
Abstract
Caveolin-3, the principal scaffold protein in sarcolemmal caveolae, regulates signal transduction and vesicular trafficking. Dominant-negative mutations in the caveolin-3 gene (CAV3) cause autosomal dominant limb-girdle muscular dystrophy 1C (LGMD1C) and autosomal dominant rippling muscle disease (AD-RMD). Myostatin, a member of the muscle-specific transforming growth factor (TGF)-β family, negatively regulates muscle growth and volume. We recently showed that wild-type caveolin-3 binds to and inhibits TGF-β type I receptor (TβRI), thereby suppressing intracellular TGF-β signaling. In contrast, LGMD1C-causing mutant caveolin-3 activates TβRI, resulting in muscle atrophy. Recently, small-molecule compounds suppressing activation of TβRI, also known as activin receptor-like kinase 5 (ALK5), have been developed as anticancer agents. Oral administration of a TβRI inhibitor, Ki26894, ameliorates muscle atrophy and weakness in a caveolin-3-deficient LGMD1C mouse model. The therapeutic effect of Ki26894 is associated with a reduction in TGF-β signaling and an increase in the number of muscle precursor satellite cells. This suggests that the caveolin-3/TβRI signaling pathway plays an important role in the pathogenesis of LGMD1C and that it regulates skeletal muscle size by controlling the number of muscle precursor cells. Consequently, drugs that target the TGF-β pathway may have therapeutic potential for diseases characterized by muscle atrophy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
5.1 Mutations in the Caveolin-3 Gene Cause Autosomal Dominant Limb-Girdle Muscular Dystrophy 1C and Autosomal Dominant Rippling Muscle Disease
Caveolins, 18–24-kDa integral membrane proteins, play a crucial role in the formation of caveolae, flask-shaped pits in the plasma membrane, in terminally differentiated cells, such as adipocytes, endothelial cells, and skeletal muscle cells [1, 2]. Caveolin-1 and caveolin-2 are co-expressed and form hetero-oligomers in non-muscle cells, whereas caveolin-3 forms homo-oligomers in muscle cells [3, 4]. De novo synthesized caveolins assemble to form ~350-kDa oligomers in the endoplasmic reticulum (ER) and are subsequently targeted to the plasma membrane via the trans-Golgi network. Caveolins participate in diverse cellular processes, including vesicular trafficking, lipid metabolism, and signal transduction [1–4]. They bind to and regulate specific lipid and lipid-modified signaling proteins, including cholesterol, G-proteins, G-protein-coupled receptors, Src family kinases, Ha-Ras, and nitric oxide synthases [3, 4]. The interaction between caveolins and these molecules is mediated by a caveolin-binding motif on the target protein and a scaffolding domain in caveolins [5]. Numerous recent in vitro studies have demonstrated an important role of caveolins in regulating signaling by these various effectors. However, very few studies have examined the role of caveolins in signal transduction pathways in vivo [2, 3].
