
Abstract
Protein folding and clearance machineries are required for the maintenance and function of the proteome. Quality control systems and activation of stress signaling pathways have, therefore, profound consequences on the long-term health of the cell and, by extension, on lifespan. Aging is associated with loss of cellular function, increased vulnerability to stress, and enhanced susceptibility to disease. Over the course of a lifespan, proteome stability is substantially impacted by mutations, by processing errors, and by the acute effects of environmental stresses. Recently, the function of cellular protein quality control networks, as well as stress signaling pathways, was shown to be differentially regulated over the course of life, leading to reduced proteostasis capacity and decreased stress response activation during adulthood. Proteostatic collapse can be partially mitigated by overexpression of stress response transcription factors, such as HSF1, or by enhancing the activity of quality control systems, which can have significant beneficial effects on lifespan extension and suppression of age-related misfolding diseases. However, the link between proteostasis and lifespan can also be uncoupled, for example, by cell-nonautonomous stress signaling. Here, we will examine how proteostasis changes with age. We will then focus on HSF1 and review its roles in lifespan regulation, as well as how HSF1 function is modulated with age. Finally, we will examine the cell-nonautonomous regulation of HSF1, specifically during aging.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Aging
- Chaperone
- Dietary restriction
- Germline stem cells signaling
- Insulin/IGF-1 signaling
- Heat shock
- HSF1
- Longevity
- Sirtuins
- Stress
1 Introduction
Cellular quality control networks are comprised of molecular chaperones, degradation machineries, and cytoprotective stress responses, such as the heat shock response (HSR) and the unfolded protein response (UPR) (Akerfelt et al. 2010; Haynes et al. 2013; Hetz 2012). The HSR enables the cell to adjust the expression of chaperones and other cytoprotective genes under proteotoxic conditions, thereby ensuring stress survival, recovery, and adaptation (Akerfelt et al. 2010). At the molecular level, this involves transcriptional regulation of heat shock (HS) genes in a manner proportional to the intensity, duration, and type of stress (Abravaya et al. 1991a, 2010; Gasch et al. 2000). HSF1, as the “master regulator” of the HSR, functions both as a stress sensor and transcriptional regulator of downstream stress response genes that are essential for protecting and remodeling the cell (Akerfelt et al. 2010).
The HSR has been considered a universal molecular response to various stress stimuli. Nonetheless, there are several examples in which the HSR is poorly or incompletely activated, such as the limited response seen in early development, as well as in different tissues of aged animals and in late-onset neurodegenerative diseases, such as Huntington’s disease and Alzheimer’s disease (Ben-Zvi et al. 2009; Bienz 1984; Heydari et al. 1993; Labbadia et al. 2011; Prahlad et al. 2008; Sprang and Brown 1987; Taylor and Dillin 2011). These observations suggest that the HSR can be spatially and temporally regulated during the lifetime of a multicellular organism.
In the past decade, lifespan-enhancing pathways were shown to be potent modifiers of proteostasis and to suppress protein aggregation and toxicity. Moreover, HSF1 has been shown to be an important factor influencing lifespan (Kenyon 2010b; Labbadia and Morimoto 2014; Tissenbaum 2012). Recent studies mainly conducted in Caenorhabditis elegans have uncovered several cell-nonautonomous pathways that modulate cellular quality control systems during the lifespan of the organism, in particular, HSF1. These pathways include cell-nonautonomous regulation of the heat shock response by neurons and germline stem cells, as well as transcellular signaling of proteostatic deficiencies in which the expression of misfolded proteins in one tissue can induce a systemic HSR (Prahlad and Morimoto 2009; Shai et al. 2014; van Oosten-Hawle and Morimoto 2014). Several of these pathways are also linked to lifespan, suggesting that proteostasis networks may show differential sensitivity at different stages over the lifespan of an organism. Here, we review changes in proteostatic function with age, focusing on cell-nonautonomous regulation of HSF1 during aging (mainly in C. elegans) to offer insight into how multicellular organisms adjust stress responses during key life stages and how HSF1 function, in turn, can impact lifespan.
2 The Loss of Proteostasis Is a Hallmark of Aging
Aging is often associated with the time-dependent loss of cellular function, increased susceptibility to environmental and physiological insults, and increased vulnerability to disease (Gems 2015; Partridge 2014; Riera and Dillin 2015). One of the hallmarks of aging is age-dependent loss of proteostasis. In agreement, aggregation and toxicity of aggregation-prone proteins associated with disease, such as huntingtin, is enhanced with age, while different protein quality control machineries show decreased function (Labbadia and Morimoto 2015a; Shai et al. 2014; Taylor and Dillin 2011). Proteostasis loss and age-dependent aggregation can be recapitulated in different model systems in a time frame related to their aging. In fact, a subnetwork of chaperones was shown to be required in both human brain aging, as well as in brain samples, and C. elegans models of age-associated diseases (Brehme et al. 2014). Thus, quality control systems and, specifically, the decline in proteostasis capacity are suggested to play important roles in aging and age-associated diseases (Brehme et al. 2014; Labbadia and Morimoto 2015a).
2.1 When Does Proteostasis Collapse?
The failure of proteostasis networks to maintain the proteome of aged animals is commonly explained by the slow accumulation of damage over time. Protein damage and misfolding in aged individuals is, therefore, suggested to result from a constant difference between the error rate and the efficiency of cellular quality control network in repairing or removing misfolded proteins over the course of an organism’s life, leading to a gradual accumulation of damaged proteins over time. An alternative explanation for the failure of proteostatic networks in old individuals is that the function of cellular proteostatic networks decline with age (Taylor and Dillin 2011). For example, translation fidelity may decline with age, resulting in an increased load of damaged proteins as the individual ages (Kirstein-Miles et al. 2013). Likewise, the expression and function of cellular quality control networks may be differentially regulated over the lifespan of the organism (Bar-Lavan et al. 2012). Such changes can remodel cellular folding capacity and stress tolerance, thus increasing the risk for age-associated pathology. What distinguishes between these mechanisms is the rate of damage accumulation during adulthood. While the first scenario suggests a passive accumulation of damage over time that can be modulated by the efficacy of the proteostatic network, the second suggests that quality control function is differentially regulated over the lifespan of the organism.
2.2 Age-Dependent Changes in HSR Activation
Recent studies mostly conducted with C. elegans demonstrated that cellular quality control networks are modified as animals undergo transition to a reproductively mature state. Specifically, rapid changes in the regulation and activation of stress responses were identified. When thermoresistance and induction of heat shock genes were monitored over time in adult C. elegans, both declined sharply 12 h following transition to adulthood (Shemesh et al. 2013). Thus, within hours of reaching adulthood, most animals lost the ability to mount an effective HSR and survive the insult. This finding suggests the existence of very strong negative regulation of the HSR upon transition to adulthood, inhibiting HSF1 function. When other stress responses were examined, such as activation of UPR in the endoplasmic reticulum (UPRER), UPR in the mitochondria (UPRmt), and oxidative stress in response to different perturbations, an early decline in the mounting of a stress response was also observed (Ben-Zvi et al. 2009; Labbadia and Morimoto 2015b; Taylor and Dillin 2013). Likewise, survival from acute oxidative stress and activation of an NRF2-dependent oxidative stress response in adult Drosophila melanogaster was strongly dampened, as compared to young animals (Rahman et al. 2013). Altered temporal regulation was also apparent for C. elegans c-Jun N-terminal kinase (JNK) signaling. While the JNK homolog KGB-1 enhances DAF-16 nuclear localization and transcriptional regulation during C. elegans development, this function was reversed upon transition to adulthood (Twumasi-Boateng et al. 2012). Changes in the activation of JNK signaling were also observed in adult Drosophila and old mice, although expression modulation was not monitored early in adulthood in either case (Girardot et al. 2006; Hsieh et al. 2003; Simonsen et al. 2008; Tsakiri et al. 2013). These data suggest that stress-induced transcriptional activation is strongly dampened early in adulthood and exhibits switch-like behavior associated with reproduction onset (Fig. 5.1).

