
Abstract
Heat-shock factors (HSFs) are a family of transcription factors whose name is derived from the discovery of their activation following heat shock. Of these, HSF1 is the most well studied and is the master transcriptional regulator of cellular responses to not only heat but also a wide variety of other stressors. During the progression of many types of cancer, HSF1 becomes co-opted by mechanisms that are evidently triggered in the emerging tumor cell. Concerted activation of HSF1 participates in many of the traits (initiation, promotion, proliferation, invasion, metabolism, anti-apoptosis, and inhibition of replicative senescence) that permit the malignant phenotype. Indeed, overexpression of HSF1 has been found in many organ tumor tissues and is associated with poor prognosis. In this chapter we describe (1) the biology of HSF1-associated cancers; (2) the role of HSF1 in cancer initiation, promotion, and progression; (3) cancer-related pathways regulated by HSF1; and (4) HSF1 and its targets as prognostic factors for cancer patients.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The mammalian heat-shock factor (HSF) family consists of four members (HSF1, HSF2, HSF3, and HSF4), with HSF1 being the main regulator of the heat-shock response, which is involved in protecting cells and organisms from heat, ischemia, inflammation, oxidative stress, and other noxious conditions (Pirkkala et al. 2001; Sorger 1991). Under various forms of physiological stress, HSF1 drives the production of heat-shock proteins (HSPs) such as HSP27, HSP70, and HSP90, which act as protein chaperones (Guertin and Lis 2010). The function of HSF1 is not limited to increasing the expression of chaperones; HSF1 also modulates the expression of hundreds of non-chaperone genes that are critical for survival under an array of potentially lethal stressors (Mendillo et al. 2012; Page et al. 2006). As a result, HSF1 influences fundamental cellular processes such as cell-cycle control, protein translation, glucose metabolism, and proliferation (Dai et al. 2007; Hayashida et al. 2006; Jacobs and Marnett 2007; Vydra et al. 2006). In human tumors, constitutive expression of HSP27, HSP70, and HSP90 at high levels predicts poor prognosis and resistance to therapy (Neckers and Workman 2012; Khalil et al. 2011). These effects are often attributable to HSF1-dependent mechanisms (Cai and Zhu 2003). Thus, as a master regulator of cellular processes, the roles of HSF1 in carcinogenesis and tumor progression are now emerging. Several recent investigations using mouse models have suggested that HSF1 is involved in carcinogenesis (Dai et al. 2007; Min et al. 2007). In clinical samples, HSF1 is often constitutively expressed at high levels in a variety of tumors. In this chapter we describe (1) the biology of HSF1-associated cancers; (2) the role of HSF1 in cancer initiation, promotion, and progression; (3) cancer-related pathways regulated by HSF1; and (4) HSF1 and its targets as prognostic factors of cancer patients.
2 Biology of HSF1-Associated Cancers
2.1 Cancer Cells Are Generally Exposed to Stressful Environments That Promote HSF1 Activity
In humans, tumorigenesis is a multistep process that includes genome instabilities (mutation and chromosomal deletion) and epigenetic changes (abnormal histone acetylation and DNA methylation). During tumorigenesis, mutated nonnative proteins are synthesized and deregulated and abnormal signal transduction activity is observed. Moreover, abnormal conditions observed within the tumor microenvironment include hypoxia, acidity, lack of nutrients, ATP depletion, and low glucose levels. Therefore, cancer cells are generally exposed to more stresses compared to normal cells (Hanahan and Weinberg 2011), and these stresses promote HSF1 activity.
HSF1 is the most well-studied HSF and has been found at elevated levels in tumors with high metastatic potential and is associated with poor prognosis. Nevertheless, HSF1 operates as a major multifaceted enhancer of tumorigenesis through the induction of classical heat-shock genes, as well as “nonclassical” targets (Fujimoto and Nakai 2010). In this chapter, we focus on describing the different physiological roles of HSF1 in tumorigenesis.
2.2 Oncogene Versus Non-oncogene Addiction
Cancer cells harbor vast numbers of genetic and epigenetic alterations, whereas a select few arise nonrandomly and drive the cancer phenotype. Among this category are activating mutations in oncogenes. Because many cancers require increased activity of these oncogenes for tumor initiation and maintenance, this dependence has been coined “oncogene addiction” (Weinstein 2002).
Despite the focus on oncogenes as targets of cancer therapeutics, there are solid genetic arguments, based on experimental evidence, for a larger class of non-oncogene drug targets. Many, if not all, tumor-promoting proteins can be rate limiting to tumor-promoting pathways and represent potential drug targets. Solimini (Solimini et al. 2007) termed this phenomenon “non-oncogene addiction” in reference to the increased dependence of cancer cells on the normal cellular functions of certain genes, which themselves are not classical oncogenes.
