, Song K, Kondo Y, Woodruff MC, Gogineni A, Onder L, Ludewig B, Weimer RM, Carroll MC, Mooney DJ, Xia L, Turley SJ (2015) The CLEC-2-podoplanin axis controls the contractility of fibroblastic reticular cells and lymph node microarchitecture. Nat Immunol 16(1):75–84
Bender M, May F, Lorenz V, Thielmann I, Hagedorn I, Finney BA, Vogtle T, Remer K, Braun A, Bosl M, Watson SP, Nieswandt B (2013) Combined in vivo depletion of glycoprotein VI and C-type lectin-like receptor 2 severely compromises hemostasis and abrogates arterial thrombosis in mice. Arterioscler Thromb Vasc Biol 33(5):926–934
Benezech C, Nayar S, Finney BA, Withers DR, Lowe K, Desanti GE, Marriott CL, Watson SP, Caamano JH, Buckley CD, Barone F (2014) CLEC-2 is required for development and maintenance of lymph nodes. Blood 123(20):3200–3207
Bergmeier W, Bouvard D, Eble JA, Mokhtari-Nejad R, Schulte V, Zirngibl H, Brakebusch C, Fassler R, Nieswandt B (2001) Rhodocytin (aggretin) activates platelets lacking alpha(2)beta(1) integrin, glycoprotein VI, and the ligand-binding domain of glycoprotein Ibalpha. J Biol Chem 276(27):25121–25126
Bertozzi CC, Schmaier AA, Mericko P, Hess PR, Zou Z, Chen M, Chen CY, Xu B, Lu MM, Zhou D, Sebzda E, Santore MT, Merianos DJ, Stadtfeld M, Flake AW, Graf T, Skoda R, Maltzman JS, Koretzky GA, Kahn ML (2010) Platelets regulate lymphatic vascular development through CLEC-2-SLP-76 signaling. Blood 116(4):661–670
Boulaftali Y, Hess PR, Getz TM, Cholka A, Stolla M, Mackman N, Owens AP 3rd, Ware J, Kahn ML, Bergmeier W (2013) Platelet ITAM signaling is critical for vascular integrity in inflammation. J Clin Invest 123(2):908–916
Chaipan C, Soilleux EJ, Simpson P, Hofmann H, Gramberg T, Marzi A, Geier M, Stewart EA, Eisemann J, Steinkasserer A, Suzuki-Inoue K, Fuller GL, Pearce AC, Watson SP, Hoxie JA, Baribaud F, Pohlmann S (2006) DC-SIGN and CLEC-2 mediate human immunodeficiency virus type 1 capture by platelets. J Virol 80(18):8951–8960
Chang CH, Chung CH, Hsu CC, Huang TY, Huang TF (2010) A novel mechanism of cytokine release in phagocytes induced by aggretin, a snake venom C-type lectin protein, through CLEC-2 ligation. J Thromb Haemost 8(11):2563–2570
Colonna M, Samaridis J, Angman L (2000) Molecular characterization of two novel C-type lectin-like receptors, one of which is selectively expressed in human dendritic cells. Eur J Immunol 30(2):697–704
Finney BA, Schweighoffer E, Navarro-Nunez L, Benezech C, Barone F, Hughes CE, Langan SA, Lowe KL, Pollitt AY, Mourao-Sa D, Sheardown S, Nash GB, Smithers N, Reis ESC, Tybulewicz VL, Watson SP (2011) CLEC-2 and Syk in the megakaryocytic/platelet lineage are essential for development. Blood 119(7):1747–1756
Fu J, Gerhardt H, McDaniel JM, Xia B, Liu X, Ivanciu L, Ny A, Hermans K, Silasi-Mansat R, McGee S, Nye E, Ju T, Ramirez MI, Carmeliet P, Cummings RD, Lupu F, Xia L (2008) Endothelial cell O-glycan deficiency causes blood/lymphatic misconnections and consequent fatty liver disease in mice. J Clin Invest 118(11):3725–3737
Fuller GL, Williams JA, Tomlinson MG, Eble JA, Hanna SL, Pohlmann S, Suzuki-Inoue K, Ozaki Y, Watson SP, Pearce AC (2007) The C-type lectin receptors CLEC-2 and Dectin-1, but not DC-SIGN, signal via a novel YXXL-dependent signaling cascade. J Biol Chem 282(17):12397–12409