Proteostasis is remodeled upon transition to adulthood. Changes in stress response activation, expression and function of chaperones, the ubiquitin-proteasome system, and autophagy modulate cellular proteostasis capacity, resulting in accumulation of misfolded and aggregated proteins during adulthood. These changes coincide with the onset of oocyte biomass production and can be modulated by GSC arrest
It is important to note that stress response activation can also be remodeled at other stages over the lifespan of an organism. For example, activation of UPRmt can be transmitted to other tissues, albeit only during development (Durieux et al. 2011). Likewise, heat shock response can be modulated during C. elegans development by including 5-fluoro-2′-deoxyuridine in the growth medium (Feldman et al. 2014). These observations strongly support the hypothesis that quality control networks can be remodeled during development and with age and suggest that many different pathways modulate cellular responses to stress.
2.3 Age-Dependent Changes in Proteostatic Network Composition
Changes in proteostatic function over time are associated with changes in expression of genes encoding quality control machinery components, such as ribosomal proteins, chaperones, and proteasome- and autophagy-associated proteins (Taylor and Dillin 2011). Relying on a quantitative mass spectrometry proteomics-based approach designed to follow changes in protein expression during C. elegans adulthood, it was uncovered that ribosomal abundance strongly declined with age and that ribosome stoichiometry was disrupted, while the abundance of specific small HS proteins (sHSP), proteasome subunits, and some E3 ligases increased with age. These changes were even observed early in adulthood (Walther et al. 2015). In agreement, C. elegans epidermal growth factor (EGF)-mediated signaling was shown to upregulate the expression of genes associated with the ubiquitin-proteasome system and downregulate the expression of some chaperones upon transition to reproductive adulthood (Liu et al. 2011). Additional studies observed changes in the expression of genes encoding proteostasis-mediating components during aging. For example, in adult Drosophila, changes in the activation of JNK signaling and in the levels of proteasome subunits were observed (Girardot et al. 2006; Simonsen et al. 2008; Tsakiri et al. 2013). Likewise, expression levels of autophagy-associated genes declined with age in adult Drosophila and rats (Kiffin et al. 2007; Simonsen et al. 2008). Thus, the composition of the proteostatic network is also remodeled in early adulthood (Fig. 5.1).
2.4 Age-Dependent Changes in Protein Folding and Aggregation
If proteostatic networks are remodeled during adulthood, then one would expect proteostatic capacity to decline within a specific window of time during adulthood. Changes in the activity of quality control networks would impact their interactions with the proteome and, therefore, change rates of protein misfolding. One such example is the onset of aggregation and toxicity in a C. elegans model of Huntington’s disease. In this model, animals expressing 35 repeats of poly-glutamine (polyQ) start to accumulate aggregates following the onset of reproduction (Morley et al. 2002). For animals cultivated at 20 °C, this occurs ~4 days postembryo, while for animals cultivated at 25 °C, this transpires 2.5 days postembryo (Morley et al. 2002; Shemesh et al. 2013). Thus, aggregation-prone proteins quickly became insoluble upon transition to adulthood.
The functions and folding of metastable temperature-sensitive (ts) mutant proteins are also disrupted early in adulthood. Temperature-sensitive mutant proteins assayed for changes in their folding capacity, such as ras, the acetylcholine receptor, and perlecan (UNC-52), all showed a rapid loss of function early in adulthood (days 2–6 of adulthood) (Ben-Zvi et al. 2009; Shemesh et al. 2013). In C. elegans muscle, expression of a ts version of myosin gave rise to age-dependent mislocalization of myosin and resulted in uncoordinated movement. Here, myosin misfolding was detected as early as day 3 of adulthood (Ben-Zvi et al. 2009). Likewise, neurons and coelomocytes of animals expressing a ts version of DYN-1 showed age-dependent dysfunction and DYN-1 mislocalization as early as day 2 of adulthood (Ben-Zvi et al. 2009). In such animals, the misfolding of metastable proteins observed early in adulthood was thus general but affected different proteins in different tissues.
Age-dependent protein misfolding associated with functional decline was also apparent for wild-type proteins, although their aggregation started, for the most part, later in adulthood (Ben-Zvi et al. 2009; Haithcock et al. 2005; Shemesh et al. 2013). When wild-type myosin was monitored over time, it showed age-dependent mislocalization and misfolding, similar to what was seen with metastable myosin, albeit later in adulthood than the nonnative protein (Ben-Zvi et al. 2009; Shemesh et al. 2013). An unbiased examination of age-dependent aggregation during C. elegans adulthood supported this finding. Using mass spectrometry to systematically examine age-dependent changes in protein solubility, David et al. identified several hundred proteins that became insoluble over time. Some wild-type proteins aggregated as early as day 3 of adulthood (David et al. 2010). Many of these proteins were also identified in another independent study (Reis-Rodrigues et al. 2012). A recent study using quantitative mass spectrometry to analyze changes in the proteome during C. elegans adulthood showed that many proteins underwent more than twofold changes in abundance, linked to widespread aggregation (Walther et al. 2015). Thus, upon transition to reproductive adulthood, there is an onset of age-dependent decline in cellular proteostasis, coinciding with a decline in HSR activation (Fig. 5.1).
The rate of protein clearance is also modulated early in C. elegans adulthood. Monitoring the clearance of GFP tagged with an uncleavable ubiquitin tag as a degradation reporter (UbG76V-GFP) demonstrated that there is a sharp increase in protein degradation upon transition to adulthood. While UbG76V-GFP was completely degraded during development, it started to accumulate when animals became reproductive adults (Liu et al. 2011). At later stages, protein degradation was shown to decrease in worms, flies, and rats (Hamer et al. 2010; Keller et al. 2000; Liu et al. 2011; Tonoki et al. 2009). Changes in autophagy early in adulthood were not monitored, although reduced autophagic activity was observed in adult Drosophila and rats (Kiffin et al. 2007; Simonsen et al. 2008). Given that the two systems are linked for the removal of aggregated proteins (Cha-Molstad et al. 2015), these observations raises interesting questions about how such interactions are modulated with age to change protein clearance.