Mutations in the caveolin-3 gene (CAV3) have been identified in autosomal dominant limb-girdle muscular dystrophy 1C (LGMD1C) and autosomal dominant rippling muscle disease (AD-RMD) [6]. These mutations cause a significant reduction in sarcolemmal caveolin-3 protein levels and, to a lesser extent, mistargeting of the mutant protein to the Golgi complex from the sarcolemma [6–8]. The loss of caveolin-3 by mutations in CAV3 in LGMD1C/AD-RMD patients may also impact the function of caveolin-3-binding molecules. Indeed, we and another group have shown that the enzymatic activity of neuronal nitric oxide synthase, which is suppressed by caveolin-3 in vitro, is elevated in skeletal muscles in a transgenic mouse model of LGMD1C and in LGMD1C/AD-RMD patients [7, 8]. Cytokine-induced nitric oxide production is increased in C2C12 myoblast cells transfected with LGMD1C/AD-RMD-causing mutant caveolin-3, compared with cells transfected with wild-type caveolin-3 [8]. Src, a membrane tyrosine kinase that regulates the balance between cell survival and cell death, is highly activated and accumulates in the perinuclear region, instead of the plasma membrane, in cells transfected with mutant LGMD1C/AD-RMD-causing caveolin-3 [9]. Furthermore, muscle-specific phosphofructokinase, a glycolytic enzyme, is significantly reduced in cells transfected with LDMD1C/AD-RMD-causing mutant caveolin-3, likely because of enhanced ubiquitin-mediated proteasomal degradation [10]. Of note, dysferlin, a protein involved in skeletal muscle repair, is deficient in LGMD2B/Miyoshi myopathy and is mistargeted to the cytoplasm in skeletal muscle of LGMD1C/AD-RMD patients, probably due to misdelivery of mutant caveolin-3 to the trans-Golgi network [11]. We previously showed that the ER stress molecule GRP78 is upregulated in parallel with the gene dosage of mutant caveolin-3 (CAV3P104L) in skeletal muscle in a rodent model of LGMD1C [12]. Despite the recent insight into the function of wild-type and mutant caveolin-3, the pathogenetic mechanisms underlying LGMD1C/AD-RMD remain fully understood.
5.2 Myostatin is a Promising Therapeutic Target for Muscle Wasting Disorders
Myostatin, a muscle-specific TGF-β family member, was discovered as a negative regulator of muscle growth and muscle volume. Overexpression of myostatin causes severe muscle atrophy, whereas targeted disruption of the myostatin gene increases skeletal muscle mass in mice [13, 14]. Like most other TGF-β family members, myostatin is synthesized as a precursor that undergoes proteolytic processing to generate a peptide with an N-terminal prodomain and a biologically active C-terminal disulfide-linked dimer [15, 16] (Fig. 5.1a). In the circulating inactive state, the N-terminal prodomain suppresses the biological activity of the C-terminal dimer [17, 18]. Once the prodomain is cleaved by BMP-1/tolloid-like proteases in the extracellular matrix, the C-terminal active dimer binds to and phosphorylates the activin receptor IIB/IIA (ActRIIB/IIA), a receptor type II serine/threonine kinase [18, 19]. This, in turn, recruits and phosphorylates activin receptor-like kinase 4/5 (ALK4/5), a receptor type I serine/threonine kinase, at the plasma membrane [16, 18–20]. The activation of a heteromeric complex of receptor type II and type I serine/threonine kinases results in the phosphorylation of intracellular effectors, including the receptor-regulated Smads (R-Smads), such as Smad2/3 [16, 21]. Phosphorylated R-Smads translocate to the nucleus from the cytoplasm, where they regulate the transcription of specific target genes leading to skeletal muscle atrophy [16]. In 2002, Khurana’s group reported that administration of a neutralizing antibody against myostatin ameliorated symptoms in dystrophin-deficient mdx mice, including muscle atrophy, muscle weakness, and dystrophic changes [22]. However, a clinical trial using a humanized neutralizing antibody against myostatin showed no significant improvement in muscle strength in patients with Becker-type muscular dystrophy (BMD), facioscapulohumeral muscular dystrophy (FSHD), or LGMD [23]. Recently, highly potent myostatin signal blockers have been developed for clinical application in patients with muscle wasting disorders. A clinical trial of an antibody against the receptor type II serine/threonine kinase is currently ongoing in patients with sporadic inclusion body myositis (sIBM). This antibody, named bimagrumab, was designated as a breakthrough therapy by the US Food and Drug Administration (FDA) in 2013 [24, 25].