Herzog BH, Fu J, Wilson SJ, Hess PR, Sen A, McDaniel JM, Pan Y, Sheng M, Yago T, Silasi-Mansat R, McGee S, May F, Nieswandt B, Morris AJ, Lupu F, Coughlin SR, McEver RP, Chen H, Kahn ML, Xia L (2013) Podoplanin maintains high endothelial venule integrity by interacting with platelet CLEC-2. Nature 502(7469):105–109
Hess PR, Rawnsley DR, Jakus Z, Yang Y, Sweet DT, Fu J, Herzog B, Lu M, Nieswandt B, Oliver G, Makinen T, Xia L, Kahn ML (2014) Platelets mediate lymphovenous hemostasis to maintain blood-lymphatic separation throughout life. J Clin Invest 124(1):273–284
Huang TF, Liu CZ, Yang SH (1995) Aggretin, a novel platelet-aggregation inducer from snake (Calloselasma rhodostoma) venom, activates phospholipase C by acting as a glycoprotein Ia/IIa agonist. Biochem J 309(Pt 3):1021–1027
Hughes CE, Pollitt AY, Mori J, Eble JA, Tomlinson MG, Hartwig JH, O’Callaghan CA, Futterer K, Watson SP (2010a) CLEC-2 activates Syk through dimerization. Blood 115(14):2947–2955
Hughes CE, Navarro-Nunez L, Finney BA, Mourao-Sa D, Pollitt AY, Watson SP (2010b) CLEC-2 is not required for platelet aggregation at arteriolar shear. J Thromb Haemost 8(10):2328–2332
Ichise H, Ichise T, Ohtani O, Yoshida N (2009) Phospholipase Cgamma2 is necessary for separation of blood and lymphatic vasculature in mice. Development 136(2):191–195
Inoue K, Ozaki Y, Satoh K, Wu Y, Yatomi Y, Shin Y, Morita T (1999) Signal transduction pathways mediated by glycoprotein Ia/IIa in human platelets: comparison with those of glycoprotein VI. Biochem Biophys Res Commun 256(1):114–120
Katagiri Y, Hayashi Y, Baba I, Suzuki H, Tanoue K, Yamazaki H (1991) Characterization of platelet aggregation induced by the human melanoma cell line HMV-I: roles of heparin, plasma adhesive proteins, and tumor cell membrane proteins. Cancer Res 51(4):1286–1293
Kato Y, Fujita N, Kunita A, Sato S, Kaneko M, Osawa M, Tsuruo T (2003) Molecular identification of Aggrus/T1alpha as a platelet aggregation-inducing factor expressed in colorectal tumors. J Biol Chem 278(51):51599–51605
Kato Y, Sasagawa I, Kaneko M, Osawa M, Fujita N, Tsuruo T (2004) Aggrus: a diagnostic marker that distinguishes seminoma from embryonal carcinoma in testicular germ cell tumors. Oncogene 23(52):8552–8556
Kato Y, Kaneko M, Sata M, Fujita N, Tsuruo T, Osawa M (2005) Enhanced expression of Aggrus (T1alpha/podoplanin), a platelet-aggregation-inducing factor in lung squamous cell carcinoma. Tumour Biol 26(4):195–200
Kato Y, Kaneko MK, Kuno A, Uchiyama N, Amano K, Chiba Y, Hasegawa Y, Hirabayashi J, Narimatsu H, Mishima K, Osawa M (2006) Inhibition of tumor cell-induced platelet aggregation using a novel anti-podoplanin antibody reacting with its platelet-aggregation-stimulating domain. Biochem Biophys Res Commun 349(4):1301–1307
Kato Y, Kaneko MK, Kunita A, Ito H, Kameyama A, Ogasawara S, Matsuura N, Hasegawa Y, Suzuki-Inoue K, Inoue O, Ozaki Y, Narimatsu H (2008) Molecular analysis of the pathophysiological binding of the platelet aggregation-inducing factor podoplanin to the C-type lectin-like receptor CLEC-2. Cancer Sci 99(1):54–61
Kerrigan AM, Dennehy KM, Mourao-Sa D, Faro-Trindade I, Willment JA, Taylor PR, Eble JA, Reis e Sousa C, Brown GD (2009) CLEC-2 is a phagocytic activation receptor expressed on murine peripheral blood neutrophils. J Immunol 182(7):4150–4157