Cancer cells are known to exhibit high levels of reactive oxygen species, spontaneous DNA damage, and aneuploidy, each of which represents a form of cellular stress. These cellular stresses might burden the chaperone and proteasome pathways, thus compromising folding of essential cellular proteins and leading to the elevated levels of heat-shock proteins observed in many tumors (Whitesell and Lindquist 2005). Thus, cancer cells are highly dependent upon these general stress responses, which exemplify non-oncogene addiction.
2.3 HSF1 Is Implicated in the Hallmarks of Cancer
It has been suggested that there are key traits, described as “the hallmarks of cancer,” required for the emergence of a complete malignant phenotype (Hanahan and Weinberg 2011). HSF1 has been implicated in causing these hallmarks (Ciocca et al. 2013), specifically we describe five key traits: (1) maintenance of sustained proliferative signaling, (2) inhibition of replicative senescence, (3) activation of invasion and metastasis, (4) reprogramming of energy metabolism, and (5) resisting cell death (anti-apoptosis). In this chapter, we first describe the role of HSF1 in cancer initiation/promotion and subsequently describe the cancer-related pathways (pertaining to the five key traits) regulated by HSF1.
3 HSF Supports Cancer Initiation, Promotion, and Progression
3.1 HSF1 Plays a Role in Enabling the Initiation and Maintenance of Cancer
HSF1 by itself does not act as a classical oncogene or tumor suppressor. Neither induced overexpression nor knockout directly drives transformation. However, recent studies have reported that HSF1 supports the malignant phenotype by initiating and promoting oncogenic signal transduction pathways, proliferation, survival, protein synthesis, and glucose metabolism (Dai et al. 2012a; Whitesell and Lindquist 2009; Vydra et al. 2014). HSF1 is dispensable for viability and growth of non-transformed human cells, as well as in whole animals (Dai et al. 2007; Hunt et al. 2004; Meng et al. 2011). The specific dependence of cancer cells on HSF1 has been termed “non-oncogene addiction” (Solimini et al. 2007). HSF1 has itself been implicated in the promotion of tumorigenesis. Dai et al. (2007) found that HSF1 plays a major role in enabling the initiation and maintenance of cancer in several mouse tumor models. Knockdown of HSF1 inhibited cell viability in several cancer cell lines, whereas it had no effect on normal human fibroblasts. They concluded that HSF1 function helps maintain the growth and survival of human cancer cells.
HSF1 is also required for epidermal growth factor receptor-2 (HER2)-induced transformation of MCF-10A cells (Meng et al. 2010) and cooperates with HER2 to promote mammary tumorigenesis (Xi et al. 2012). In addition, HSF1 promotes lymphomas in p53-deficient mice (Min et al. 2007). These findings suggest that HSF1 plays a role in enabling the initiation and maintenance of cancer.
3.2 HSF1 Affects Oncogene-Induced Senescence
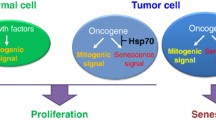
Cellular senescence was originally described as a limit to the number of divisions that a normal cell can undergo. Senescence can be triggered by telomere shortening or stressful treatments and is associated with activation of p53 and accumulation of the cell-cycle inhibitors p21 and/or p16 (Ben-Porath and Weinberg 2005). Although tumor cells are immortal and divide indefinitely, they often retain functional senescence programs and can become senescent in response to various DNA-damaging drugs and radiation (Chang et al. 2002), which is especially relevant to solid tumors of epithelial origin. Importantly, cell senescence can be triggered upon activation of oncogenes, e.g., Ras or Raf (Bihani et al. 2004; Lowe et al. 2004), which has been observed in various systems. Therefore, a novel concept has emerged whereby oncogene-induced senescence (OIS) represents the major block on the path toward neoplastic transformation, and cells in emerging tumors must acquire adaptations/mutations that allow the bypass of senescence programs (Braig and Schmitt 2006). Gabai et al. (2009) have found that HSF1 plays an important role in the evasion of OIS (Gabai et al. 2009; Meng et al. 2010, 2011), which is critical in the early stages of neoplastic transformation. HSF1 is involved in prevention of the OIS caused by HER2, phosphoinositide 3-kinase catalytic subunit (PIK3CA), and Ras oncogenes (Fig. 13.1).

Schematic model of the role of HSF1 in cancer. Stressful environments in cancer cells promote HSF1 activity/overexpression. The HSF1 transcriptome regulates multiple traits associated with malignant transformation, tumorigenesis (proliferation, inhibition of replicative senescence, invasion and metastasis, energy metabolism, and resistance to cell death), and related molecule pathways. BAG3 Bcl-2-associated athanogene domain 3, HSP70 heat-shock protein 70, XAF-1 XIAP-associated factor 1, ERK extracellular signal-regulated kinase, MAPK mitogen-activated protein kinase, PI3K phosphatidylinositol 3-kinase, AMPK AMP-activated protein kinase, MTA1 metastasis-associated factor 1, FBXW7 F-box/WD40 repeat-containing protein 7
3.3 Progression
HSF1 supports cancer progression by promoting proliferation, invasion, and metastasis. These processes are described as “cancer-related pathways” in the next chapter.