2.5 Modulating HSF1 Levels Can Affect Proteostatic Collapse
If the levels and functions of proteostasis-related genes are limiting in adulthood, could modifying the levels of quality control transcription factors, such as HSF1, modulate proteostasis? When HSF1 levels were diminished using RNAi, the loss of function of metastable proteins, such as paramyosin and dynamin, and of aggregation-prone proteins, such as polyQ and Aβ, was exacerbated, resulting in an earlier decline in proteostasis. In contrast, overexpression of HSF1 proved to be protective for metastable and aggregation-prone proteins (Ben-Zvi et al. 2009; Cohen et al. 2006; Hsu et al. 2003; Morley and Morimoto 2004). Modulating HSF1 levels also affected the aggregation of wild-type proteins (Walther et al. 2015). The switch-like behavior of the HSR and the fact that over-expression of HSF1 can rescue proteostasis in adulthood (Ben-Zvi et al. 2009; Labbadia and Morimoto 2015b; Shemesh et al. 2013) suggest that HSF1 and other stress transcription factors are likely differentially regulated over the lifespan of the organism.
3 HSF1 Is a Lifespan Regulator
The involvement of HSF1 in setting the function of proteostasis in adulthood suggests that HSF1 can impact aging and lifespan. In agreement, HSF1 was shown to modulate age-associated phenotypes and shorten lifespan in C. elegans (Garigan et al. 2002). HSF1 knockdown induced tissue integrity decline of both somatic and germline stem cells, and wild-type animals treated with HSF1 RNAi were short-lived (Garigan et al. 2002). Conversely, the lifespan of animals overexpressing HSF1 was extended by 22–40 % (Hsu et al. 2003; Morley and Morimoto 2004). Thus, HSF1 is a lifespan regulator and it can promote longevity.
3.1 HSF1 Is Required for Insulin/IGF-1 Signaling (IIS) Pathway Activity
Downregulation of the insulin/IGF-1 signaling (IIS) pathway promotes longevity and suppresses protein aggregation and toxicity. Occupation of the IIS receptor (DAF-2 in C. elegans) initiates a conserved cascade of kinases that results in the phosphorylation and subsequent inactivation of the FOXO transcription factor (DAF-16 in C. elegans). In different model organisms, activation of the FOXO transcription factor, as occurs upon reduced IIS receptor function or levels, resulted in an increase in lifespan (Kenyon 2010b). In C. elegans, DAF-16 is required for DAF-2-mediated extended lifespan and improved proteostasis (Cohen et al. 2006, 2009; Hsu et al. 2003; Morley et al. 2002), as is HSF1. RNAi knockdown of HSF1 in a DAF-2 mutant background shortened the lifespan of the animal and resulted in a loss of the expected proteostatic improvement (Ben-Zvi et al. 2009; Cohen et al. 2006, 2009; Hsu et al. 2003; Morley et al. 2002).
3.2 HSF1 Is Required for Germline Stem Cell (GSC) Signaling
Endocrine signaling from germline stem cells (GSCs) and the somatic gonad links reproduction to aging. Removal of GSCs by laser ablation or induction of GSC arrest through mutations extended the lifespan of C. elegans (Arantes-Oliveira et al. 2002; Hsin and Kenyon 1999) and other model animals, such as the nematode Pristionchus pacificus (Hsin and Kenyon 1999; Rae et al. 2012) and Drosophila (Flatt et al. 2008). Transplantation of young ovaries into old mice also promoted longevity (Cargill et al. 2003; Mason et al. 2009). These studies established a role for the reproductive system, and specifically signals from the somatic gonad and GSCs, in lifespan (Kenyon 2010a). Because removal of the entire gonad did not extend lifespan, it would appear that longevity is not a simple consequence of sterility but rather depends on communication between the reproductive system and somatic tissues that modulate different signaling pathways, including those involved in metabolism and quality control, to extend lifespan (Antebi 2012). GSC-dependent extension of lifespan requires the activation of many different downstream signaling pathways, one of which includes HSF1 (Antebi 2012; Libina et al. 2003).
3.3 HSF1 Is Required in Part for Dietary Restriction (DR)
Dietary restriction (DR) is another manipulation that extends life in organisms spanning from yeast to mammals. In fact, DR was the first manipulation described that increased lifespan (McCay et al. 1989). There are many different protocols to induce DR, including limiting food intake (Greer and Brunet 2009). For example, mutant C. elegans with a defective eat-2 pharyngeal pump-encoding gene showed reduced food intake and thus serve as a genetic model of DR (Avery 1993). While different DR regimes all extend lifespan, they activate different nutrient-sensing pathways, including those involving TOR, AMP kinase, and sirtuins (Kenyon 2010b). Only some of these pathways, however, require HSF1 for longevity and reduction of proteotoxicity (Greer and Brunet 2009; Mouchiroud et al. 2011; Sutphin and Kaeberlein 2008; Vora et al. 2013; Zhang et al. 2009). Thus, HSF1 is required for lifespan extension in several different models of longevity that are linked but not always coupled to improved proteostasis.
3.4 HSF1 Is Differentially Regulated in Development and in Adulthood
Similar to the observed changes in proteostatic composition and function in adulthood, HSF1 regulation by the IIS pathway is also modulated upon transition to adulthood. Using conditional RNAi, Cohen et al. showed that HSF1 function in IIS-dependent reduction of aggregates toxicity was mostly required during larval development. Knocking-down HSF1 levels against a background of reduced IIS signaling (as achieved upon knockdown of the daf-2 insulin-like receptor-encoding gene) on the first day of adulthood did not reduce IIS-mediated protective effects. In contrast, downregulation of DAF-16 was essential only in adulthood (Cohen et al. 2010). Moreover, HSF1 knockdown during development also abolished IIS-mediated extension of lifespan, while HSF1 downregulation during early or late adulthood had only a mild or no effect on lifespan, respectively (Volovik et al. 2012). Thus, HSF1 function in proteostasis and lifespan when part of the IIS pathway shows switch-like behavior upon transition to adulthood (Fig. 5.1).
4 Cell-Autonomous and Cell-Nonautonomous Regulation of HSF1
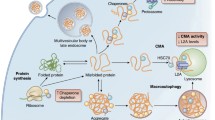
HSF1 regulation has been studied in depth in both in vitro and in single-cell systems (see Chaps. 1, 2, 3 and 4). In short, upon accumulation of misfolded and damaged protein, the equilibrium of molecular chaperones is altered such that HSF1 monomers are released from repressive complexes that include molecular chaperones (Abravaya et al. 1992; Shi et al. 1998; Zou et al. 1998). Once free, HSF1 acquires DNA-binding activity through trimerization in a process that is also regulated by extensive posttranslational modifications that include phosphorylation (Guettouche et al. 2005; Holmberg et al. 2001; Kline and Morimoto 1997; Knauf et al. 1996; Sorger and Pelham 1988), sumoylation (Anckar et al. 2006; Hietakangas et al. 2003), and acetylation (Westerheide et al. 2009) that can enhance or suppress HSF1 DNA-binding activity (Akerfelt et al. 2010). In the nucleus, HSF1 binds to promoters of target loci (heat shock elements (HSEs)) found in promoter regions of stress-inducible genes, which are maintained in a state that is permissive for HSF1 binding and transcription regulation (Fujimoto et al. 2012; Guertin and Lis 2013; Shopland et al. 1995; Zelin et al. 2012).