Kitagawa H, Yamamoto N, Yamamoto K, Tanoue K, Kosaki G, Yamazaki H (1989) Involvement of platelet membrane glycoprotein Ib and glycoprotein IIb/IIIa complex in thrombin-dependent and -independent platelet aggregations induced by tumor cells. Cancer Res 49(3):537–541
May F, Hagedorn I, Pleines I, Bender M, Vogtle T, Eble J, Elvers M, Nieswandt B (2009) CLEC-2 is an essential platelet-activating receptor in hemostasis and thrombosis. Blood 114(16):3464–3472
Michelson AD (2013) Platelets, 3rd edn. Academic Press/Elsevier Science, Amsterdam
Mishima K, Kato Y, Kaneko MK, Nakazawa Y, Kunita A, Fujita N, Tsuruo T, Nishikawa R, Hirose T, Matsutani M (2006a) Podoplanin expression in primary central nervous system germ cell tumors: a useful histological marker for the diagnosis of germinoma. Acta Neuropathol 111(6):563–568
Mishima K, Kato Y, Kaneko MK, Nishikawa R, Hirose T, Matsutani M (2006b) Increased expression of podoplanin in malignant astrocytic tumors as a novel molecular marker of malignant progression. Acta Neuropathol 111(5):483–488
Navdaev A, Clemetson JM, Polgar J, Kehrel BE, Glauner M, Magnenat E, Wells TN, Clemetson KJ (2001) Aggretin, a heterodimeric C-type lectin from Calloselasma rhodostoma (malayan pit viper), stimulates platelets by binding to alpha 2beta 1 integrin and glycoprotein Ib, activating Syk and phospholipase Cgamma 2, but does not involve the glycoprotein VI/Fc receptor gamma chain collagen receptor. J Biol Chem 276(24):20882–20889
Nieswandt B, Hafner M, Echtenacher B, Mannel DN (1999) Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res 59(6):1295–1300
Osada M, Inoue O, Ding G, Shirai T, Ichise H, Hirayama K, Takano K, Yatomi Y, Hirashima M, Fujii H, Suzuki-Inoue K, Ozaki Y (2012) Platelet activation receptor CLEC-2 regulates blood/lymphatic vessel separation by inhibiting proliferation, migration, and tube formation of lymphatic endothelial cells. J Biol Chem 287(26):22241–22252
Ozaki Y, Suzuki-Inoue K, Inoue O (2009) Novel interactions in platelet biology: CLEC-2/podoplanin and laminin/GPVI. J Thromb Haemost 7(Suppl 1):191–194
Schacht V, Dadras SS, Johnson LA, Jackson DG, Hong YK, Detmar M (2005) Up-regulation of the lymphatic marker podoplanin, a mucin-type transmembrane glycoprotein, in human squamous cell carcinomas and germ cell tumors. Am J Pathol 166(3):913–921
Severin S, Pollitt AY, Navarro-Nunez L, Nash CA, Mourao-Sa D, Eble JA, Senis YA, Watson SP (2011) Syk dependent phosphorylation of CLEC-2: a novel mechanism of hemitam signalling. J Biol Chem 286(6):4107–4116
Shin Y, Morita T (1998) Rhodocytin, a functional novel platelet agonist belonging to the heterodimeric C-type lectin family, induces platelet aggregation independently of glycoprotein Ib. Biochem Biophys Res Commun 245(3):741–745
Shirai T, Inoue O, Hirayama K, Endo H, Fujii H, Utida-Sato H, K S-I YO (2013) A role of CLEC-2 in tumor growth and metastasis. J Thromb Haemost 11(Suppl 2):139
Spalton JC, Mori J, Pollitt AY, Hughes CE, Eble JA, Watson SP (2009) The novel Syk inhibitor R406 reveals mechanistic differences in the initiation of GPVI and CLEC-2 signaling in platelets. J Thromb Haemost 7(7):1192–1199
Suzuki-Inoue K, Ozaki Y, Kainoh M, Shin Y, Wu Y, Yatomi Y, Ohmori T, Tanaka T, Satoh K, Morita T (2001) Rhodocytin induces platelet aggregation by interacting with glycoprotein Ia/IIa (GPIa/IIa, Integrin alpha 2beta 1). Involvement of GPIa/IIa-associated src and protein tyrosine phosphorylation. J Biol Chem 276(2):1643–1652