4 Cancer-Related Pathways Regulated by HSF1
4.1 HSF1-Bound Genes Are Involved in Many Facets of Tumorigenesis
HSF1 binds genes that are involved in regulating the cell cycle, apoptosis, energy metabolism, and other processes. Mendillo et al. (2012) used chromatin immunoprecipitation coupled with massively parallel DNA sequencing (ChIP-Seq) to identify genes bound by HSF1. These authors defined a distinct genome-wide transcriptional program coordinated by HSF1 in malignancy. They established an “HSF1-cancer signature” of 456 genes that were bound by HSF1 near their transcription start sites and found that this HSF1 cancer program is active in several types of tumors and is strongly associated with metastasis and mortality (Mendillo et al. 2012).
4.2 Signaling Pathways of Cancer Alter HSF1 Activity
Dysregulation of signaling pathways in cancer could also drive posttranslational modifications to HSF1. Some of these will likely be shared with heat-shocked cells, while others will likely be unique to cancer. Indeed, it seems extremely likely that different mechanisms of activation will operate in different cancers. Several pathways activated in cancer have been reported to alter HSF1 activity, including the epidermal growth factor receptor (EGFR)/HER2 axis (Zhao et al. 2009), the RAS/mitogen-activated protein kinase (MAPK) (Stanhill et al. 2006), and the insulin growth factor system (Chiang et al. 2012). Additional modes of cancer-specific regulation might include epigenetic states common to cancer and proliferating cells and transcriptional coregulators.
4.3 Maintenance of Sustained Proliferative Signaling
HSF1 supports cancer cell proliferation via many signaling pathways. The MAPK and phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathways govern fundamental physiological processes, such as proliferation and survival (Baines and Molkentin 2005; Mullonkal and Toledo‐Pereyra 2007). The extracellular signal-regulated kinase (ERK)/MAPK and PI3K/Akt signaling pathways are frequently constitutively activated in many human cancers and are associated with carcinogenesis (Rho et al. 2011). Growth and survival of many cancer cells critically depend on aberrant signaling by the ERK/MAPK and PI3K/Akt pathways, which are also involved in intensive cross talk (Aksamitiene et al. 2012). Several investigations have reported on the role of HSF1 in supporting MAPK signaling (Dai et al. 2012b; O’Callaghan-Sunol and Sherman 2006; Chuma et al. 2014). Yang demonstrated that HSF1 promotes breast cancer cell proliferation by regulating the ERK/MAPK and PI3K/Akt signaling pathway (Yang et al. 2014). Tang showed MEK phosphorylate HSF1 (Ser326) and MEK guard proteome stability and inhibit tumor-suppressive amyloidogenesis via HSF1 (Tang et al. 2015) (Fig. 13.1).
One of the mechanisms by which Ras proteins influence cell growth is through the regulation of intracellular levels of reactive oxygen species (ROS) molecules that affect a variety of cellular processes including proliferation. Recently it was shown that repression of the sestrin 3 (SESN3) gene can play a critical role in Ras-induced ROS upregulation. Zamkova et al. (2013) found that Ras-induced repression of SESN3 expression and ROS upregulation is mediated via alterations in HSF1 transcriptional activity, thereby affecting proliferation.
4.4 Activation of Invasion and Metastasis Pathways
Cancer metastasis is a multistep process that involves cell detachment from the primary tumor, entry into the vascular or lymphatic system, dispersal through the circulation, and proliferation after extravasation in target organs (Poste and Fidler 1980). Many studies have examined the importance and mechanisms of invasion, one of the first steps in metastasis. Loss of cell–cell contact caused by self-regulation of E-cadherin (Hirohashi 1998) and increased cell motility or migration (Genda et al. 1999; Takamura et al. 2001) is a critical step in this process. There is a growing evidence that actin polymerization and the dynamic reorganization of the actin cytoskeleton play important roles in the regulation of cell migration (Chen et al. 2009). During the invasion process, tumor cells become detached from the substratum and each other and dispersed through the circulation where they are liberated from the signals associated with cell growth and cell survival generated by cell–substratum interaction (Meredith et al. 1993; Frisch and Francis 1994; Frisch and Ruoslahti 1997). Some cancer cells can survive without these signals, a process which is termed anchorage-independent growth (AIG).