In nonstressed metazoan cells, HSF1 is predominantly found in a monomeric non-DNA-binding state, repressed by transient interactions with chaperones including Hsp90, Hsp70, and Hsp40 (Abravaya et al. 1992; Shi et al. 1998; Zou et al. 1998). When the stress signal is dampened, or when damaged proteins are no longer expressed, the HSR is attenuated. Attenuation is associated with the release of HSF1 trimers from DNA and subsequent conversion of the trimers to HSF1 monomers (Anckar and Sistonen 2007; Morimoto and Santoro 1998; Wu 1995). HSR attenuates, in part, due to the accumulation of chaperones that bind to HSF1 and acetylation of HSF1 in the DNA-binding domain (Abravaya et al. 1991b, 1992; Shi et al. 1998; Yao et al. 2006). The combination of these posttranslational modifications and chaperone interactions thus subjects HSF1 to multiple levels of control and feedback loops so as to precisely regulate chaperone levels in the cell. Regulation of the HSR, therefore, occurs through HSF1-dependent and HSF1-independent mechanisms (Fig. 5.2a). However, the mode of regulation whereby HSR is linked to chaperone levels in the cell cannot explain the poor or incomplete activation of the HSR during development and aging. Such variability in HSR induction suggests the existence of additional, cell-nonautonomous modes of HSR regulation in multicellular organisms. Accordingly, we discuss below five novel pathways that are suggested to regulate HSF1 activation, efficacy, or attenuation in a cell-nonautonomous manner (Fig. 5.2b).

Cell-autonomous and cell-nonautonomous regulation of HSF1. (a) Cell-autonomous HSR activation is a stepwise process. (i–ii) Stress-induced appearance of misfolded and damaged proteins, resulting in (iii–v) an equilibrium shift of chaperones from HSF1 to unfolded species, (vi) leading to formation of HSF1 trimers, which (vii) translocate to the nucleus, (viii) bind to HS gene promoters, and induce transcription of HS genes, leading to (ix) elevated levels of chaperones that bind to damaged proteins until the stress signal is alleviated. (b) Cell-nonautonomous signals can integrate into the cell-autonomous response. (1) HSF1 monomers are sequestered by the DHIC complex (1), an event that is regulated by IIS signaling. (2) HSF1 function is modulated by posttranslational modifications, such as acetylation that can be activated by cell-nonautonomous signals. (3) HSF1 binding can be modified by chromatin remodeling, while (4) HSF1 activation can be impacted by other transcription factors that modulate HSP levels and thus their repressive effects on HSF1 trimerization
4.1 Neuronal Regulation of HSF1
The first example of cell-nonautonomous regulation of HSF1 was identified as a novel cross talk between an animal’s ability to perceive ambient temperature and the ability of the cell to mount a HSR (Prahlad et al. 2008; Prahlad and Morimoto 2011; Tatum et al. 2015). In C. elegans, two neurons termed AFD neurons are required to sense temperature and regulate temperature-dependent behaviors, such as feeding and reproduction (Lee and Kenyon 2009; Mori et al. 2007). For example, AFD neurons can modulate foraging behavior according to the history of food availability at a given temperature (Mori et al. 2007). Mutations in gcy-8, encoding AFD-specific receptor-type guanylyl cyclases, can disrupt thermotactic behavior (Inada et al. 2006). Work by Prahlad et al. demonstrated that the ability of gcy-8 mutant animals exposed to an acute HS treatment to mount an effective HSR was blocked in different somatic cells. This resulted in low expression of HS genes and reduced thermo-survival (Prahlad et al. 2008). In agreement, excitation of AFD neurons in the absence of stress using optogenetic tools was sufficient to activate HSF1, resulting in reorganization of HSF1 into nuclear puncta in germ cells (Tatum et al. 2015). In contrast, gcy-8 mutant animals were able to mount a HSF1-dependent heavy-metal stress response, revealing specificity to a given sensory input (Prahlad et al. 2008). While the ability to respond to acute HS was inhibited, the ability of gcy-8 mutant animals to upregulate chaperones and maintain proteostasis was enhanced in different somatic cells under chronic stress conditions, such as upon expression of aggregation-prone proteins. Activation of the AFD neurons by a single optogenetic event was sufficient to retard protein aggregation, suggesting that activation of HSR resulted in improved proteostasis (Tatum et al. 2015). Neuronal regulation of the HSR is mediated by the serotonergic neurons ADF and NSM as activated by signals from AFD neurons. Temperature increase resulted in serotonin release in an AFD-dependent manner, leading to preemptive activation of the HSR (Tatum et al. 2015). Thus, in C. elegans, the HSR and proteostatic maintenance of somatic cells depend on sensory neuronal function that modulates the organismal response to stress and can distinguish between acute and chronic stresses (Prahlad et al. 2008; Prahlad and Morimoto 2011; Tatum et al. 2015). Somatic tissues can also send feedback information on the cultivation temperature to the AFD neurons. This response requires HSF1 in both neuronal and nonneuronal cells (Sugi et al. 2011). It remains unknown how these signals are integrated into the cellular response to stress or, specifically, how these signals modulate HSF1. Given that C. elegans exposed to high concentrations of the dauer pheromone that signal crowdedness present reduced HSR activation, it would appear that other signals may also feed into the neuronal pathway, either directly or via a different route (Prahlad et al. 2008). Thus, signals are also likely to be sent to the AFD neurons, integrating environmental signals into the decision of whether or not to activate the HSR in response to temperature shift (Prahlad et al. 2008; Prahlad and Morimoto 2011; Sugi et al. 2011; Tatum et al. 2015).
4.2 IIS-Mediated Regulation of HSF1
IIS-dependent suppression of protein aggregation, as well as lifespan extension, required HSF1 activity, suggesting that cell-nonautonomous regulation of HSF1 plays a role in both proteostasis and lifespan modulation (Alavez et al. 2011; Ben-Zvi et al. 2009; Cohen et al. 2006, 2009; Gidalevitz et al. 2011; Hsu et al. 2003; Morley et al. 2002; Taylor and Dillin 2011). Indeed, Chiang et al. showed that HSF1 is not only held in repressive complexes by chaperones preventing its activation (Abravaya et al. 1992; Shi et al. 1998; Zou et al. 1998) (Fig. 5.1a ii–v) but that HSB-1 and DDL-1/2 also form repressive complexes with HSF1 (Chiang et al. 2012). Formation of these inhibitory complexes, termed DDL-1/2 HSF1 inhibitory complexes (DHIC), is regulated by phosphorylation of DDL-1. DDL-1 phosphorylation, in turn, is regulated by IIS signaling (Chiang et al. 2012). Thus, HSF1 activation can be regulated by its active sequestration into a repressive complex regulated by cell-nonautonomous signals. This mode of regulation may be shared by other regulators, either via phosphorylation of DDL-1 or by assembly of other repressive complexes that affect HSF1 activation in a specific tissue (Fig. 5.2b).
