
Abstract
Myotonic dystrophy (DM) is the most common form of muscular dystrophy in adults, caused by unstable genomic expansions of simple tandem repeats. Myotonic dystrophy type 1 (DM1) results from expansion of a CTG repeat in the 3′ untranslated region of DMPK. In myotonic dystrophy type 2 (DM2), the expanded repeat is a CCTG tetramer in intron 1 of CNBP/ZNF9. The mRNA transcripts containing expanded repeats form ribonuclear inclusions, thereby retained in the nucleus. The mutant RNA gives rise to a toxic gain of function by perturbing splicing factors, resulting in misregulation of alternative pre-mRNA splicing that may underlie the multisystemic symptoms of DM. Although no curative treatment exists, recent advances in basic and translational research and pharmacological approaches provide clues for therapeutic intervention in DM. In this review, we describe the RNA-dominant mechanism in DM and summarize potential therapeutic approaches that address RNA toxicity and current progress toward translational research.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
3.1 Myotonic Dystrophy
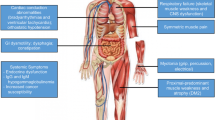
Myotonic dystrophy (DM) is the most common type of muscular dystrophy in adults, with a prevalence of 1 per 8,000 individuals [1]. DM is a dominantly inherited multisystemic disorder caused by the expansion of CTG (myotonic dystrophy type 1, DM1) or CCTG (myotonic dystrophy type 2, DM2) repeats [2, 3]. The common molecular basis of DM1 and DM2 is a toxic RNA transcript containing the expanded repeats. DM is the first disease proven to result from by toxic gain of function of the mutant mRNA and is now recognized as an RNA-mediated disease [4]. The clinical expression of both types of DM is quite similar, including myotonia, progressive muscle weakness, cataracts, insulin resistance, and cardiac conduction defects, although some differences exist between DM1 and DM2 (Fig. 3.1).

Multisystemic symptoms of DM
3.2 Clinical Features
3.2.1 Adult-Onset DM1
3.2.1.1 Muscle Weakness
The predominant symptom of adult-onset DM1 is facial and distal muscle weakness. Facial weakness is an early feature and atrophy of temporalis and masseter leads to the characteristic facial appearance, so-called hatchet face. Mild ptosis is often seen, but ophthalmoplegia is rare. The neck flexors and sternocleidomastoid muscles are commonly involved. Weakness in the limbs is initially distal, especially finger and wrist flexors and ankle dorsiflexors, leading to difficulty with opening caps and foot drop. Muscle weakness progresses gradually and proximal limb weakness occurs later in the disease course, leading to loss of ambulation. Swallowing and respiratory muscles are often involved in later disease stages, with resultant difficulty in swallowing and breathing.
3.2.1.2 Myotonia
Myotonia, defined as delayed muscle relaxation following voluntary contraction or mechanical percussion, is caused by abnormal muscle fiber membrane activity [5]. Grip myotonia, which is difficulty in relaxing the grip, is often observed in adult-onset DM1. Improvement in myotonia by repeated contractions is termed “warm-up phenomenon.” Myotonia can be elicited by percussion of the thenar or tongue. Myotonia can be recorded with electromyography (EMG) as bursts of repetitive muscle fiber discharges, giving a characteristic sound called “dive bomber” or “motorcycle.”
3.2.1.3 Cardiac Involvement
Cardiac involvement is the second most frequent cause of death in DM1 [6]. Conduction disturbance is quite common, such as first-degree atrioventricular (AV) block and QRS widening, sometimes progressing to third-degree AV block. Atrial tachyarrhythmia and fatal ventricular tachyarrhythmia can occur. Regular ECG monitoring and early implantation of a pacemaker or cardiac defibrillator are critical for the prevention of sudden unexpected death. In contrast to cardiac arrhythmia, cardiomyopathy is not common, though some DM1 patients suffer from severe dilated cardiomyopathy and heart failure.
3.2.1.4 CNS Involvement
Central nervous system (CNS) involvement is frequently seen in adult-onset DM1. The characteristic neuropsychiatric features are mild cognitive impairment, inertia, apathy, and reduced perception of disease symptoms. Hypersomnia is extremely common even in the absence of respiratory involvement. Polysomnography shows nonobstructive apneas and central hypoventilation. Brain MRI scan frequently shows diffuse white matter lesions, particularly in the frontal or anterior temporal lobes.
3.2.1.5 Endocrine Involvement
Although overt diabetes is not frequent, hyperinsulinemia and insulin resistance are common in adult-onset DM1. Hypothyroidism, dyslipidemia, and male infertility because of testicular atrophy are also seen.
3.2.1.6 Other Multisystemic Symptoms
In addition to striated muscle, smooth muscle is affected in DM1. Gastrointestinal problems, such as diarrhea and constipation, are particularly frequent and may lead to pseudo-obstruction and megacolon. Cholecystitis and gallstones are also common because of increased tone of the gallbladder sphincter. Cataracts (typically iridescent posterior subcapsular forms) develop in most adult-onset DM1 patients and are sometimes the only symptom of very late-onset mild DM1. Early frontal baldness is often seen in male and even in some female DM1 patients. Increased risks of cancers of endometrium, ovary, thyroid, and colon have been reported [7].
3.2.2 Congenital DM1
Congenital DM1 (CDM) is not merely a severe early form of DM1 but rather a distinct clinical phenotype. CDM patients are severely affected at birth with hypotonia and generalized muscle weakness resulting in difficulty of sucking, swallowing, and breathing. Some CDM patients present symptoms even in utero, such as reduced fetal movements and hydramnios in later pregnancy. Talipes and tented upper lip (so-called carp mouth) are also seen in CDM. Despite neonatal intensive support, the mortality from respiratory failure remains high in affected infants. Surviving infants gradually improve in motor function, but nevertheless show delayed motor and mental development. Children with CDM can achieve independent walking, but develop myotonia and progressive muscle weakness in early adulthood. CDM is almost exclusively maternally inherited, even from mothers with mild forms of DM (see Sect. 3.3.2).
3.2.3 DM2
Although DM1 is common in populations of European and Asian descent, most DM2 patients are of northern and eastern European descent. DM2 patients also present myotonia and muscle weakness; however, the weakness typically affects the proximal muscles including the neck, elbow extension, and hip flexors. Significant muscle pain and fatigue are common in DM2. Cardiac conduction defects, cataracts, and insulin resistance are seen, whereas cognitive manifestation is rare in DM2. In contrast to DM1, a severe congenital form of DM2 has not been reported to date.