O’Callaghan-Sunol and Sherman (2006) showed that immortalized mouse embryonic fibroblast cells (MEF) derived from HSF1−/− mice were deficient in both basal and EGF-induced migration. Fang et al. (2012) demonstrated the effects of HSF1 on hepatocellular carcinoma (HCC) cell migration and invasion using in vitro migration and invasion assays. Furthermore, induced expression of HSF1 can stimulate the reorganization of actin, leading to the formation of stress fiber-like structures in HCC cells, which can be ablated with HSF1 knockdown. These findings indicate that HSF1 promotes HCC cell migration and invasion, possibly by regulating actin reorganization.
Expression of active HSF1 promotes AIG in vitro, as well as metastases in animal models. Furthermore, active HSF1 was found to enhance cell motility, reduce the adherence of cells to a fibronectin-coated surface or a wound healing assay (Nakamura et al. 2014), and affect the actin cytoskeleton. Concerning the molecular mechanism of HSF1 in cancer invasion, Toma-Jonik et al. (2015) showed that HSF1-induced downregulation of the focal adhesion protein vinculin increased motility and reduced cellular adherence. Khaleque et al. (2008) showed that HSF1 could be involved in promoting cancer cell invasion and metastasis through its ability to associate with metastasis-associated factor 1 (MTA1) in mammary carcinoma cells activated by the transforming cytokine heregulin. MTA1 is a gene corepressor and may function to decrease expression of genes that inhibit invasion and metastasis (Mazumdar et al. 2001) (Fig. 13.1).
Protein stability controlled by the ubiquitin proteasome pathway is emerging in human cancer. FBXW7 (F-box/WD40 repeat-containing protein 7), a substrate-targeting subunit of the SCF (Skp1–Cul1–F-box) ubiquitin ligase complex (Skaar et al. 2013), targets several key regulators of proliferation, growth, and apoptosis for proteasomal degradation (Davis et al. 2014; Inuzuka et al. 2011). FBXW7 is mutated in a significant portion of diverse human cancers (Akhoondi et al. 2007). Kourtis et al. show that FBXW7 controls the stability of nuclear HSF1 and modulates the attenuation phase of the heat-shock response (Kourtis et al. 2015). Moreover, FBXW7 deficiency enhances the metastaticability of melanoma through HSF1 stabilization and alteration of the HSF1 malignant transcriptional program. Together, they concluded that a tumor suppressor, FBXW7, regulates heat-shock response and cancer cell stress response and metastatic potential through modification of HSF1.
These results suggest that HSF1 may be involved in the regulation of cancer metastasis.
4.5 HSF1 Regulates Anabolic Metabolism
Cancer cells undergo profound changes in metabolism involving a switch from oxidative phosphorylation to glycolysis, as first observed in the classical studies of Warburg (1956). This leads to elevated glucose consumption and an increased rate of metabolism in malignant cells (Gullino 1966). A role for HSF1 in this metabolic switch was suggested by the findings that glucose uptake in cancer cells is decreased in HSF1 knockout mice and that HSF1 is required for the expression of the glycolytic intermediate lactate dehydrogenase in cells expressing the oncogene HER2 (Dai et al. 2007; Zhao et al. 2009). HSF1 activation stimulates hepatocyte lipid biosynthesis and perpetuates chronic hepatic metabolic disease induced by carcinogens through the promotion of premalignant cell growth in a mouse model (Jin et al. 2011). Furthermore, inactivation of HSF1 impairs cancer progression, mitigating adverse effects of carcinogens on hepatic metabolism by enhancing insulin sensitivity and sensitizing activation of AMP-activated protein kinase (AMPK), an important regulator of energy homeostasis and inhibitor of lipid synthesis (Fig. 13.1). These studies imply that HSF1 has a central role in HCC development, as it modulates proteostasis and metabolic pathways by regulating access to two critical elements, glucose and lipids.
4.6 HSF1 Plays a Role in Apoptosis Pathways
HSF1 plays a role in regulating apoptosis in several cancer cell lines, including multiple myeloma (Heimberger et al. 2013), gastrointestinal cancer (Wang et al. 2006), colorectal cancer (Wang et al. 2006; Kim et al. 2013; Benderska et al. 2014), HCC (Chuma et al. 2014), and pancreatobiliary cancer cells (Dudeja et al. 2011). Silencing HSF1 is thought to reduce anti-apoptotic proteins and/or induce apoptosis-related protein. Dudeja et al. (2011) showed that downregulation of HSF1 expression in pancreatobiliary tumors induces annexin V, TdT-mediated dUTP nick end labeling (TUNEL) positivity, and caspase-3 activation, which is indicative of activation of a caspase-dependent apoptotic pathway.