4.3 SIRT1-Mediated Regulation of HSF1
Sirtuins are NAD+-dependent deacetylases that are implicated in lifespan regulation, although the mechanism of such action is not clear (Kenyon 2010b). Given that NAD+/NADH levels are determined by energy homeostasis, sirtuin function is linked to nutrient availability. SIRT1 was shown to deacetylate HSF1 and enhance DNA binding, suggesting that diet can modulate HSF1 release from DNA by removing the acetylation from the K80 acetylation site in the DNA-binding domain of HSF1. This deacetylation promotes the occupancy of HSF1 at the HS promoter sites, enhances HSR activation, and increases thermotolerance (Westerheide et al. 2009). In support of this idea, DR was shown to induce HS gene expression in a SIRT1- (sir-2.1 in C. elegans) dependent manner (Raynes et al. 2012). This suggests that signals that modulate SIRT1 function can affect HSF1. Likewise, other posttranslational modifications of HSF1 could, potentially, be regulated by cell-nonautonomous signals (Fig. 5.2b).
4.4 Germline Stem Cell-Mediated Regulation of HSF1
The coincidence of proteostatic collapse and the onset of reproduction, whereby the activation of different stress responses is strongly inhibited after egg laying begins, supports a link between proteostatic remodeling and reproduction. Several studies have examined the effects of inhibiting reproduction, and specifically GSC arrest, on proteostasis in C. elegans. The functions of several stress transcription factors, such as HSF1 and DAF-16, were modulated by GSC ablation or arrest (Arantes-Oliveira et al. 2002; Berman and Kenyon 2006; Ghazi et al. 2009; Hansen et al. 2005; Hsin and Kenyon 1999). Moreover, germline-less mutant animals were shown to have enhanced expression of proteostasis machinery genes, such as lgg-1, bec-1, and unc-51 required for autophagy (Lapierre et al. 2011) and the rpn-6.1 subunit of the proteasome (Vilchez et al. 2012). Whole genome microarray analysis of P. pacificus in germline-ablated animals identified ~3000 differentially expressed genes, including ribosomal and translation-, proteasome-, and protein folding- and refolding-associated genes (Rae et al. 2012). Moreover, the expression of stress-inducible genes, such as heat shock genes, in response to different assaults was modulated in germline-less animals (Ghazi et al. 2009; Shemesh et al. 2013). Thus, the proteostatic network, including the translational, chaperone, autophagic, and proteasomal machineries, is remodeled by GSC proliferation (Ghazi et al. 2009; Lapierre et al. 2011; Shemesh et al. 2013; Vilchez et al. 2012). The expression of proteostatic components is dependent on different downstream signaling pathways (Lapierre et al. 2011; Shemesh et al. 2013; Vilchez et al. 2012), suggesting that GSC signaling activates different regulatory programs to modulate somatic functions. This change in proteostatic composition is strongly associated with altered proteostatic function. Germline-less animals showed increased autophagosome numbers (Lapierre et al. 2011), increased levels of chymotrypsin-like proteasome activity, and decreased levels of highly polyubiquitinated proteins (Vilchez et al. 2012). GSC proliferation also modulated protein misfolding and aggregation in somatic cells. Germline-less animals displayed reduced aggregation, as well as reduced toxicity of polyQ proteins (Shemesh et al. 2013). Likewise, the functions of metastable and wild-type proteins that are compromised with age, such as UNC-52(ts) and myosin (Ben-Zvi et al. 2009), were rescued in germline-less animals (Shemesh et al. 2013). Finally, stress survival and the activation of stress responses, such as HS, were also rescued by GSC arrest (Alper et al. 2010; Arantes-Oliveira et al. 2002; Ermolaeva et al. 2013; Libina et al. 2003; Shemesh et al. 2013; Stiernagle 2006; TeKippe and Aballay 2010). Thus, inhibition of GSC proliferation reversed the decline in somatic protein quality control upon transition to adulthood.
Labbadia and Morimoto directly examined how HSF1 function is remodeled upon transition to adulthood and in GSC-arrested animals. They examined different regulation nodes in the HSF1 activation cycle, such as HSF 1 levels, nuclear localization, and DNA binding (Fig. 5.2a). Using chromatin immunoprecipitation coupled to qPCR to address specific HS genes, they found that HSF1 and RNA polymerase II association with HS gene promoters following HS was reduced between the first and second days of adulthood (Labbadia and Morimoto 2015b). At the same time, proteostasis is remodeled (Labbadia and Morimoto 2015b; Shemesh et al. 2013). HSF1 transcriptional repression was associated with reduced chromatin accessibility and specifically, with an increased histone methylation repressive marker, H3K27me3, on HS, as well as other stress genes. Moreover, changes in the expression levels of the H3K27me3 demethylase jmjd-3.1 were correlated with the onset of proteostasis collapse (Labbadia and Morimoto 2015b; Shemesh et al. 2013). Thus, the promoter regions of HSF1 target genes are more repressed on the second day of adulthood. In contrast, the levels of jmjd-3.1 and H3K27me3 markers on the promoter of HS genes in GSC-arrested animals remained low (Labbadia and Morimoto 2015b), in agreement with increased HS gene activation and HS survival collapse (Labbadia and Morimoto 2015b; Shemesh et al. 2013). Taken together, these data strongly suggest that HS activation is actively repressed upon transition to adulthood by cell-nonautonomous signals from the reproductive system (Fig. 5.2b). These signals globally change the chromatin accessibility of HSF1 and likely of other transcription factors, resulting in remodeling of several different signaling pathways upon transition to adulthood by GSC inhibition, with these signaling pathways drastically remodeling somatic proteostasis. This switch-like mechanism links reproduction to maintenance of the soma. However, the nature of the signals sent from the reproductive system to the soma and how they modulate jmjd-3.1 levels remain unknown.
4.5 Tissue-to-Tissue HS Signaling
As noted above, HSF1 is regulated by chaperones, such as Hsp90 and Hsp70 that form a repressive complex with HSF1 and inhibit its trimerization (Abravaya et al. 1992; Shi et al. 1998; Zou et al. 1998). However, chaperone levels are also regulated cell-nonautonomously by transcellular chaperone signaling that is regulated by the FOXA transcription factor PHA-4 in C. elegans (van Oosten-Hawle et al. 2013). Enhanced expression of Hsp90 in one tissue was sufficient to elevate the levels of Hsp90 in other tissues and thus block the induction of thermo-protective HSR in the distal tissues. Conversely, knocking down the expression of Hsp90 in a single tissue was sufficient to induce HS genes in distal tissues and protect the animals from stress. Such transcellular chaperone signaling was dependent on PHA-4 regulation of chaperone expression (van Oosten-Hawle et al. 2013). Thus, PHA-4 and possibly other transcription factors that modulate chaperone levels can impact the cell’s ability to mount an effective stress response (Fig 5.2b).
5 Perspectives
The findings that proteostasis is actively remodeled upon transition to adulthood and that HSF1 plays a role in proteostatic maintenance and lifespan open many questions. For example, how are regulation of proteostasis and lifespan linked? How does the decline in stress activation impact lifespan? While we do not yet know the answers to these questions, it is interesting to note that downregulation of any of the GSC-dependent signaling pathways is required for increased lifespan (Antebi 2012), even though these pathways differentially modulated proteostatic functions and stress response regulation (Lapierre et al. 2011; Shemesh et al. 2013; Vilchez et al. 2012). This suggests that only the combined actions of many protective pathways is sufficient for lifespan enhancement, while proteostatic function can be modulated by partial activation of these pathways. This is in agreement with chemical and genetic manipulation of lifespan-extending pathways in which lifespan, proteostasis, and stress resistance were mechanistically dissociated (El-Ami et al. 2014; Tissenbaum 2012; Van Raamsdonk and Hekimi 2012; Yu and Driscoll 2011).