Suzuki-Inoue K, Fuller GL, Garcia A, Eble JA, Pohlmann S, Inoue O, Gartner TK, Hughan SC, Pearce AC, Laing GD, Theakston RD, Schweighoffer E, Zitzmann N, Morita T, Tybulewicz VL, Ozaki Y, Watson SP (2006) A novel Syk-dependent mechanism of platelet activation by the C-type lectin receptor CLEC-2. Blood 107(2):542–549
Suzuki-Inoue K, Kato Y, Inoue O, Kaneko MK, Mishima K, Yatomi Y, Yamazaki Y, Narimatsu H, Ozaki Y (2007) Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J Biol Chem 282(36):25993–26001
Suzuki-Inoue K, Inoue O, Ding G, Nishimura S, Hokamura K, Eto K, Kashiwagi H, Tomiyama Y, Yatomi Y, Umemura K, Shin Y, Hirashima M, Ozaki Y (2010) Essential in vivo roles of the C-type lectin receptor CLEC-2: embryonic/neonatal lethality of CLEC-2-deficient mice by blood/lymphatic misconnections and impaired thrombus formation of CLEC-2-deficient platelets. J Biol Chem 285(32):24494–24507
Suzuki-Inoue K, Inoue O, Ozaki Y (2011) Novel platelet activation receptor CLEC-2: from discovery to prospects. J Thromb Haemost 9(Suppl 1):44–55
Tammela T, Alitalo K (2010) Lymphangiogenesis: molecular mechanisms and future promise. Cell 140(4):460–476
Tang T, Li L, Tang J, Li Y, Lin WY, Martin F, Grant D, Solloway M, Parker L, Ye W, Forrest W, Ghilardi N, Oravecz T, Platt KA, Rice DS, Hansen GM, Abuin A, Eberhart DE, Godowski P, Holt KH, Peterson A, Zambrowicz BP, de Sauvage FJ (2010) A mouse knockout library for secreted and transmembrane proteins. Nat Biotechnol 28(7):749–755
Tsuruo T, Fujita N (2008) Platelet aggregation in the formation of tumor metastasis. Proc Jpn Acad Ser B Phys Biol Sci 84(6):189–198
Uhrin P, Zaujec J, Breuss JM, Olcaydu D, Chrenek P, Stockinger H, Fuertbauer E, Moser M, Haiko P, Fassler R, Alitalo K, Binder BR, Kerjaschki D (2010) Novel function for blood platelets and podoplanin in developmental separation of blood and lymphatic circulation. Blood 115(19):3997–4005
Watson SP, Auger JM, McCarty OJ, Pearce AC (2005) GPVI and integrin alphaIIb beta3 signaling in platelets. J Thromb Haemost 3(8):1752–1762
Watson AA, Brown J, Harlos K, Eble JA, Walter TS, O’Callaghan CA (2007) The crystal structure and mutational binding analysis of the extracellular domain of the platelet-activating receptor CLEC-2. J Biol Chem 282(5):3165–3172
Watson AA, Christou CM, James JR, Fenton-May AE, Moncayo GE, Mistry AR, Davis SJ, Gilbert RJ, Chakera A, O’Callaghan CA (2009) The platelet receptor CLEC-2 is active as a dimer. Biochemistry 48(46):10988–10996
Yuan P, Temam S, El-Naggar A, Zhou X, Liu DD, Lee JJ, Mao L (2006) Overexpression of podoplanin in oral cancer and its association with poor clinical outcome. Cancer 107(3):563–569
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this chapter
Cite this chapter
Suzuki-Inoue, K. (2016). C-Type Lectin-Like Receptor 2 (CLEC-2). In: Yamasaki, S. (eds) C-Type Lectin Receptors in Immunity. Springer, Tokyo. https://doi.org/10.1007/978-4-431-56015-9_6
Download citation
DOI: https://doi.org/10.1007/978-4-431-56015-9_6
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-56013-5
Online ISBN: 978-4-431-56015-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)