HSF1 induces HSP70 (heat-shock protein 70) and BAG3 (Bcl-2-associated athanogene domain 3) protein. HSP70 and its co-chaperone BAG3 have been reported to protect cells from apoptosis by stabilizing anti-apoptotic Bcl-2 family proteins. Silencing of HSF1 suppresses HSP70 and BAG3, as well as significantly decreasing the amounts of BCL-2 and BCL-xL protein, thereby inducing apoptotic cell death (Jacobs and Marnett 2009; Kim et al. 2013; Chuma et al. 2014). Another report found that HSF1-mediated BAG3 expression attenuates apoptosis in 4-hydroxynonenal-treated colon cancer cells via stabilization of anti-apoptotic Bcl-2 proteins (Jacobs and Marnett 2009) (Fig. 13.1).
XAF1 (XIAP-associated factor 1) is an inhibitor of apoptosis interacting proteins that antagonizes the cytoprotective role of XIAP. The expression of XAF1 and HSF1 was negatively correlated in gastrointestinal cancer cell lines. Upregulation of HSF1 suppresses XAF1 expression, implicating the synergistic effects of HSP and inhibitors of apoptosis in cytoprotection under conditions of cellular stress (Wang et al. 2006).
Endogenous stress in cancer cells sustains the elevated expression levels of HSF1 and subsequently suppresses apoptosis-related proteins, implicating the synergistic effects of anti-apoptotic proteins.
On the other hand, there are reports that activation or overexpression of HSF1 leads to enhanced induction of apoptosis. Benderska et al. (2014) reported that transient overexpression of HSF1 protein accelerates apoptosis. Death-associated protein kinase (DAPK) is a serine-threonine kinase that functions in tumor suppression. The DAPK-HSF1 interaction is a positive feedback mechanism that stimulates tumor necrosis factor (TNF)-induced apoptosis in colorectal cancer cells. They showed that transient overexpression of HSF1 protein led to an increase in DAPK mRNA levels and consequently to increased apoptosis. Further studies are needed to clarify the involvement of HSF1 in apoptosis.
5 HSF and Its Targets as Prognostic Factors in Cancer Patients
The role of altered HSF1 expression in various cancers appears to be tissue specific (Table 13.1).
HSF1 is elevated in a number of cells and tissues from a variety of cancers, including breast carcinoma (Santagata et al. 2011; Mendillo et al. 2012; Gabai et al. 2012), endometrial carcinoma (Engerud et al. 2014), pancreatic cancer (Dudeja et al. 2011), prostate carcinoma (Hoang et al. 2000), oral squamous cell carcinoma (OSCC) (Ishiwata et al. 2012), and HCC (Fang et al. 2012; Zhang et al. 2013; Chuma et al. 2014; Li et al. 2014). Studies of breast carcinoma, endometrial carcinoma, and HCC have shown the upregulation of HSF1 in cancers and its association with increased aggressiveness and lower survival rates. High expression of HSF1 mRNA in human breast cancer was correlated with Elston grade, metastasis, and poor prognosis (Gabai et al. 2012). HSF1 protein expression has been investigated in large samples. Santagata et al. (2011) showed that HSF1 expression in breast cancer samples (1,841 participants) was associated with high histologic grade, larger tumor size, and nodal involvement, and that increased levels of HSF1 are closely correlated with poor prognosis in estrogen receptor (ER)-positive breast carcinomas. On the other hand, at the time of diagnosis, the majority of breast cancer patients have ER-positive tumors and early-stage disease (ER-positive/lymph node-negative tumors). These patients will experience a recurrence and might benefit from more aggressive treatment, but it is currently very difficult to identify them in advance. Mendillo et al. (2012) found that HSF1 expression was significantly associated with metastatic recurrence in women initially diagnosed with ER-positive/lymph node-negative tumors and showed association with high HSF1 protein expression and poor prognosis. In endometrial carcinoma, high expression of HSF1 protein is significantly associated with aggressive disease (histological type, high-grade metastatic nodes, and aneuploidy) and poor survival (Engerud et al. 2014). In OSCC, higher nuclear HSF1 expression was closely related to tumor size and histopathologic types (Ishiwata et al. 2012). However, the correlation between HSF1 expression and prognosis in OSCC was not described in this study. HSF1 expression in HCC has been associated with aggressive disease, tumor number, tumor size, poor differentiation, venous invasion, Edmondson grade, etc. Four studies demonstrated that high expression of HSF1 protein is significantly associated with poor survival (Fang et al. 2012; Zhang et al. 2013; Chuma et al. 2014; Li et al. 2014). These reports indicated that HSF1 is a prognostic factor for cancer patients, and targeting HSF1 represents a potentially attractive treatment strategy for several cancers.