Recent studies have identified several signaling pathways that can regulate HSF1, as well as defining points in HSF1 regulation that can be modulated by cell-nonautonomous signals. Still, many parts of this puzzle are missing. For example, how does neuronal regulation modulate HSF1 function? How are the different cell-nonautonomous signals integrated at the level of the cell, as well as at the level of the organism, to mount an effective HSR? Finally, focusing on lifespan modulation and treatment of age-dependent diseases, the most important question asks whether the age-dependent decline in stress activation and proteostasis can be reversed.
References
Abravaya K, Phillips B, Morimoto RI (1991a) Attenuation of the heat shock response in HeLa cells is mediated by the release of bound heat shock transcription factor and is modulated by changes in growth and in heat shock temperatures. Genes Dev 5(11):2117–2127
Abravaya K, Phillips B, Morimoto RI (1991b) Heat shock-induced interactions of heat shock transcription factor and the human hsp70 promoter examined by in vivo footprinting. Mol Cell Biol 11(1):586–592
Abravaya K, Myers MP, Murphy SP, Morimoto RI (1992) The human heat shock protein hsp70 interacts with HSF, the transcription factor that regulates heat shock gene expression. Genes Dev 6(7):1153–1164
Akerfelt M, Morimoto RI, Sistonen L (2010) Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol 11(8):545–555
Alavez S, Vantipalli MC, Zucker DJ, Klang IM, Lithgow GJ (2011) Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 472(7342):226–229
Alper S, McElwee MK, Apfeld J, Lackford B, Freedman JH, Schwartz DA (2010) The Caenorhabditis elegans germ line regulates distinct signaling pathways to control lifespan and innate immunity. J Biol Chem 285(3):1822–1828
Anckar J, Sistonen L (2007) Heat shock factor 1 as a coordinator of stress and developmental pathways. Adv Exp Med Biol 594:78–88
Anckar J, Hietakangas V, Denessiouk K, Thiele DJ, Johnson MS, Sistonen L (2006) Inhibition of DNA binding by differential sumoylation of heat shock factors. Mol Cell Biol 26(3):955–964
Antebi A (2012) Regulation of longevity by the reproductive system. Exp Gerontol 48(7):596–602
Arantes-Oliveira N, Apfeld J, Dillin A, Kenyon C (2002) Regulation of life-span by germ-line stem cells in Caenorhabditis elegans. Science 295(5554):502–505
Avery L (1993) The genetics of feeding in Caenorhabditis elegans. Genetics 133(4):897–917
Bar-Lavan Y, Kosolapov L, Frumkin A, Ben-Zvi A (2012) Regulation of cellular protein quality control networks in a multicellular organism. FEBS J 279(4):526–531
Ben-Zvi A, Miller EA, Morimoto RI (2009) Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci U S A 106(35):14914–14919
Berman JR, Kenyon C (2006) Germ-cell loss extends C. elegans life span through regulation of DAF-16 by kri-1 and lipophilic-hormone signaling. Cell 124(5):1055–1068
Bienz M (1984) Developmental control of the heat shock response in Xenopus. Proc Natl Acad Sci U S A 81(10):3138–3142
Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, Orton K, Villella A, Garza D, Vidal M, Ge H, Morimoto RI (2014) A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep 9(3):1135–1150
Cargill SL, Carey JR, Muller HG, Anderson G (2003) Age of ovary determines remaining life expectancy in old ovariectomized mice. Aging Cell 2(3):185–190
Cha-Molstad H, Sung KS, Hwang J, Kim KA, Yu JE, Yoo YD, Jang JM, Han DH, Molstad M, Kim JG, Lee YJ, Zakrzewska A, Kim SH, Kim ST, Kim SY, Lee HG, Soung NK, Ahn JS, Ciechanover A, Kim BY, Kwon YT (2015) Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat Cell Biol 17(7):917–929
Chiang WC, Ching TT, Lee HC, Mousigian C, Hsu AL (2012) HSF-1 regulators DDL-1/2 link insulin-like signaling to heat-shock responses and modulation of longevity. Cell 148(1–2):322–334
Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A (2006) Opposing activities protect against age-onset proteotoxicity. Science 313(5793):1604–1610
Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW, Masliah E, Dillin A (2009) Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 139(6):1157–1169
Cohen E, Du D, Joyce D, Kapernick EA, Volovik Y, Kelly JW, Dillin A (2010) Temporal requirements of insulin/IGF-1 signaling for proteotoxicity protection. Aging Cell 9(2):126–134
David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, Kenyon C (2010) Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol 8(8):e1000450
Durieux J, Wolff S, Dillin A (2011) The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144(1):79–91
El-Ami T, Moll L, Carvalhal Marques F, Volovik Y, Reuveni H, Cohen E (2014) A novel inhibitor of the insulin/IGF signaling pathway protects from age-onset, neurodegeneration-linked proteotoxicity. Aging Cell 13(1):165–174
Ermolaeva MA, Segref A, Dakhovnik A, Ou HL, Schneider JI, Utermohlen O, Hoppe T, Schumacher B (2013) DNA damage in germ cells induces an innate immune response that triggers systemic stress resistance. Nature 501(7467):416–420
Feldman N, Kosolapov L, Ben-Zvi A (2014) Fluorodeoxyuridine improves Caenorhabditis elegans proteostasis independent of reproduction onset. PLoS One 9(1):e85964
Flatt T, Min KJ, D'Alterio C, Villa-Cuesta E, Cumbers J, Lehmann R, Jones DL, Tatar M (2008) Drosophila germ-line modulation of insulin signaling and lifespan. Proc Natl Acad Sci U S A 105(17):6368–6373
Fujimoto M, Takaki E, Takii R, Tan K, Prakasam R, Hayashida N, Iemura S, Natsume T, Nakai A (2012) RPA assists HSF1 access to nucleosomal DNA by recruiting histone chaperone FACT. Mol Cell 48(2):182–194
Garigan D, Hsu AL, Fraser AG, Kamath RS, Ahringer J, Kenyon C (2002) Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat-shock factor and bacterial proliferation. Genetics 161(3):1101–1112
Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO (2000) Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11(12):4241–4257
Gems D (2015) The aging-disease false dichotomy: understanding senescence as pathology. Front Genet 6:212
Ghazi A, Henis-Korenblit S, Kenyon C (2009) A transcription elongation factor that links signals from the reproductive system to lifespan extension in Caenorhabditis elegans. PLoS Genet 5(9):e1000639
Gidalevitz T, Prahlad V, Morimoto RI (2011) The stress of protein misfolding: from single cells to multicellular organisms. Cold Spring Harb Perspect Biol 3(6):a009704. doi:10.1101/cshperspect.a009704
Girardot F, Lasbleiz C, Monnier V, Tricoire H (2006) Specific age-related signatures in Drosophila body parts transcriptome. BMC Genomics 7:69
Greer EL, Brunet A (2009) Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell 8(2):113–127