3.3 Genetics
3.3.1 Repeat Expansion in DM1 and DM2
Short DNA tandem repeats are observed ubiquitously across the human genome [8]. A small fraction of these repetitive elements are prone to become mutable and highly expanded. Such genomic expansions of simple tandem repeats cause various neurogenetic disorders, including DM. DM1 is caused by an expansion of CTG repeats in the 3′ untranslated region (UTR) of the DMPK gene [2] (Fig. 3.2). The length of CTG repeats is between 50 and several thousand in DM1, whereas the normal repeat number is between 5 and 37. In general, the length of expanded CTG repeats in blood cells correlates with the age of onset in DM1 [9]. DM2 results from a similar expansion of CCTG repeats in the first intron of the CNBP/ZNF9 gene [3] (Fig. 3.2). The expanded CCTG repeat in DM2 is much longer than that in DM1, ranging from 75 to over 11,000. Importantly, unlike that seen in DM1, the size of expanded CCTG repeats does not correlate with the age of onset in DM2.

Location of unstable CTG (DM1) or CCTG (DM2) repeats within their respective genes
3.3.2 Repeat Instability
The expanded CTG and CCTG repeats in DM are not stable [10]. These mutations exhibit an exceptional degree of genetic instability in germline and somatic cells. For instance, the size of the repeat is prone to change during transmission from one generation to the next. In fact, intergenerational increments in the order of hundreds of repeats are often seen in DM1 (intergenerational instability). These mutations are also unstable in somatic cells, leading to an age-dependent growth of repeat expansion during the life of an individual (somatic instability).
3.3.2.1 Intergenerational Instability
The expanded CTG repeats in DM1 often increase in size when transmitted from parent to offspring, resulting in earlier onset and more severe clinical symptoms in subsequent generations, a phenomenon called “anticipation.” A maternal expansion bias is evident in DM1 and cases with CDM are almost exclusively maternal in origin. There are a few cases of repeat contraction through paternal transmission [11]. In contrast to DM1, anticipation is less evident, both clinically and genetically, in DM2.
3.3.2.2 Somatic Instability
The expansion process continues throughout life in somatic cells, at rates that are variable between tissues in DM. This can lead to over tenfold variations of expansion length in different tissues of a DM individual [12]. Progenitor alleles that are estimated in leukocytes in adult-onset DM1 are usually between 100 and 800 CTG repeat, whereas adult biopsy and autopsy samples show typically 2,000–6,000 CTG repeats in the muscle or heart. In DM2, the CCTG expansion length is also considerably longer in the muscle than in leukocytes from the same individual [13]. Since the heterogeneity in fetal tissues is much less extensive, somatic instability causing these highly expanded alleles occurs mainly during postnatal life. The functional significance of somatic instability remains unclear; however, the progression of DM may be depending on the growth of the expanded repeat over time.
3.3.2.3 Mechanism of Repeat Instability
Instability of the expanded repeats is associated with DNA replication, repair, or transcription, which instigate separation of double-stranded DNA, thus promoting the formation of extrahelical slipped strand structures that are substrates for error-prone repair (Fig. 3.3a). Recent studies have shown that the effects of transcription on repeat stability are particularly pertinent for DM1 [14]. The predominance of transcription-induced instability in DM is reasonable as the organs most affected, such as the skeletal muscle, heart, and brain, are those with low rates of cell proliferation but the highest levels of DMPK expression. Recent studies also indicate a role for RNA repeats in transcription-induced repeat instability. RNAs comprised of expanded CUG or CCUG repeats bind to the template strand of DNA, then form RNA:DNA hybrids (R-loops) (Fig. 3.3b). The presence of R-loops triggers the formation of extrahelical looped-out structures on the non-template strand and thereby stimulates error-prone repair, leading to further expansion or contraction [15].

(a) Mechanism of repeat instability. Extrahelical slipped strand structures form in (CTG)•(CAG) repeats, resulting in error-prone processing by mismatch repair proteins. (b) R-loop formation at the DMPK locus. In DM1, expanded CUG RNA binds to the template strand of DNA, then forms RNA:DNA hybrids (R-loops). The presence of R-loops instigates the formation of slipped strand structures on the non-template strand, thereby leading to further repeat instability. RNAP: RNA polymerase
3.3.3 Genotype-Phenotype Correlation
Although the size of expanded CTG repeats in leukocytes correlates with the age of onset in DM1, there was no correlation between muscle weakness and CTG expansion size in either leukocytes or muscle cells [16]. This suggests that some DM1 patients may tolerate longer expansions better than others and raises the possibility that DM1 severity is modulated by additional factors, such as modifier genes, sequence interruptions in the CTG repeat tract, or epigenetic changes at the DMPK locus. The CTG expansion length in the muscle also did not correlate with that in leukocyte from the same subjects [16, 17]. However, the difference in repeat size between leukocytes and muscle is correlated with age, suggesting an age-dependent process of somatic expansion that is more pronounced in the muscle than in leukocytes.
3.4 Pathomechanism
3.4.1 Unstable Repeat Expansion
More than 20 neurogenetic disorders are caused by unstable genomic expansions of simple tandem repeats [8]. Such unstable repeat expansion disorders fall in two categories according to the position of the repeat element within the mutant gene. In most cases the repeat tract is located in protein-coding regions, the motif is CAG, and the orientation in the reading frame produces expanded polyglutamine proteins, resulting in neurotoxicity (e.g., Huntington disease, spinocerebellar ataxia types 1, 2, 3, and 6). The second major group of unstable repeat expansion disorders results from repeats in non-protein-coding regions of genes, such as expanded CGG repeats in the 5′ UTR of FMR1 in fragile X syndrome (FXS) [18], GAA repeats in the first intron of FXN in Friedreich’s ataxia (FRDA) [19], and CTG/CCTG repeats in DM. In cases with FXS and FRDA, the critical effect of the expanded repeats is transcriptional silencing of FMR1 and FXN, respectively. However, there is no proven effect on transcription of DMPK or CNBP/ZNF9 in DM, indicating that the main pathogenic effect is a deleterious gain of function by the mutant mRNA.