Caveolins regulate myostatin and related TGF-β signaling at the type I receptor level. (a) Schematic representation of myostatin and related TGF-β signaling and caveolin-3. Type I serine/threonine receptor (R) controls intramuscular signaling by activating R-Smad (Smad2/3). Caveolin-3 suppresses the type I receptor. (b) In vitro autophosphorylation of constitutively active type I TGF-β receptors, ALK4 and ALK5. Cell lysates from COS-7 cells cotransfected with FLAG-tagged wild-type or mutant caveolin-3 (CAV3P104L) and HA-tagged constitutively active ALK4 (HA-caALK4) or ALK5 (HA-caALK5) were immunoprecipitated with anti-HA agarose. The in vitro kinase reaction was initiated by the addition of kinase reaction buffer and [γ-32P] ATP. Phosphorylated caALK4 and caALK5 were detected by autoradiography. Immunoprecipitated caALK4, caALK5, and caveolin-3 were analyzed by immunoblotting with anti-HA (α-HA) or anti-FLAG (α-FLAG) mAb. Bands corresponding to phosphorylated type I receptor (in vitro kinase assay), total type I receptor (α-HA), and caveolin-3 (α-FLAG) are shown
5.3 Ceveolin-3 Suppresses the Type I TGF-β Receptor: Relationship to the Pathogenesis of Caveolin-3-Deficient LGMD1C
Recently, caveolin-1 has drawn considerable attention as a critical regulator of TGF-β signaling. Caveolin-1 binds to and suppresses activation of the TGF-β1 receptor type I serine/threonine kinase (TβRI), which induces growth arrest in non-muscle cells [26]. Caveolin-1 facilitates ligand-bound TβRI internalization and ubiquitin-mediated degradation [27]. In addition, caveolin-1 interacts with type II and type I receptors for bone morphogenic proteins (BMPs) in vivo [28]. These findings indicate that caveolin-1 regulates TGF-β signaling at the receptor level.
Based on the role of caveolin-1 in non-muscle cells, we postulated that caveolin-3 likely inhibits myostatin and TGF-β signaling in muscle cells. Indeed, we found several caveolin-3 binding motifs in the cytoplasmic kinase domain of the TβRI [29]. We cotransfected caveolin-3 and a constitutively active TβRI in COS-7 monkey kidney cells (Fig. 5.1b). We found that caveolin-3 colocalized and co-immunoprecipitated with the TβRI. Furthermore, phosphorylation of the TβRI was decreased by wild-type caveolin-3 and, conversely, increased by the P104L disease-causing mutant caveolin-3 in vitro [3]. These results indicate that wild-type caveolin-3 inhibits activation of the TβRI, whereas mutant caveolin-3 enhances activation. Genetic introduction of the myostatin prodomain or administration of type II decoy receptor (ActRIIB-Fc) drastically rescues muscle atrophy in a LGMD1C mouse model (CAV3P104L). This therapeutic effect is associated with a decrease in TGF-β signaling, which is elevated in mutant mouse muscle [29]. Thus, caveolin-3 normally suppresses myostatin and TGF-β signaling at the type I receptor level, thereby preventing muscular atrophy. These findings suggest that excessive TGF-β signaling, triggered by caveolin-3 deficiency, could participate in the pathogenesis of muscular atrophy in LGMD1C.
5.4 TβRI Inhibitors Potentially Suppress Multiple TGF-β Signaling Pathways
Small-molecule compounds antagonizing TGF-β signaling at type I receptors (TβRIs) have recently been developed as targeted drugs for advanced cancer [30]. Tumor cells in advanced stages become refractory to TGF-β-induced growth arrest, but often overexpress TGF-β1, TGF-β2, and TGF-β3 [31]. TGF-β family members promote the epithelial–mesenchymal transition, immunosuppression, and angiogenesis, resulting in tumor growth and metastasis [31]. Each member of the TGF-β family binds to the type II receptor, which, in turn, recruits a TβRI. Seven different TβRIs—activin receptor-like kinases 1–7 (ALK1–7)—determine the intracellular signaling specificity of the 33 members of the TGF-β family. Small-molecule TβRI inhibitors were originally developed to compete with the binding of adenosine triphosphate (ATP) to the kinase domain of ALK5, the TβRI for TGF-β1–3 [15, 16]. TβRI inhibitors have been found to suppress tumor enlargement and metastasis in the advanced stages of cancer in animals [32, 33]. Notably, these inhibitors also suppress a similar kinase domain in ALK4, the type I receptor for activin, and potentially block ALK4/5, the type I receptor for myostatin [30]. However, the effects of TβRI inhibitors on activin, myostatin, and TGF-β1–3 signaling in muscle remain unclear.