Guertin MJ, Lis JT (2013) Mechanisms by which transcription factors gain access to target sequence elements in chromatin. Curr Opin Genet Dev 23(2):116–123
Guettouche T, Boellmann F, Lane WS, Voellmy R (2005) Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem 6:4
Haithcock E, Dayani Y, Neufeld E, Zahand AJ, Feinstein N, Mattout A, Gruenbaum Y, Liu J (2005) Age-related changes of nuclear architecture in Caenorhabditis elegans. Proc Natl Acad Sci U S A 102(46):16690–16695
Hamer G, Matilainen O, Holmberg CI (2010) A photoconvertible reporter of the ubiquitin-proteasome system in vivo. Nat Methods 7(6):473–478
Hansen M, Hsu AL, Dillin A, Kenyon C (2005) New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet 1(1):119–128
Haynes CM, Fiorese CJ, Lin YF (2013) Evaluating and responding to mitochondrial dysfunction: the mitochondrial unfolded-protein response and beyond. Trends Cell Biol 23(7):311–318
Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13(2):89–102
Heydari AR, Wu B, Takahashi R, Strong R, Richardson A (1993) Expression of heat shock protein 70 is altered by age and diet at the level of transcription. Mol Cell Biol 13(5):2909–2918
Hietakangas V, Ahlskog JK, Jakobsson AM, Hellesuo M, Sahlberg NM, Holmberg CI, Mikhailov A, Palvimo JJ, Pirkkala L, Sistonen L (2003) Phosphorylation of serine 303 is a prerequisite for the stress-inducible SUMO modification of heat shock factor 1. Mol Cell Biol 23(8):2953–2968
Holmberg CI, Hietakangas V, Mikhailov A, Rantanen JO, Kallio M, Meinander A, Hellman J, Morrice N, MacKintosh C, Morimoto RI, Eriksson JE, Sistonen L (2001) Phosphorylation of serine 230 promotes inducible transcriptional activity of heat shock factor 1. EMBO J 20(14):3800–3810
Hsieh CC, Rosenblatt JI, Papaconstantinou J (2003) Age-associated changes in SAPK/JNK and p38 MAPK signaling in response to the generation of ROS by 3-nitropropionic acid. Mech Ageing Dev 124(6):733–746
Hsin H, Kenyon C (1999) Signals from the reproductive system regulate the lifespan of C. elegans. Nature 399(6734):362–366
Hsu AL, Murphy CT, Kenyon C (2003) Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300(5622):1142–1145
Inada H, Ito H, Satterlee J, Sengupta P, Matsumoto K, Mori I (2006) Identification of guanylyl cyclases that function in thermosensory neurons of Caenorhabditis elegans. Genetics 172(4):2239–2252
Keller JN, Huang FF, Markesbery WR (2000) Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience 98(1):149–156
Kenyon C (2010a) A pathway that links reproductive status to lifespan in Caenorhabditis elegans. Ann N Y Acad Sci 1204:156–162
Kenyon CJ (2010b) The genetics of ageing. Nature 464(7288):504–512
Kiffin R, Kaushik S, Zeng M, Bandyopadhyay U, Zhang C, Massey AC, Martinez-Vicente M, Cuervo AM (2007) Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J Cell Sci 120(Pt 5):782–791
Kirstein-Miles J, Scior A, Deuerling E, Morimoto RI (2013) The nascent polypeptide-associated complex is a key regulator of proteostasis. EMBO J 32(10):1451–1468
Kline MP, Morimoto RI (1997) Repression of the heat shock factor 1 transcriptional activation domain is modulated by constitutive phosphorylation. Mol Cell Biol 17(4):2107–2115
Knauf U, Newton EM, Kyriakis J, Kingston RE (1996) Repression of human heat shock factor 1 activity at control temperature by phosphorylation. Genes Dev 10(21):2782–2793
Labbadia J, Morimoto RI (2014) Proteostasis and longevity: when does aging really begin? F1000prime Rep 6:7
Labbadia J, Morimoto RI (2015a) The biology of proteostasis in aging and disease. Annu Rev Biochem 84:435–464
Labbadia J, Morimoto RI (2015b) Repression of the heat shock response is a programmed event at the onset of reproduction. Mol Cell 59(4):639–50
Labbadia J, Cunliffe H, Weiss A, Katsyuba E, Sathasivam K, Seredenina T, Woodman B, Moussaoui S, Frentzel S, Luthi-Carter R, Paganetti P, Bates GP (2011) Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J Clin Invest 121(8):3306–3319
Lapierre LR, Gelino S, Melendez A, Hansen M (2011) Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr Biol 21(18):1507–1514
Lee SJ, Kenyon C (2009) Regulation of the longevity response to temperature by thermosensory neurons in Caenorhabditis elegans. Curr Biol 19(9):715–722
Libina N, Berman JR, Kenyon C (2003) Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 115(4):489–502
Liu G, Rogers J, Murphy CT, Rongo C (2011) EGF signalling activates the ubiquitin proteasome system to modulate C. elegans lifespan. EMBO J 30(15):2990–3003
Mason JB, Cargill SL, Anderson GB, Carey JR (2009) Transplantation of young ovaries to old mice increased life span in transplant recipients. J Gerontol A Biol Sci Med Sci 64(12):1207–1211
McCay CM, Crowell MF, Maynard LA (1989) The effect of retarded growth upon the length of life span and upon the ultimate body size. Nutrition 5(3):155–171
Mori I, Sasakura H, Kuhara A (2007) Worm thermotaxis: a model system for analyzing thermosensation and neural plasticity. Curr Opin Neurobiol 17(6):712–719
Morimoto RI, Santoro MG (1998) Stress-inducible responses and heat shock proteins: new pharmacologic targets for cytoprotection. Nat Biotechnol 16(9):833–838
Morley JF, Morimoto RI (2004) Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell 15(2):657–664
Morley JF, Brignull HR, Weyers JJ, Morimoto RI (2002) The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A 99(16):10417–10422
Mouchiroud L, Molin L, Kasturi P, Triba MN, Dumas ME, Wilson MC, Halestrap AP, Roussel D, Masse I, Dalliere N, Segalat L, Billaud M, Solari F (2011) Pyruvate imbalance mediates metabolic reprogramming and mimics lifespan extension by dietary restriction in Caenorhabditis elegans. Aging Cell 10(1):39–54
Partridge L (2014) Intervening in ageing to prevent the diseases of ageing. Trends Endocrinol Metab 25(11):555–557
Prahlad V, Morimoto RI (2009) Integrating the stress response: lessons for neurodegenerative diseases from C. elegans. Trends Cell Biol 19(2):52–61
Prahlad V, Morimoto RI (2011) Neuronal circuitry regulates the response of Caenorhabditis elegans to misfolded proteins. Proc Natl Acad Sci U S A 108(34):14204–14209
Prahlad V, Cornelius T, Morimoto RI (2008) Regulation of the cellular heat shock response in Caenorhabditis elegans by thermosensory neurons. Science 320(5877):811–814
Rae R, Sinha A, Sommer RJ (2012) Genome-wide analysis of germline signaling genes regulating longevity and innate immunity in the nematode Pristionchus pacificus. PLoS Pathog 8(8):e1002864
Rahman MM, Sykiotis GP, Nishimura M, Bodmer R, Bohmann D (2013) Declining signal dependence of Nrf2-MafS-regulated gene expression correlates with aging phenotypes. Aging Cell 12(4):554–562