6 Future Perspectives
HSF1 thus acts as multifaceted enhancer of tumorigenesis by regulating diverse functions that include initiation, promotion, proliferation, invasion, metabolism, apoptosis, and replicative senescence. Furthermore, overexpression of HSF1 in clinical samples is associated with aggressive tumor characteristics and poor prognosis. Therefore, HSF1 is an attractive potential target for cancer therapy. However, several factors must be considered when targeting HSF1 for therapeutics. Firstly, inhibition of HSF1 might, in parallel, accelerate neurodegenerative processes and aging. Indeed, HSF1 displays a protective effect against neurodegenerative diseases, partly through the induction of chaperones, such as HSP27, that inhibit protein aggregation (Lu et al. 2002). Therefore, in the development of new drugs that target HSF1, it would appear important that such compounds are not capable of crossing the blood-brain barrier, so that neurodegenerative risks are minimized. Secondly, it remains to be convincingly demonstrated whether HSF1 is a useful molecular marker for predicting which cancer types and patients in particular will most benefit from an individualized anticancer strategy. Thirdly, the challenge is to design the most appropriate approach to pharmacologically perturbing HSF1 in order to promote cancer cell death while minimizing adverse effects on normal cells. Future studies are needed to understand the mechanisms whereby the HSF1 transcriptional system is activated in cancer, which may permit rational approaches to therapies based on this target.
References
Akhoondi S, Sun D, von der Lehr N et al (2007) FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res 67:9006–9012
Aksamitiene E, Kiyatkin A, Kholodenko BN (2012) Cross‐talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: a fine balance. Biochem Soc Trans 40:139–146
Baines CP, Molkentin JD (2005) Stress signaling pathways that modulate cardiac myocyte apoptosis. J Mol Cell Cardiol 38:47–62
Benderska N, Ivanovska J, Rau TT et al (2014) DAPK-HSF1 interaction as a positive-feedback mechanism stimulating TNF-induced apoptosis in colorectal cancer cells. J Cell Sci 127:5273–5287
Ben-Porath I, Weinberg RA (2005) The signals and pathways activating cellular senescence. Int J Biochem Cell Biol 37:961–976
Bihani T, Mason DX, Jackson TJ et al (2004) Differential oncogenic ras signaling and senescence in tumor cells. Cell Cycle 3:1201–1207
Braig M, Schmitt CA (2006) Oncogene-induced senescence: putting the brakes on tumor development. Cancer Res 66:2881–2884
Cai L, Zhu JD (2003) The tumor-selective over-expression of the human Hsp70 gene is attributed to the aberrant controls at both initiation and elongation levels of transcription. Cell Res 13:93–109
Chang BD, Swift ME, Shen M et al (2002) Molecular determinants of terminal growth arrest induced in tumor cells by a chemotherapeutic agent. Proc Natl Acad Sci USA 99:389–394
Chen HF, Xie LD, Xu CS (2009) Role of heat shock protein 27 phosphorylation in migration of vascular smooth muscle cells. Mol Cell Biochem 327:1–6
Chiang WC, Ching TT, Lee HC et al (2012) HSF-1 regulators DDL-1/2 link insulin-like signaling to heat-shock responses and modulation of longevity. Cell 148:322–334
Chuma M, Sakamoto N, Nakai A et al (2014) Heat shock factor 1 accelerates hepatocellular carcinoma development by activating nuclear factor-κB/mitogen-activated protein kinase. Carcinogenesis 35:272–281
Ciocca DR, Arrigo AP, Calderwood SK (2013) Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: an update. Arch Toxicol 87:19–48
Dai C, Whitesell L, Rogers AB et al (2007) Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130:1005–1018
Dai C, Dai S, Cao J (2012a) Proteotoxic stress of cancer: implication of the heat-shock response in oncogenesis. J Cell Physiol 227:2982–2989
Dai C, Santagata S, Tang Z et al (2012b) Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J Clin Invest 122:3742–3754
Davis RJ, Welcker M, Clurman BE (2014) Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell 26:455–464
Dudeja V, Chugh RK, Sangwan V et al (2011) Prosurvival role of heat shock factor 1 in the pathogenesis of pancreatobiliary tumors. Am J Physiol Gastrointest Liver Physiol 300:948–955
Engerud H, Tangen IL, Berg A et al (2014) High level of HSF1 associates with aggressive endometrial carcinoma and suggests potential for HSP90 inhibitors. Br J Cancer 111:78–84
Fang F, Chang R, Yang L (2012) Heat shock factor 1 promotes invasion and metastasis of hepatocellular carcinoma in vitro and in vivo. Cancer 118:1782–1794