3.4.2 RNA Dominance
Although the mutant mRNA is fully processed in DM1, repeat expansion blocks the nuclear export of mRNA with the expanded repeats of CUG (CUGexp) [20]. Thus, the mRNA containing expanded repeats is retained in the nucleus and accumulates in discrete foci. Since DM1 is an autosomal dominant disease, nuclear retention of the mutant mRNA could theoretically lead to a 50 % reduction in DMPK protein. However, several lines of evidence strongly indicate that symptoms of DM1 result not from haploinsufficiency but from the toxic gain of function by RNA transcripts containing the CUGexp. First, DMPK knockout mice, in which DMPK protein can be completely eliminated, show only minor symptoms in the skeletal muscle [21, 22]. Second, muscular features of DM1 can be reproduced by the expression of CUGexp RNA at the 3′ UTR of human skeletal actin transgene in a mouse model [23]. Finally, in DM2, the highly expanded CCUG repeat in CNBP/ZNF9 is fully transcribed, but the intron containing the CCUG expansion is retained and accumulated in nuclear foci similar to that seen in DM1 [3, 24].
3.4.3 RNA-Binding Proteins: MBNL Family and CELF1
The mutant RNA retained in the nucleus affects at least two RNA-binding protein families: the MBNL and CELF. MBNL1, a member of the MBNL family protein, has a strong affinity for CUGexp and CCUGexp [25]. When CUGexp or CCUGexp RNA accumulates in the nucleus, MBNL1 is sequestered in nuclear RNA foci and depleted from the nucleoplasm [26, 27] (Fig. 3.4). Since MBNL1 is a regulator of alternative splicing [28], MBNL1 sequestration in foci leads to the misregulation of alternative splicing of its target exons. One clear example of the functional consequence in DM is the splicing misregulation of CLCN1 [29, 30]. This gene encodes a chloride ion channel that stabilizes the transmembrane potential in skeletal muscle. When alternative splicing of CLCN1 is misregulated in response to expression of CUGexp RNA, the channel activity is lost, causing electrical instability of the membrane, which triggers repetitive action potentials and delay of relaxation (myotonia). The loss of MBNL1 has additional effects on gene expression, including transcriptional alterations, mRNA decay, transport of mRNA, and micro-RNA biogenesis [31–33]. Mbnl1 knockout mice exhibit myotonia and splicing alteration of Clcn1, supporting the evidence for MBNL1 dysfunction in DM1 [34]. Two other members of the MBNL family, MBNL2 and MBNL3, may also be involved in DM [34, 35]. Similar to MBNL1, MBNL2 is expressed in the skeletal muscle, heart, and brain, whereas MBNL3 is expressed mainly in the placenta. MBNL2 and MBNL3 are closely related to MBNL1 and can also be sequestered in nuclear foci. These suggest a role for the combinatorial deficiency of all three MBNL family members in DM, depending on the expression of CUGexp or CCUGexp RNA relative to the distinct levels of MBNL proteins in different tissues. Indeed, a recent report showed compound loss of MBNL1 and MBNL2 functions exaggerated cardiac and skeletal muscle symptoms in a mouse model [36].
Expression of CUGexp also causes perturbations in cell signaling via several kinases, including protein kinase C (PKC), Akt kinase, cyclin-dependent kinase 4 (CDK4), and glycogen synthase kinase 3 beta (GSK3B) [37–39]. While the mechanisms of activation of these pathways have not yet been determined, upregulation of PKC enhances the phosphorylation of CELF1, a member of CELF RNA-binding protein family. The phosphorylation of CELF1 extends its half-life, resulting in the upregulation of its steady-state level [37] (Fig. 3.4). Since CELF1 is a multifunctional RNA-binding protein involved in the regulation of alternative splicing and translation, upregulation of CELF1 protein in DM1 results in misregulated alternative splicing of several pre-mRNAs, as well as altered translation or decay of mRNAs that are bound by CELF1 [40–43]. Transgenic mice overexpressing Celf1 show splicing misregulation and cardiac conduction defect similar to DM1 [44]. As CELF1 plays an important role in developmental splicing switch in the heart, increased CELF1 expression may underlie the cardiac pathology observed in DM1. However, CELF1 upregulation has not been confirmed in DM2 [27, 45].
3.4.4 Spliceopathy in DM
RNA splicing is the mechanism by which intervening introns are removed, while functional exons are ligated together to form a mature mRNA transcript [46]. There are two types of RNA splicing: constitutive and alternative splicing. Alternative splicing serves to generate a wide variety of unique mRNA transcripts by selecting and pairing discrete exons, resulting in distinct but similar proteins from the same gene (Fig. 3.5).

Patterns of alternative splicing. Constitutive exons present in all final mRNAs are indicated by white boxes. Alternative RNA segments that may or may not be included in the final mRNA are indicated by gray boxes
Although many human genetic disorders have been identified as the consequences of mutations on RNA splicing, in almost all cases, these are cis-acting effects that affect the splicing of a single pre-mRNA. In contrast, DM is caused by a trans-effect of alternative splicing factors on many RNAs and now is recognized as the first example of a human genetic disease resulting from “spliceopathy” [4]. The spliceopathy in DM shares common features in splicing misregulation. First, it does not involve constitutive exons, but selectively affects a group of exons that are subject to alternative splicing. Second, it usually affects alternative exons that normally undergo a developmental splicing switch; therefore, the fetal or neonatal splicing isoforms are expressed in mature muscle fibers in DM1. Third, most target exons in the spliceopathy in DM are regulated by MBNL1, CELF1, or both.