Raynes R, Leckey BD Jr, Nguyen K, Westerheide SD (2012) Heat shock and caloric restriction have a synergistic effect on the heat shock response in a sir2.1-dependent manner in Caenorhabditis elegans. J Biol Chem 287(34):29045–29053
Reis-Rodrigues P, Czerwieniec G, Peters TW, Evani US, Alavez S, Gaman EA, Vantipalli M, Mooney SD, Gibson BW, Lithgow GJ, Hughes RE (2012) Proteomic analysis of age-dependent changes in protein solubility identifies genes that modulate lifespan. Aging Cell 11(1):120–127
Riera CE, Dillin A (2015) Tipping the metabolic scales towards increased longevity in mammals. Nat Cell Biol 17(3):196–203
Shai N, Shemesh N, Ben-Zvi A (2014) Remodeling of proteostasis upon transition to adulthood is linked to reproduction onset. Curr Genomics 15(2):122–129
Shemesh N, Shai N, Ben-Zvi A (2013) Germline stem cell arrest inhibits the collapse of somatic proteostasis early in Caenorhabditis elegans adulthood. Aging Cell 12(5):814–822
Shi Y, Mosser DD, Morimoto RI (1998) Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev 12(5):654–666
Shopland LS, Hirayoshi K, Fernandes M, Lis JT (1995) HSF access to heat shock elements in vivo depends critically on promoter architecture defined by GAGA factor, TFIID, and RNA polymerase II binding sites. Genes Dev 9(22):2756–2769
Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD (2008) Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4(2):176–184
Sorger PK, Pelham HR (1988) Yeast heat shock factor is an essential DNA-binding protein that exhibits temperature-dependent phosphorylation. Cell 54(6):855–864
Sprang GK, Brown IR (1987) Selective induction of a heat shock gene in fibre tracts and cerebellar neurons of the rabbit brain detected by in situ hybridization. Brain Res 427(1):89–93
Stiernagle T (2006) Maintenance of C. elegans. WormBook 1–11
Sugi T, Nishida Y, Mori I (2011) Regulation of behavioral plasticity by systemic temperature signaling in Caenorhabditis elegans. Nat Neurosci 14(8):984–992
Sutphin GL, Kaeberlein M (2008) Dietary restriction by bacterial deprivation increases life span in wild-derived nematodes. Exp Gerontol 43(3):130–135
Tatum MC, Ooi FK, Chikka MR, Chauve L, Martinez-Velazquez LA, Steinbusch HW, Morimoto RI, Prahlad V (2015) Neuronal serotonin release triggers the heat shock response in C. elegans in the absence of temperature increase. Curr Biol 25(2):163–174
Taylor RC, Dillin A (2011) Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol 3(5):a004440. doi:10.1101/cshperspect.a004440
Taylor RC, Dillin A (2013) XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153(7):1435–1447
TeKippe M, Aballay A (2010) C. elegans germline-deficient mutants respond to pathogen infection using shared and distinct mechanisms. PLoS One 5(7):e11777
Tissenbaum HA (2012) Genetics, life span, health span, and the aging process in Caenorhabditis elegans. J Gerontol A Biol Sci Med Sci 67(5):503–510
Tonoki A, Kuranaga E, Tomioka T, Hamazaki J, Murata S, Tanaka K, Miura M (2009) Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process. Mol Cell Biol 29(4):1095–1106
Tsakiri EN, Sykiotis GP, Papassideri IS, Gorgoulis VG, Bohmann D, Trougakos IP (2013) Differential regulation of proteasome functionality in reproductive vs. somatic tissues of Drosophila during aging or oxidative stress. FASEB J 27(6):2407–2420
Twumasi-Boateng K, Wang TW, Tsai L, Lee KH, Salehpour A, Bhat S, Tan MW, Shapira M (2012) An age-dependent reversal in the protective capacities of JNK signaling shortens Caenorhabditis elegans lifespan. Aging Cell 11(4):659–667
van Oosten-Hawle P, Morimoto RI (2014) Transcellular chaperone signaling: an organismal strategy for integrated cell stress responses. J Exp Biol 217(Pt 1):129–136
van Oosten-Hawle P, Porter RS, Morimoto RI (2013) Regulation of organismal proteostasis by transcellular chaperone signaling. Cell 153(6):1366–1378
Van Raamsdonk JM, Hekimi S (2012) Superoxide dismutase is dispensable for normal animal lifespan. Proc Natl Acad Sci U S A 109(15):5785–5790
Vilchez D, Morantte I, Liu Z, Douglas PM, Merkwirth C, Rodrigues AP, Manning G, Dillin A (2012) RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature 489(7415):263–268
Volovik Y, Maman M, Dubnikov T, Bejerano-Sagie M, Joyce D, Kapernick EA, Cohen E, Dillin A (2012) Temporal requirements of heat shock factor-1 for longevity assurance. Aging Cell 11(3):491–499
Vora M, Shah M, Ostafi S, Onken B, Xue J, Ni JZ, Gu S, Driscoll M (2013) Deletion of microRNA-80 activates dietary restriction to extend C. elegans healthspan and lifespan. PLoS Genet 9(8):e1003737
Walther DM, Kasturi P, Zheng M, Pinkert S, Vecchi G, Ciryam P, Morimoto RI, Dobson CM, Vendruscolo M, Mann M, Hartl FU (2015) Widespread proteome remodeling and aggregation in aging C. elegans. Cell 161(4):919–932
Westerheide SD, Anckar J, Stevens SM Jr, Sistonen L, Morimoto RI (2009) Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 323(5917):1063–1066
Wu C (1995) Heat shock transcription factors: structure and regulation. Annu Rev Cell Dev Biol 11:441–469
Yao J, Munson KM, Webb WW, Lis JT (2006) Dynamics of heat shock factor association with native gene loci in living cells. Nature 442(7106):1050–1053
Yu S, Driscoll M (2011) EGF signaling comes of age: promotion of healthy aging in C. elegans. Exp Gerontol 46(2–3):129–134
Zelin E, Zhang Y, Toogun OA, Zhong S, Freeman BC (2012) The p23 molecular chaperone and GCN5 acetylase jointly modulate protein-DNA dynamics and open chromatin status. Mol Cell 48(3):459–470
Zhang M, Poplawski M, Yen K, Cheng H, Bloss E, Zhu X, Patel H, Mobbs CV (2009) Role of CBP and SATB-1 in aging, dietary restriction, and insulin-like signaling. PLoS Biol 7(11):e1000245
Zou J, Salminen WF, Roberts SM, Voellmy R (1998) Correlation between glutathione oxidation and trimerization of heat shock factor 1, an early step in stress induction of the Hsp response. Cell Stress Chaperones 3(2):130–141
Acknowledgments
A.B-Z. was supported by the Israeli Council for Higher Education Alon Fellowship, by a Marie Curie International Reintegration grant, and by grants from the Binational Science Foundation and the Israel Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this chapter
Cite this chapter
Shemesh, N., Ben-Zvi, A. (2016). HSF1 Regulation in Aging and Its Role in Longevity. In: Nakai, A. (eds) Heat Shock Factor. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55852-1_5
Download citation
DOI: https://doi.org/10.1007/978-4-431-55852-1_5
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55850-7
Online ISBN: 978-4-431-55852-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)