Frisch SM, Francis H (1994) Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol 124:619–626
Frisch SM, Ruoslahti E (1997) Integrins and anoikis. Curr Opin Cell Biol 9:701–706
Fujimoto M, Nakai A (2010) The heat shock factor family and adaptation to proteotoxic stress. FEBS J 277:4112–4125
Gabai VL, Yaglom JA, Waldman T et al (2009) Heat shock protein Hsp72 controls oncogene-induced senescence pathways in cancer cells. Mol Cell Biol 29:559–569
Gabai VL, Meng L, Kim G et al (2012) Heat shock transcription factor Hsf1 is involved in tumor progression via regulation of hypoxia-inducible factor 1 and RNA-binding protein HuR. Mol Cell Biol 32:929–940
Genda T, Sakamoto M, Ichida T et al (1999) Cell motility mediated by rho and Rho-associated protein kinase plays a critical role in intrahepatic metastasis of human hepatocellular carcinoma. Hepatology 30:1027–1036
Guertin MJ, Lis JT (2010) Chromatin landscape dictates HSF binding to target DNA elements. PLoS Genet 6(9):e1001114
Gullino PM (1966) The internal milieu of tumors. Prog Exp Tumor Res 8:1–25
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Hayashida N, Nouye S, Fujimoto M et al (2006) A novel HSF1-mediated death pathway that is suppressed by heat shock proteins. EMBO J 25:4773–4783
Heimberger T, Andrulis M, Riedel S et al (2013) The heat shock transcription factor 1 as a potential new therapeutic target in multiple myeloma. Br J Haematol 160:465–476
Hirohashi S (1998) Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am J Pathol 153:333–339
Hoang AT, Huang J, Rudra-Ganguly N et al (2000) A novel association between the human heat shock transcription factor 1 (HSF1) and prostate adenocarcinoma. Am J Pathol 156:857–864
Hunt CR, Dix DJ, Sharma GG et al (2004) Genomic instability and enhanced radiosensitivity in Hsp70.1- and Hsp70.3-deficient mice. Mol Cell Biol 24:899–911
Inuzuka H, Shaik S, Onoyama I et al (2011) SCF (FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471:104–109
Ishiwata J, Kasamatsu A, Sakuma K et al (2012) State of heat shock factor 1 expression as a putative diagnostic marker for oral squamous cell carcinoma. Int J Oncol 40:47–52
Jacobs AT, Marnett LJ (2007) Heat shock factor 1 attenuates 4-Hydroxynonenal-mediated apoptosis: critical role for heat shock protein 70 induction and stabilization of Bcl-XL. J Biol Chem 282:33412–33420
Jacobs AT, Marnett LJ (2009) HSF1-mediated BAG3 expression attenuates apoptosis in 4-hydroxynonenal-treated colon cancer cells via stabilization of anti-apoptotic Bcl-2 proteins. J Biol Chem 284:9176–9183
Jin X, Moskophidis D, Mivechi NF (2011) Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metab 14:91–103
Khaleque MA, Bharti A, Gong J et al (2008) Heat shock factor 1 represses estrogen-dependent transcription through association with MTA1. Oncogene 27:1886–1893
Khalil AA, Kabapy NF, Deraz SF et al (2011) Heat shock proteins in oncology: diagnostic biomarkers or therapeutic targets? Biochim Biophys Acta 1816:89–104
Kim JA, Kim Y, Kwon BM et al (2013) The natural compound cantharidin induces cancer cell death through inhibition of heat shock protein 70 (HSP70) and Bcl-2-associated athanogene domain 3 (BAG3) expression by blocking heat shock factor 1 (HSF1) binding to promoters. J Biol Chem 288:28713–28726
Kourtis N, Moubarak RS, Aranda-Orgilles B et al (2015) FBXW7 modulates cellular stress response and metastatic potential through HSF1 post-translational modification. Nat Cell Biol 17:322–332
Li S, Ma W, Fei T et al (2014) Upregulation of heat shock factor 1 transcription activity is associated with hepatocellular carcinoma progression. Mol Med Rep 10:2313–2321
Lowe SW, Cepero E, Evan G (2004) Intrinsic tumour suppression. Nature 432:307–315
Lu A, Ran R, Parmentier-Batteur S et al (2002) Geldanamycin induces heat shock proteins in brain and protects against focal cerebral ischemia. J Neurochem 81:355–364
Mazumdar A, Wang RA, Mishra SK et al (2001) Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nat Cell Biol 1:30–37
Mendillo ML, Santagata S, Koeva M et al (2012) HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150:549–562
Meng L, Gabai VL, Sherman MY (2010) Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene 29:5204–5213
Meng L, Hunt C, Yaglom JA et al (2011) Heat shock protein Hsp72 plays an essential role in Her2-induced mammary tumorigenesis. Oncogene 30:2836–2845
Meredith JE Jr, Fazeli B, Schwartz MA (1993) The extracellular matrix as a cell survival factor. Mol Biol Cell 4:953–961