More than 70 mis-splicing events have been reported in DM. Table 3.1 lists the transcripts known to be affected by spliceopathy in DM. In case of CLCN1, the exon 7a is negatively regulated by MBNL1 [34]. When MBNL1 is depleted in DM muscle, the exon 7a is retained in the mRNA. The inclusion of exon 7a induces a premature termination codon, which causes loss of mature CLCN1 protein in muscle membrane in DM, resulting in myotonia [29]. Misregulated splicing of insulin receptor (INSR) is also reported in DM muscle [41]. Alternative splicing of exon 11 is an important factor dictating the function of this receptor. The receptor lacking the exon 11 (non-muscle form) has a higher affinity for insulin than the receptor including the exon (muscle form). Because the inclusion of exon 11 is negatively regulated by CELF1, the non-muscle form lacking exon 11 is increased in DM muscle, leading to insulin resistance. However, whereas numerous mis-splicing events have been identified in DM, the underlying cause of progressive muscle wasting, the main disabling symptom in DM, has not been well defined. One possible candidate is mis-splicing of a group of genes regulating Ca2+ homeostasis, such as ryanodine receptor (RYR1), sarcoplasmic/endoplasmic reticulum calcium ATPases (SERCAs), voltage-dependent Ca2+ channel (CACNA1S), and T-tubule membrane scaffolding protein (BIN1) [47–49]. Because Ca2+ concentration is increased in DM1 cells and impaired Ca2+ homeostasis triggers muscle damage, misregulated splicing of these transcripts may cause muscle wasting in DM [50]. Another candidate is splicing misregulation of genes encoding cytoskeletal proteins, such as dystrophin (DMD), dystrobrevin (DTNA), titin (TTN), and ZASP (LDB3), as mutations or deletions in these genes causes other types of muscular dystrophies and myopathies [27, 51, 52].
The spliceopathy in DM also involves the heart and brain. Although the cause of cardiac symptoms in DM is not well elucidated, mis-splicing of cardiac cytoskeletal proteins (cardiac troponin T, DMD, DTNA, TTN, and LDB3) may contribute to cardiomyopathy [51–54]. In addition, conduction block and arrhythmia in DM may be caused by mis-splicing of cardiac ion channels, such as K+ channel beta subunit (KCNAB1) and voltage-gated Na+ channel (SCN5A) [54] (Charlet-Berguerand & Takahashi, manuscript in preparation). In DM1 brain, several splicing misregulations have been reported, including Tau (MAPT), APP, and CAMK2D [26, 55]. However, neither the functional consequences of these mis-splicing events nor the cause of cognitive impairment in DM1 has been determined.
3.4.5 Other Possible Mechanisms
Besides MBNL and CELF1, other RNA-binding proteins, such as hnRNP H and staufen, are implicated in DM pathogenesis [56, 57]. Dysregulation of micro-RNAs and MEF2 transcription factor proteins are also suggested to contribute to cardiac symptoms in DM [33, 58]. Furthermore, repeat-associated non-ATG translation of the DMPK antisense transcript was shown to give rise to polyglutamine aggregates [59]. Future studies are needed to elucidate the role of each mechanism in DM pathogenesis.
3.5 Therapeutic Strategies
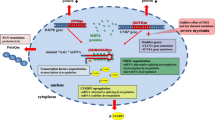
Although no curative treatment exists in DM at present, recent advances in research and pharmacological approaches provide clues for potential future therapeutic intervention. Along with a better understanding of disease mechanisms such as the spliceopathy because of RNA toxicity, several experimental approaches have been developed and applied to translational research in DM (Fig. 3.4).

Schematic illustration of the RNA-mediated disease mechanism (left) and possible therapeutic strategies (right) in DM1. Expanded CUG repeats in the mutant DMPK mRNA form hairpin structures and nuclear foci, sequester MBNL1 in the nucleus, and phosphorylate and stabilize CELF1. Loss of functional MBNL1 and upregulation of CELF1 cause misregulation of alternative splicing. Mis-splicing of CLCN1 exon 7a induces a frameshift and premature termination codon in exon 7, resulting in loss of functional CLCN1 protein on the sarcolemma and myotonia in DM1. Therapeutic strategies are currently being developed for DM, including (1) stabilization of expanded repeats, (2) degradation of the toxic RNA by ASOs, (3) neutralization of CUGexp toxicity by preventing MBNL sequestration with ASOs or small molecules, (4) modulation of MBNL or CELF1, and (5) induction of exon skipping of individual targets (Figure modified from Ref. [82])
3.5.1 Splicing Correction
Since splicing misregulation is the molecular basis of DM, correction of individual mis-splicing events is the most direct way to alleviate the symptoms. Recent advances in technology of antisense oligonucleotides (ASOs) has enabled this strategy. ASOs are short, synthetic, modified nucleic acids that bind complementary RNA and regulate its function [60]. ASOs modulate splicing events by hiding specific sites essential for exon inclusion from the splicing machinery. ASOs blocking target splice sites can skip the target exon(s) and restore normal splicing. This approach has been applied as a treatment for Duchenne muscular dystrophy (DMD). The exon skipping achieved by ASOs restores dystrophin expression in DMD patients and is currently evaluated by clinical trials [61]. In a mouse model of DM1, a morpholino ASO targeting the 3′ splice site of Clcn1 exon 7a reversed the defect of alternative splicing [62]. The morpholino ASOs repressed the inclusion of exon 7a, restored the normal Clcn1 mRNA, increased the expression of CLCN1 protein on sarcolemma, stabilized the membrane potential and, thereby, alleviated myotonia. These findings suggest that ASO-induced exon skipping is a powerful tool for correcting mis-splicing in DM. However, there are several hurdles to overcome in the application of this approach in DM patients: (1) alternative splicing of CLCN1 is more complicated in humans than in mice and (2) mis-splicing events responsible for other symptoms, especially muscle wasting, are not known.
3.5.2 Modulation of Alternative Splicing Factors
In DM, MBNL1 sequestration leads to multiple mis-splicing events, such as CLCN1 and ATP2A1 [34]. One therapeutic approach is the restoration of MBNL1 protein in the nucleoplasm. Overexpression of MBNL1 by adeno-associated viral gene delivery in skeletal muscle of mice expressing CUGexp reversed MBNL1-dependent mis-splicing of exons and rescued myotonia [63]. However, the MBNL1 overexpression in a DM1 mouse model did not improve muscle pathology. In addition, Mbnl1 knockout mice neither reproduce the developmental features of CDM nor display the severe muscle wastings that occur in adult-onset DM1 [34]. Thus, as MBNL1 sequestration does not provide a unifying explanation for the disease process, simultaneous modulation of other splicing factors, such as MBNL2 and CELF1, may be required for successful treatment of DM.