5.5 A TβRI Inhibitor Ameliorates Muscle Atrophy and Weakness in a Rodent Model of Caveolin-3-Deficient LGMD1C by Impacting Muscle Precursor Cells
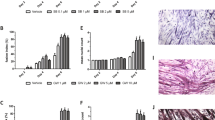
Myostatin suppresses the differentiation of C2C12 myoblasts exposed to low-serum conditions [34]. Using an efficient retrovirus-mediated gene transfer system [35], we assessed the effect of a TβRI inhibitor, Ki26894 [33], on the differentiation of C2C12 myoblasts expressing TGF-β1 or activin A, as well as myostatin (Fig. 5.2a) [36]. Transferring C2C12 myoblasts expressing an empty vector from high-serum (growth) to low-serum (differentiation) media caused them to fuse and form multinucleated myotubes. We then stained the cells with antibodies against myosin heavy chain (MyHC). Adding Ki26894 to the culture media enhanced myoblast fusion and myotube formation (Fig. 5.2a, green). In comparison, myotube formation was impaired in C2C12 myoblasts expressing myostatin, activin A, or TGF-β1 (Fig. 5.2a, blue), compared with controls harboring an empty vector. Notably, Ki26894 alleviated the impairment in myotube formation induced by the TGF-β family members (Fig. 5.2a, red). TβRI inhibitors enhance myoblast differentiation in vitro by suppressing the activity of several members of the anti-myogenic TGF-β family, including activin, TGF-β1, and myostatin. Ki26894 also prevented the impairment in myotube formation induced by LGMD-causing mutant caveolin-3 [36].

TβRI inhibitor Ki26894 (Ki) prevents muscular atrophy in a caveolin-3-deficient LGMD1C mouse model. (a) Ki26894 suppresses the impairment in myogenesis produced by myostatin, activin A, and TGF-β1. C2C12 myoblasts expressing an empty vector were grown in DMEM containing 10 % FBS (growth medium) and then differentiated in DMEM containing 2 % horse serum (differentiation medium), without (−) or with (+) 10 nM Ki26894 for 6 days. The cells were then stained with the muscle differentiation marker myosin heavy chain (MyHC). (b) Oral administration of Ki26894 ameliorates muscular atrophy. Appearance of de-skinned hind limb from 16-week-old mice treated without (−) or with (+) Ki26894. Scale bar, 5 mm. (c) Fluorescence images of satellite cells attached to single myofibers isolated from the EDL muscles of a rodent model of caveolin-3-deficient LGMD (CAV3P104L) at 16 weeks of age. The white arrow indicates caveolin-1-positive satellite cells
We orally administered Ki26894 to a rodent model of caveolin-3-deficient LGMD (CAV3P104L) from 6 to 16 weeks of age. Muscle atrophy was reduced in Ki26894-treated animals compared with untreated mice (Fig. 5.2b). Ki26894 increased muscle weight, muscle strength, and myofiber size, as well as centrally nucleated regenerative myofiber number in the mouse [36]. Furthermore, Ki26894 ameliorated the reduction in the number of satellite cells attached to isolated single myofibers in the mice (Fig. 5.2c). Ki26894 also increased muscle mass and strength and elevated the number of satellite cells, in wild-type littermate mice. These findings indicate that TβRI inhibitors strongly antagonize anti-myogenic TGF-β signaling and alleviate muscle atrophy.