Min JN, Huang L, Zimonjic DB et al (2007) Selective suppression of lymphomas by functional loss of Hsf1 in a p53-deficient mouse model for spontaneous tumors. Oncogene 26:5086–5097
Mullonkal CJ, Toledo‐Pereyra LH (2007) Akt in ischemia and reperfusion. J Invest Surg 20:195–203
Nakamura Y, Fujimoto M, Fukushima S et al (2014) Heat shock factor 1 is required for migration and invasion of human melanoma in vitro and in vivo. Cancer Lett 354:329–335
Neckers L, Workman P (2012) Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 18:64–76
O’Callaghan-Sunol C, Sherman MY (2006) Heat shock transcription factor (HSF1) plays a critical role in cell migration via maintaining MAP kinase signaling. Cell Cycle 13:1431–1437
Page TJ, Sikder D, Yang L (2006) Genome-wide analysis of human HSF1 signaling reveals a transcriptional program linked to cellular adaptation and survival. Mol Biosyst 2:627–639
Pirkkala L, Nykänen P, Sistonen L et al (2001) Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J 15:1118–1131
Poste G, Fidler IJ (1980) The pathogenesis of cancer metastasis. Nature 283:139–146
Rho O, Kim DJ, Kiguchi K et al (2011) Growth factor signaling pathways as targets for prevention of epithelial carcinogenesis. Mol Carcinog 50:264–279
Santagata S, Hu R, Lin NU et al (2011) High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc Natl Acad Sci USA 108:18378–18383
Skaar JR, Pagan JK, Pagano M (2013) Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol 14:369–381
Solimini NL, Luo J, Elledge SJ (2007) Non-oncogene addiction and the stress phenotype of cancer cells. Cell 130:986–988
Sorger PK (1991) Heat shock factor and the heat shock response. Cell 65:363–366
Stanhill A, Levin V, Hendel A et al (2006) Ha-ras(val12) induces HSP70b transcription via the HSE/HSF1 system, but HSP70b expression is suppressed in Ha-ras(val12)-transformed cells. Oncogene 25:1485–1495
Takamura M, Sakamoto M, Genda T et al (2001) Inhibition of intrahepatic metastasis of human hepatocellular carcinoma by Rho associated protein kinase inhibitor Y-27632. Hepatology 33:577–581
Tang Z, Dai S, He Y et al (2015) MEK guards proteome stability and inhibits tumor-suppressive amyloidogenesis via HSF1. Cell 160:729–744
Toma-Jonik A, Widlak W, Korfanty J et al (2015) Active heat shock transcription factor 1 supports migration of the melanoma cells via vinculin down-regulation. Cell Signal 2:394–401
Vydra N, Malusecka E, Jarzab M (2006) Spermatocyte-specific expression of constitutively active heat shock factor 1 induces HSP70i-resistant apoptosis in male germ cells. Cell Death Differ 13:212–222
Vydra N, Toma A, Widlak W (2014) Pleiotropic role of HSF1 in neoplastic transformation. Curr Cancer Drug Targets 14:144–155
Wang J, He H, Yu L et al (2006) HSF1 down-regulates XAF1 through transcriptional regulation. J Biol Chem 281:2451–2459
Warburg O (1956) On the origin of cancer cells. Science 123:309–314
Weinstein IB (2002) Cancer: addiction to oncogenes – the achilles heal of cancer. Science 297:63–64
Whitesell L, Lindquist SL (2005) HSP90 and the chaperoning of cancer. Nat Rev Cancer 5:761–772
Whitesell L, Lindquist S (2009) Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin Ther Targets 13:469–478
Xi C, Hu Y, Buckhaults P et al (2012) Heat shock factor Hsf1 cooperates with ErbB2 (Her2/Neu) protein to promote mammary tumorigenesis and metastasis. J Biol Chem 287:35646–35657
Yang X, Wang J, Liu S et al (2014) HSF1 and Sp1 regulate FUT4 gene expression and cell proliferation in breast cancer cells. J Cell Biochem 115:168–178
Zamkova M, Khromova N, Kopnin BP et al (2013) Ras-induced ROS upregulation affecting cell proliferation is connected with cell type-specific alterations of HSF1/SESN3/p21Cip1/WAF1 pathways. Cell Cycle 12:826–836
Zhang JB, Guo K, Sun HC et al (2013) Prognostic value of peritumoral heat-shock factor-1 in patients receiving resection of hepatocellular carcinoma. Br J Cancer 109:1648–1656
Zhao YH, Zhou M, Liu H et al (2009) Up regulation of lactate dehydrogenase a by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 28:3689–3701
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this chapter
Cite this chapter
Chuma, M. (2016). HSF Supports Cancer. In: Nakai, A. (eds) Heat Shock Factor. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55852-1_13
Download citation
DOI: https://doi.org/10.1007/978-4-431-55852-1_13
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55850-7
Online ISBN: 978-4-431-55852-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)