In addition to MBNL1 sequestration, increased steady-state level of CELF1 protein is another important molecular event involved in DM1 pathogenesis [64]. Overexpression of CELF1 in the mouse heart and skeletal muscle causes DM1-associated splicing changes and results in disease phenotypes [44, 65, 66]. Activation of the PKC signaling pathway is suggested to leads to the hyperphosphorylation and stabilization of CELF1 [37]. A recent report showed that PKC inhibitors reduced phosphorylation of CELF1, decreased the steady-state levels, reversed misregulation of splicing events regulated by CELF1, and ameliorated the cardiac conduction defects and contraction abnormalities in a heart-specific DM1 mouse model [67]. Although this approach may be promising for the treatment of cardiac symptoms in DM, there remain important unanswered questions: (1) why is CELF1 not consistently increased in DM2 despite cardiac involvement similar to DM1, and (2) how do CUGexp activate the PKC signaling pathway?
3.5.3 Targeting Toxic RNA
In DM, the mutant RNA exerts a pivotal role in trans-dominant effects on different transcripts and spliceopathy. Thus, interventional strategies against toxic RNA are ideal to accomplish a generalized correction of DM symptoms. Several experimental therapeutic approaches have been attempted to degrade the toxic RNA by small interfering or short hairpin RNA delivered with a retroviral vector and hammerhead ribozyme [68–70]. Although these RNA-based approaches reduced the mutant RNA in DM1 myoblast cells, normal DMPK mRNA was simultaneously decreased, raising concerns regarding the loss of endogenous DMPK function. In addition, unmodified nucleic acids are susceptible to cleavage by endogenous nucleases and are rapidly degraded in vivo. From this view point, chemically modified ASOs with high binding affinity and extreme nuclease resistance emerge as a suitable tool for the modulation of toxic RNAs in DM. Depending on the chemistry and sequence, ASO binding can modulate the target RNA metabolism by two distinct mechanisms: neutralization and degradation. Neutralizing ASOs bind to the target RNA and sterically hinder RNA-protein interactions. Therefore, CAG-repeat ASOs can be expected to bind CUGexp, compete with MBNL proteins, and prevent MBNL sequestration in DM1. In fact, recent reports showed that CAG-repeat ASOs with different chemistry (2-O-methyl, morpholino, locked nucleic acid) mitigated the RNA toxicity in DM1 model mice [15, 71, 72]. Most strikingly, CAG-morpholino ASOs dispersed nuclear foci of CUGexp, released MBNL1 from foci, corrected mis-splicing events, and eliminated myotonia [72]. One caveat with this approach is the delivery of the ASOs by direct injection into the muscle, followed by electroporation, an approach that is not practical for human patients. However, systemic administration of CAG-ASOs with peptide-linked morpholino (PPMO) modification was reported to improve myotonia in a DM1 mouse model, providing the proof-of-concept for clinical applications given the versatility of CAG-ASOs [73] (see also Sect. 3.5.4). In contrast to these neutralizing ASOs, target-degrading ASOs cleave target RNA by recruiting RNase H to the oligonucleotide-RNA heteroduplex. This degradation capability arises from a characteristic structure called “gapmer” that has chemically modified residues on either side of a stretch of unmodified residues referred to as the gap. The modified wings enhance the binding and stability of the ASOs, and the gap allows for the induction of RNase H activity. Because of the exceptional capability of target degradation, gapmer ASOs are considered as promising for DM treatment and have already been tested in DM1 model mice [15, 74, 75]. In one study, systemic administration of the gapmer ASO with 2-O-methyl modification decreased toxic RNA, reduced foci formation, corrected splicing defects, and eliminated myotonia in DM1 mouse model [74]. More importantly, these effects were sustained for up to one year after the discontinuation of treatment. This approach achieved the best therapeutic effects among all strategies in mice so far and led to the development of gapmer ASOs against human DMPK. As therapeutic effects were confirmed in a mouse model [74], the gapmer ASOs (ISIS-DMPKRx) will hopefully be applied to a clinical trial soon.
Small molecules are also appropriate tools for targeting mutant RNA. Recent studies have identified numerous small molecules that bind specifically the CUG or CCUG repeats and competitively release the sequestered MBNL1. Several of these molecules were shown to induce the dispersal of RNA foci, redistribution of MBNL1 to the nucleoplasm, and partial reversal of mis-splicing events in mice expressing CUGexp [76–78]. However, toxicity of these compounds in human has not been determined, and none of the candidates achieved a full rescue of mis-splicing of Clcn1 and myotonia in mice. Pentamidine and related compounds have been recently suggested to inhibit the transcription of CUGexp by binding to the CTG repeat DNA [79, 80]. Indeed, heptamidine, a methylene linker analogue of pentamidine, reversed splicing defects and improved myotonia in the mouse model.
3.5.4 Stabilization of Expanded Repeats
Another striking feature of DM is the marked instability of the expanded repeats. The progression of DM1 may depend on the growth of the expanded repeat throughout life. The ongoing repeat expansion also raises a concern that an initial beneficial effect of therapeutic ASOs on the toxic RNA may be lost because of ongoing expansion and increased production of CUGexp RNA. Therefore, stabilization of the repeat is also a potential approach to postpone the onset, slow the progression, or to sustain the effect of therapeutic ASOs. Several compounds that stabilize the repeats in cultured cells have been identified [81]. However, these compounds are DNA intercalators or drugs that affect DNA methylation or replication, evoking concerns about long-term safety. In DM1, the instability of expanded repeats is enhanced by transcription across the repeat and R-loop formation [14, 15]. Because the R-loop is formed with CUG RNA and CAG DNA on the template strand, CAG-ASOs can theoretically prevent its formation. Indeed, a recent report showed that CAG-ASOs suppressed the repeat instability in both a cellular model and a mouse model by reducing the formation of R-loops at the DMPK locus [15]. Thus, intervention with CAG-repeat ASOs has the dual benefit of preventing the RNA toxicity and stabilizing the repeat.
3.6 Concluding Remarks
Since the CTG expansion was discovered in DM1, the understanding of the molecular pathomechanisms in DM has been enormously advanced. Promising experimental therapeutic strategies targeting toxic RNA continue to emerge. A recent study showed that several mis-splicing events can be a good biomarker for disease severity and therapeutic response in DM [16]. National and international DM patient registries have already been established in several countries for a clinical trial. Indeed, gapmer-ASOs targeting mutant DMPK mRNA for treatment of DM1 is currently being planned for a first-in-human clinical trial. Combination therapy with small molecules is also an attractive option. These developments are opening a new, fascinating, and promissing era for the treatment of DM.