5.6 Future Directions
As mentioned above, caveolin-3 regulates skeletal muscle volume by suppressing multiple anti-myogenic TGF-β signaling pathways. Pharmacological intervention strategies based on TβRI inhibitors are expected to prevent the progression of muscle weakness better than those based on inhibitors that suppress myostatin signaling alone. An increase in the number of satellite cells and the enhancement of myoblast differentiation induced by this type of inhibitor suggests that they may hold substantial therapeutic potential for the treatment of muscle atrophy in various diseases, including muscular dystrophies, sarcopenia, and cancer cachexia. To evaluate their effect on muscle performance, TβRI inhibitors should be tested in large-animal models of muscular atrophy. However, much additional study is required before clinical application of these compounds. For example, it is necessary to define the optimal, but nontoxic, dosage of these drugs for the treatment of individual patients with muscular atrophy. To facilitate this, we have developed an ex vivo myostatin activity assay as a convenient real-time biomonitoring system to determine how myostatin signaling is affected by the administration of these drugs in individual patients [36]. Furthermore, novel delivery systems targeting skeletal muscles also require development before the full potential of these drugs can be realized.
References
Parton RG (2003) Caveolae-from ultrastructure to molecular mechanisms. Nat Rev Mol Cell Biol 4:162–167
Parton RG, Simons K (2007) The multiple faces of caveolae. Nat Rev Mol Cell Biol 8:185–194
Razani B, Schlegel A, Lisanti MP (2000) Caveolin proteins in signaling, oncogenic transformation and muscular dystrophy. J Cell Sci 113:2103–2109
Galbiati F, Razani B, Lisanti MP (2001) Emerging themes in lipid rafts and caveolae. Cell 106:403–411
Couet J, Li S, Okamoto T, Ikezu T et al (1997) Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem 272:6525–6533
Minetti C, Sotgia F, Bruno C et al (1998) Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet 18:365–368
Sunada Y, Ohi H, Hase A et al (2001) Transgenic mice expressing mutant caveolin-3 show severe myopathy associated with increased nNOS activity. Hum Mol Genet 10:173–178
Kubisch C, Schoser BG, von Düring M et al (2003) Homozygous mutations in caveolin-3 cause a severe form of rippling muscle disease. Ann Neurol 53:512–520
Smythe GM, Eby JC, Disatnik MH et al (2003) A caveolin-3 mutant that causes limb girdle muscular dystrophy type lC disrupts Src localization and activity and induces apoptosis in skeletal myotubes. J Cell Sci 116:4739–4749
Sotgia F, Bonuccelli G, Minetti C et al (2003) Phosphofructokinase muscle-specific isoform requires caveolin-3 expression for plasma membrane recruitment and caveolar targeting: implications for the pathogenesis of caveolin-related muscle diseases. Am J Pathol 163:2619–2634
Matsuda C, Hayashi YK, Ogawa M et al (2001) The sarcolemmal proteins dysferlin and caveolin-3 interact in skeletal muscle. Hum Mol Genet 10:1761–1766
Kuga A, Ohsawa Y, Okada T et al (2011) Endoplasmic reticulum stress response in P104L mutant caveolin-3 transgenic mice. Hum Mol Genet 20:2975–2983
Zimmers TA, Davies MV, Koniaris LG et al (2002) Induction of cachexia in mice by systemically administered myostatin. Science 296:1486–1488
McPherron AC, Lawler AM, Lee SJ (1997) Regulation of skeletal muscle mass in mice by a new TGFbeta superfamily member. Nature 387:83–90
Massagué J (1998) TGF-beta signal transduction. Annu Rev Biochem 67:753–791
Tsuchida K (2004) Activins, myostatin and related TGF-beta family members as novel therapeutic targets for endocrine, metabolic and immune disorders. Curr Drug Targets Immune Endocr Metabol Disord 4:157–166
Hill JJ, Davies MV, Pearson AA et al (2002) The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J Biol Chem 277:40735–40741
Lee SJ, McPherron AC (2001) Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci U S A 98:9306–9311
Wolfman NM, McPherron AC, Pappano WN et al (2003) Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc Natl Acad Sci U S A 100:15842–15846
Rebbapragada A, Benchabane H, Wrana JL et al (2003) Myostatin signals through a transforming growth factor beta-like signaling pathway to block adipogenesis. Mol Cell Biol 23:7230–7242
Moustakas A, Souchelnytskyi S, Heldin CH (2001) Smad regulation in TGF-beta signal transduction. J Cell Sci 114:4359–4369
Bogdanovich S, Krag TO, Barton ER et al (2002) Functional improvement of dystrophic muscle by myostatin blockade. Nature 420:418–421
Wagner KR, Fleckenstein JL, Amato AA et al (2008) A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol 63:561–571
Lach-Trifilieff E, Minetti GC, Sheppard K et al (2014) An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol 34:606–618
Razani B, Zhang XL, Bitzer M et al (2001) Caveolin-1 regulates transforming growth factor (TGF) -beta/SMAD signaling through an interaction with the TGF-beta type I receptor. J Biol Chem 276:6727–6738
Le Roy C, Wrana JL (2005) Clathrin- and non-clathrin-mediated endocytic regulation of cell signaling. Nat Rev Mol Cell Biol 6:112–126
Nohe A, Keating E, Underhill TM et al (2005) Dynamics and interaction of caveolin-l isoforms with BMP-receptors. J Cell Sci 118:643–650
Ohsawa Y, Hagiwara H, Nakatani M et al (2006) Muscular atrophy of caveolin-3-deficient mice is rescued by myostatin inhibition. J Clin Invest 116:2924–2934
Yingling JM, Blanchard KL, Sawyer JS (2004) Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov 3:1011–1022
Derynck R, Akhurst RJ, Balmain A (2001) TGF-beta signaling in tumor suppression and cancer progression. Nat Genet 29:117–129
Uhl M, Aulwurm S, Wischhusen J et al (2004) SD-208, a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res 64:7954–7961
Kano MR, Bae Y, Iwata C et al (2007) Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc Natl Acad Sci U S A 104:3460–3465
Rios R, Fernández-Nocelos S, Carneiro I et al (2004) Differential response to exogenous and endogenous myostatin in myoblasts suggests that myostatin acts as an autocrine factor in vivo. Endocrinology 145:2795–2803
Kitamura T, Koshino Y, Shibata F et al (2003) Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp Hematol 31:1007–1014
Ohsawa Y, Okada T, Nishimatsu S et al (2012) An inhibitor of transforming growth factor beta type I receptor ameliorates muscle atrophy in a mouse model of caveolin 3-deficient muscular dystrophy. Lab Invest 92:1100–1114
Acknowledgment
We are grateful to all collaborators involved in developing anti-myostatin therapeutics and to T. Kenmotsu, N. Naoe, M Harada, M. Kimura, F Uemura, E Sugita, and K. Tanda (Department of Neurology, Kawasaki Medical School) for providing technical assistance. This work was supported by a research grant for Neurological and Psychiatric Disorders from the National Center of Neurology and Psychiatry (23–5, 26–8) and Welfare of Japan and from the Japan Society for the Promotion of Science (JST, AS24212769) and by research project grants from Kawasaki Medical School (23-T1, 26-T1).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this chapter
Cite this chapter
Ohsawa, Y., Nishimatsu, Si., Fujino, M., Sunada, Y. (2016). Targeting the Type I TGF-β Receptor for Treating Caveolin-3-Deficient Autosomal Dominant Limb-Girdle Muscular Dystrophy Type 1C and Muscle Wasting Disorders. In: Takeda, S., Miyagoe-Suzuki, Y., Mori-Yoshimura, M. (eds) Translational Research in Muscular Dystrophy. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55678-7_5
Download citation
DOI: https://doi.org/10.1007/978-4-431-55678-7_5
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55677-0
Online ISBN: 978-4-431-55678-7
eBook Packages: MedicineMedicine (R0)