References
Harper PS (2001) Myotonic dystrophy. W.B. Saunders Company, London
Harley HG, Brook JD, Rundle SA, Crow S, Reardon W, Buckler AJ, Harper PS, Housman DE, Shaw DJ (1992) Expansion of an unstable DNA region and phenotypic variation in myotonic dystrophy. Nature 355(6360):545–546
Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP (2001) Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293(5531):864–867
Osborne RJ, Thornton CA (2006) RNA-dominant diseases. Hum Mol Genet 15(2):R162–R169. doi:10.1093/hmg/ddl181
Vicart S, Sternberg D, Fontaine B, Meola G (2005) Human skeletal muscle sodium channelopathies. Neurol Sci 26(4):194–202. doi:10.1007/s10072-005-0461-x
de Die-Smulders CE, Howeler CJ, Thijs C, Mirandolle JF, Anten HB, Smeets HJ, Chandler KE, Geraedts JP (1998) Age and causes of death in adult-onset myotonic dystrophy. Brain 121(Pt 8):1557–1563
Gadalla SM, Lund M, Pfeiffer RM, Gortz S, Mueller CM, Moxley RT 3rd, Kristinsson SY, Bjorkholm M, Shebl FM, Hilbert JE, Landgren O, Wohlfahrt J, Melbye M, Greene MH (2011) Cancer risk among patients with myotonic muscular dystrophy. JAMA 306(22):2480–2486. doi:10.1001/jama.2011.1796
Nakamori M, Thornton C (2010) Epigenetic changes and non-coding expanded repeats. Neurobiol Dis 39(1):21–27. doi:10.1016/j.nbd.2010.02.004
Harley HG, Rundle SA, MacMillan JC, Myring J, Brook JD, Crow S, Reardon W, Fenton I, Shaw DJ, Harper PS (1993) Size of the unstable CTG repeat sequence in relation to phenotype and parental transmission in myotonic dystrophy. Am J Hum Genet 52(6):1164–1174
Lopez Castel A, Cleary JD, Pearson CE (2010) Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol 11(3):165–170. doi:10.1038/nrm2854
Ashizawa T, Anvret M, Baiget M, Barcelo JM, Brunner H, Cobo AM, Dallapiccola B, Fenwick RG Jr, Grandell U, Harley H (1994) Characteristics of intergenerational contractions of the CTG repeat in myotonic dystrophy. Am J Hum Genet 54(3):414–423
Thornton CA, Johnson K, Moxley RT (1994) Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann Neurol 35:104–107
Nakamori M, Sobczak K, Moxley RT 3rd, Thornton CA (2009) Scaled-down genetic analysis of myotonic dystrophy type 1 and type 2. Neuromuscul Disord 19(11):759–762. doi:10.1016/j.nmd.2009.07.012
Nakamori M, Pearson CE, Thornton CA (2011) Bidirectional transcription stimulates expansion and contraction of expanded (CTG)•(CAG) repeats. Hum Mol Genet 20(3):580–588. doi:10.1093/hmg/ddq501
Nakamori M, Gourdon G, Thornton CA (2011) Stabilization of expanded (CTG)*(CAG) repeats by antisense oligonucleotides. Mol Ther 19(12):2222–2227. doi:10.1038/mt.2011.191
Nakamori M, Sobczak K, Puwanant A, Welle S, Eichinger K, Pandya S, Dekdebrun J, Heatwole CR, McDermott MP, Chen T, Cline M, Tawil R, Osborne RJ, Wheeler TM, Swanson MS, Moxley RT 3rd, Thornton CA (2013) Splicing biomarkers of disease severity in myotonic dystrophy. Ann Neurol 74(6):862–872. doi:10.1002/ana.23992
Zatz M, Passos-Bueno MR, Cerqueira A, Marie SK, Vainzof M, Pavanello R (1995) Analysis of the CTG repeat in skeletal muscle of young and adult myotonic dystrophy patients: when does the expansion occur? Hum Mol Genet 4(3):401–406
Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP et al (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65(5):905–914. doi:10.1016/0092-8674(91)90397-H
Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M (1996) Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271(5254):1423–1427
Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS (2000) Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 19(17):4439–4448. doi:10.1093/emboj/19.17.4439
Jansen G, Groenen PJ, Bachner D, Jap PH, Coerwinkel M, Oerlemans F, van den Broek W, Gohlsch B, Pette D, Plomp JJ, Molenaar PC, Nederhoff MG, van Echteld CJ, Dekker M, Berns A, Hameister H, Wieringa B (1996) Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat Genet 13(3):316–324. doi:10.1038/ng0796-316
Reddy S, Smith DBJ, Rich MM, Leferovich JM, Reilly P, Davis BM, Tran K, Rayburn H, Bronson R, Cros D, Balice-Gordon RJ, Housman D (1996) Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat Genet 13:325–334
Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA (2000) Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289(5485):1769–1773
Margolis JM, Schoser BG, Moseley ML, Day JW, Ranum LP (2006) DM2 intronic expansions: evidence for CCUG accumulation without flanking sequence or effects on ZNF9 mRNA processing or protein expression. Hum Mol Genet 15(11):1808–1815
Kino Y, Mori D, Oma Y, Takeshita Y, Sasagawa N, Ishiura S (2004) Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats. Hum Mol Genet 13(5):495–507
Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA (2004) Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet 13(24):3079–3088. doi:10.1093/hmg/ddh327
Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA (2006) Failure of MBNL1-dependent postnatal splicing transitions in myotonic dystrophy. Hum Mol Genet 15(13):2087–2097
Ho TH, Charlet B, Poulos MG, Singh G, Swanson MS, Cooper TA (2004) Muscleblind proteins regulate alternative splicing. EMBO J 23(15):3103–3112
Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, Cannon SC, Thornton CA (2002) Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell 10(1):35–44
Charlet-B N, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA (2002) Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell 10(1):45–53
Osborne RJ, Lin X, Welle S, Sobczak K, O’Rourke JR, Swanson MS, Thornton CA (2009) Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum Mol Genet 18(8):1471–1481. doi:10.1093/hmg/ddp058
Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, Lecuyer E, Burge CB (2012) Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 150(4):710–724. doi:10.1016/j.cell.2012.06.041
Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D, Wahbi K, Day JW, Fujimura H, Takahashi MP, Auboeuf D, Dreumont N, Furling D, Charlet-Berguerand N (2011) Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol 18(7):840–845. doi:10.1038/nsmb.2067
Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS (2003) A muscleblind knockout model for myotonic dystrophy. Science 302(5652):1978–1980
Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, Brook JD (2002) Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet 11(7):805–814
Lee KY, Li M, Manchanda M, Batra R, Charizanis K, Mohan A, Warren SA, Chamberlain CM, Finn D, Hong H, Ashraf H, Kasahara H, Ranum LP, Swanson MS (2013) Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol Med 5(12):1887–1900. doi:10.1002/emmm.201303275
Kuyumcu-Martinez NM, Wang GS, Cooper TA (2007) Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell 28(1):68–78. doi:10.1016/j.molcel.2007.07.027
Salisbury E, Sakai K, Schoser B, Huichalaf C, Schneider-Gold C, Nguyen H, Wang GL, Albrecht JH, Timchenko LT (2008) Ectopic expression of cyclin D3 corrects differentiation of DM1 myoblasts through activation of RNA CUG-binding protein, CUGBP1. Exp Cell Res 314(11–12):2266–2278. doi:10.1016/j.yexcr.2008.04.018
Jones K, Wei C, Iakova P, Bugiardini E, Schneider-Gold C, Meola G, Woodgett J, Killian J, Timchenko NA, Timchenko LT (2012) GSK3beta mediates muscle pathology in myotonic dystrophy. J Clin Invest 122(12):4461–4472. doi:10.1172/JCI64081
Charlet BN, Logan P, Singh G, Cooper TA (2002) Dynamic antagonism between ETR-3 and PTB regulates cell type-specific alternative splicing. Mol Cell 9(3):649–658, doi: S1097276502004793 [pii]
Savkur RS, Philips AV, Cooper TA (2001) Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet 29(1):40–47
Timchenko NA, Cai ZJ, Welm AL, Reddy S, Ashizawa T, Timchenko LT (2001) RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J Biol Chem 276(11):7820–7826
Vlasova IA, Tahoe NM, Fan D, Larsson O, Rattenbacher B, Sternjohn JR, Vasdewani J, Karypis G, Reilly CS, Bitterman PB, Bohjanen PR (2008) Conserved GU-rich elements mediate mRNA decay by binding to CUG-binding protein 1. Mol Cell 29(2):263–270. doi:10.1016/j.molcel.2007.11.024
Koshelev M, Sarma S, Price RE, Wehrens XH, Cooper TA (2010) Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum Mol Genet 19(6):1066–1075. doi:10.1093/hmg/ddp570
Cardani R, Bugiardini E, Renna LV, Rossi G, Colombo G, Valaperta R, Novelli G, Botta A, Meola G (2013) Overexpression of CUGBP1 in skeletal muscle from adult classic myotonic dystrophy type 1 but not from myotonic dystrophy type 2. PLoS One 8(12):e83777. doi:10.1371/journal.pone.0083777
Kornblihtt AR, Schor IE, Allo M, Dujardin G, Petrillo E, Munoz MJ (2013) Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol 14(3):153–165. doi:10.1038/nrm3525
Kimura T, Nakamori M, Lueck JD, Pouliquin P, Aoike F, Fujimura H, Dirksen RT, Takahashi MP, Dulhunty AF, Sakoda S (2005) Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2 + −ATPase in myotonic dystrophy type 1. Hum Mol Genet 14(15):2189–2200
Tang ZZ, Yarotskyy V, Wei L, Sobczak K, Nakamori M, Eichinger K, Moxley RT, Dirksen RT, Thornton CA (2012) Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of CaV1.1 calcium channel. Hum Mol Genet 21(6):1312–1324. doi:10.1093/hmg/ddr568
Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N, Kokunai Y, Tsuburaya R, de la Grange P, Dembele D, Francois V, Precigout G, Boulade-Ladame C, Hummel MC, Lopez de Munain A, Sergeant N, Laquerriere A, Thibault C, Deryckere F, Auboeuf D, Garcia L, Zimmermann P, Udd B, Schoser B, Takahashi MP, Nishino I, Bassez G, Laporte J, Furling D, Charlet-Berguerand N (2011) Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med 17(6):720–725. doi:10.1038/nm.2374
Jacobs AE, Benders AA, Oosterhof A, Veerkamp JH, van Mier P, Wevers RA, Joosten EM (1990) The calcium homeostasis and the membrane potential of cultured muscle cells from patients with myotonic dystrophy. Biochim Biophys Acta 1096(1):14–19, doi: 0925-4439(90)90006-B [pii]
Nakamori M, Kimura T, Fujimura H, Takahashi MP, Sakoda S (2007) Altered mRNA splicing of dystrophin in type 1 myotonic dystrophy. Muscle Nerve 36(2):251–257. doi:10.1002/mus.20809
Nakamori M, Kimura T, Kubota T, Matsumura T, Sumi H, Fujimura H, Takahashi MP, Sakoda S (2008) Aberrantly spliced alpha-dystrobrevin alters alpha-syntrophin binding in myotonic dystrophy type 1. Neurology 70(9):677–685. doi:10.1212/01.wnl.0000302174.08951.cf
Philips AV, Timchenko LT, Cooper TA (1998) Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 280(5364):737–741
Mankodi A, Lin X, Blaxall BC, Swanson MS, Thornton CA (2005) Nuclear RNA foci in the heart in myotonic dystrophy. Circ Res 97(11):1152–1155
Suenaga K, Lee KY, Nakamori M, Tatsumi Y, Takahashi MP, Fujimura H, Jinnai K, Yoshikawa H, Du H, Ares M Jr, Swanson MS, Kimura T (2012) Muscleblind-like 1 knockout mice reveal novel splicing defects in the myotonic dystrophy brain. PLoS One 7(3):e33218. doi:10.1371/journal.pone.0033218
Paul S, Dansithong W, Kim D, Rossi J, Webster NJ, Comai L, Reddy S (2006) Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J 25(18):4271–4283
Ravel-Chapuis A, Belanger G, Yadava RS, Mahadevan MS, DesGroseillers L, Cote J, Jasmin BJ (2012) The RNA-binding protein Staufen1 is increased in DM1 skeletal muscle and promotes alternative pre-mRNA splicing. J Cell Biol 196(6):699–712. doi:10.1083/jcb.201108113
Kalsotra A, Singh RK, Gurha P, Ward AJ, Creighton CJ, Cooper TA (2014) The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue, dramatically altering miRNA and mRNA expression. Cell Rep 6(2):336–345. doi:10.1016/j.celrep.2013.12.025
Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LP (2011) Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A 108(1):260–265. doi:10.1073/pnas.1013343108
Southwell AL, Skotte NH, Bennett CF, Hayden MR (2012) Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends Mol Med 18(11):634–643. doi:10.1016/j.molmed.2012.09.001
Koo T, Wood MJ (2013) Clinical trials using antisense oligonucleotides in Duchenne muscular dystrophy. Hum Gene Ther 24(5):479–488. doi:10.1089/hum.2012.234
Wheeler TM, Lueck JD, Swanson MS, Dirksen RT, Thornton CA (2007) Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest 117(12):3952–3957. doi:10.1172/JCI33355
Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA, Swanson MS (2006) Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc Natl Acad Sci U S A 103(31):11748–11753
Roberts R, Timchenko NA, Miller JW, Reddy S, Caskey CT, Swanson MS, Timchenko LT (1997) Altered phosphorylation and intracellular distribution of a (CUG)n triplet repeat RNA-binding protein in patients with myotonic dystrophy and in myotonin protein kinase knockout mice. Proc Natl Acad Sci U S A 94(24):13221–13226
Ho TH, Bundman D, Armstrong DL, Cooper TA (2005) Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum Mol Genet 14(11):1539–1547
Ward AJ, Rimer M, Killian JM, Dowling JJ, Cooper TA (2010) CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum Mol Genet 19(18):3614–3622. doi:10.1093/hmg/ddq277
Wang GS, Kuyumcu-Martinez MN, Sarma S, Mathur N, Wehrens XH, Cooper TA (2009) PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J Clin Invest 119(12):3797–3806. doi:10.1172/JCI37976
Furling D, Doucet G, Langlois MA, Timchenko L, Belanger E, Cossette L, Puymirat J (2003) Viral vector producing antisense RNA restores myotonic dystrophy myoblast functions. Gene Ther 10(9):795–802. doi:10.1038/sj.gt.3301955
Langlois MA, Boniface C, Wang G, Alluin J, Salvaterra PM, Puymirat J, Rossi JJ, Lee NS (2005) Cytoplasmic and nuclear retained DMPK mRNAs are targets for RNA interference in myotonic dystrophy cells. J Biol Chem 280(17):16949–16954. doi:10.1074/jbc.M501591200
Langlois MA, Lee NS, Rossi JJ, Puymirat J (2003) Hammerhead ribozyme-mediated destruction of nuclear foci in myotonic dystrophy myoblasts. Mol Ther 7(5 Pt 1):670–680, doi: S1525001603000686 [pii]
Mulders SA, van den Broek WJ, Wheeler TM, Croes HJ, van Kuik-Romeijn P, de Kimpe SJ, Furling D, Platenburg GJ, Gourdon G, Thornton CA, Wieringa B, Wansink DG (2009) Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc Natl Acad Sci U S A 106(33):13915–13920. doi:10.1073/pnas.0905780106
Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, Thornton CA (2009) Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 325(5938):336–339. doi:10.1126/science.1173110
Leger AJ, Mosquea LM, Clayton NP, Wu IH, Weeden T, Nelson CA, Phillips L, Roberts E, Piepenhagen PA, Cheng SH, Wentworth BM (2013) Systemic delivery of a Peptide-linked morpholino oligonucleotide neutralizes mutant RNA toxicity in a mouse model of myotonic dystrophy. Nucleic Acid Ther 23(2):109–117. doi:10.1089/nat.2012.0404
Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, Cheng SH, Wentworth BM, Bennett CF, Thornton CA (2012) Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 488(7409):111–115. doi:10.1038/nature11362
Lee JE, Bennett CF, Cooper TA (2012) RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc Natl Acad Sci U S A 109(11):4221–4226. doi:10.1073/pnas.1117019109
Parkesh R, Childs-Disney JL, Nakamori M, Kumar A, Wang E, Wang T, Hoskins J, Tran T, Housman D, Thornton CA, Disney MD (2012) Design of a bioactive small molecule that targets the myotonic dystrophy type 1 RNA via an RNA motif-ligand database and chemical similarity searching. J Am Chem Soc 134(10):4731–4742. doi:10.1021/ja210088v
Childs-Disney JL, Parkesh R, Nakamori M, Thornton CA, Disney MD (2012) Rational design of bioactive, modularly assembled aminoglycosides targeting the RNA that causes myotonic dystrophy type 1. ACS Chem Biol 7(12):1984–1993. doi:10.1021/cb3001606
Ofori LO, Hoskins J, Nakamori M, Thornton CA, Miller BL (2012) From dynamic combinatorial ‘hit’ to lead: in vitro and in vivo activity of compounds targeting the pathogenic RNAs that cause myotonic dystrophy. Nucleic Acids Res 40(13):6380–6390. doi:10.1093/nar/gks298
Warf MB, Nakamori M, Matthys CM, Thornton CA, Berglund JA (2009) Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc Natl Acad Sci U S A 106(44):18551–18556. doi:10.1073/pnas.0903234106
Coonrod LA, Nakamori M, Wang W, Carrell S, Hilton CL, Bodner MJ, Siboni RB, Docter AG, Haley MM, Thornton CA, Berglund JA (2013) Reducing levels of toxic RNA with small molecules. ACS Chem Biol 8(11):2528–2537. doi:10.1021/cb400431f
Gomes-Pereira M, Monckton DG (2006) Chemical modifiers of unstable expanded simple sequence repeats: what goes up, could come down. Mutat Res 598(1–2):15–34. doi:10.1016/j.mrfmmm.2006.01.011
Nakamori M, Takahashi MP (2011) Myotonic dystrophy: therapeutic approaches to RNA toxicity. Brain Nerve 63(11):1161–1168, doi: 1416101048 [pii]
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this chapter
Cite this chapter
Nakamori, M., Takahashi, M.P. (2016). Myotonic Dystrophy. In: Takeda, S., Miyagoe-Suzuki, Y., Mori-Yoshimura, M. (eds) Translational Research in Muscular Dystrophy. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55678-7_3
Download citation
DOI: https://doi.org/10.1007/978-4-431-55678-7_3
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55677-0
Online ISBN: 978-4-431-55678-7
eBook Packages: MedicineMedicine (R0)