
Abstract
The natriuretic peptide family consists of three endogenous ligands: atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and C-type natriuretic peptide (CNP). They exert their biological actions through two subtypes of particulate guanylyl cyclase (GC): GC-A for ANP and BNP, and GC-B for CNP. Among the natriuretic peptide family members, ANP and BNP are cardiac hormones that are produced and released from the atrium and ventricle of the heart, respectively, and play important roles in the regulation of the cardiovascular system. On the other hand, although CNP and its receptor, GC-B, exist ubiquitously in the body, we elucidated that the CNP/GC-B system is a crucial stimulator of endochondral bone growth, using CNP or GC-B knockout or transgenic mice. We planned to utilize the activation of the CNP/GC-B pathway as a novel therapeutic strategy for skeletal dysplasia, which consists of multiple skeletal diseases including those with impaired bone growth. We tried to investigate this effect on impaired skeletal growth in a mouse model of achondroplasia, the most common form of skeletal dysplasias, and successfully recovered the skeletal phenotype by using transgenic technology or by administration of synthetic CNP. In the future, the activation of the CNP/GC-B system may constitute a novel therapeutic strategy for the treatment of skeletal dysplasias.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others

Keywords
- Natriuretic peptide family
- C-type natriuretic peptide (CNP)
- Guanylyl cyclase-B (GC-B)
- Endochondral bone growth
- Transgenic mouse
- Knockout mouse
- Skeletal dysplasia
- Achondroplasia
Introduction
The natriuretic peptide family consists of three endogenous ligands: atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and C-type natriuretic peptide (CNP) [1]. They exert their biological actions through two subtypes of particulate guanylyl cyclase (GC): GC-A for ANP and BNP, and GC-B for CNP [2]. Among the natriuretic peptide family members, ANP and BNP are cardiac hormones that are produced and released from the atrium and ventricle of the heart, respectively, and play important roles in the regulation of the cardiovascular system [3–7]. CNP, a third member of the natriuretic peptide family, was first purified from porcine brain [8]. Therefore, CNP was thought to be the primary natriuretic peptide in the brain, but has been proved to exist ubiquitously in the body, e.g., in the blood vessels, gonads, adrenal gland, etc. [9–15] (Fig. 1). Furthermore, analysis of the CNP/GC-B system in genetically engineered mice revealed that CNP and GC-B play a pivotal role in the regulation of endochondral bone growth.

Schematic representation of the distribution of the CNP/GC-B system. CNP and its receptor, GC-B, exist ubiquitously in the body
Skeletal Phenotypes of the CNP/GC-B System in Genetically Engineered Mice
Targeted disruption of the CNP gene (Nppc) causes prominent short stature due to impaired bone growth in mice [16] (Fig. 2). Mammalian bones are formed through two different mechanisms, endochondral ossification and membranous ossification. Most mammalian bones are formed through endochondral ossification, in which the growth of bone is dependent on the growth of cartilaginous anlage as the scaffold of bone; later, it is replaced by calcified bone [17]. In the process of endochondral ossification, chondrocytes in the growth plate undergo proliferation, hypertrophy, and cell death, and finally are replaced by osteoblasts. The short-stature phenotype of CNP knockout (CNP-KO) mice results from impaired bone growth through endochondral ossification. Histological analysis of the growth plate of CNP-KO mice revealed that every chondrocyte layer of the growth plate is narrower in CNP-KO mice than in wild-type mice. Furthermore, mice depleted with the GC-B gene (Npr2) exhibit the same short-stature phenotype observed in CNP-KO mice, demonstrating that the CNP/GC-B system is a physiologically important stimulator of endochondral bone growth [18].

Gross phenotype (upper panel) and growth curve (lower panel) of the CNP-KO mouse
In contrast, cartilage-specific CNP transgenic mice under the control of type II collagen promoter (col2-CNP-Tg mice) exhibit prominent overgrowth of bones formed through endochondral ossification [19] (Fig. 3a). In contrast to CNP- or GC-B-KO mice, every chondrocyte layer, especially hypertrophic chondrocyte layer, of the growth plate of col2-CNP-Tg mice is wider than that of wild-type mice (Fig. 3b). Collectively, the CNP/GC-B system is a physiological stimulator of endochondral bone growth and can potently stimulate bone growth as a local regulator.

Cartilage-specific CNP transgenic mouse. (a) Gross appearance of female cartilage-specific CNP transgenic (col2-CNP-Tg) mouse and its wild-type litter mate at the age of 10 weeks. (b) Immunohistochemical staining for type X collagen of a histological section of the tibial growth plates
Importance of CNP/GC-B Signaling in Endochondral Bone Growth in Humans: Lessons from Rare Congenital Skeletal Disorders
Skeletal dysplasias are human genetic disorders of the skeleton, whose feature is impairment of bones. In 2004, Bartels et al., reported that acromesomelic dysplasia, type Maroteaux, one form of skeletal dysplasia with a severely impaired bone growth phenotype, is caused by a loss-of-function mutation in the human GC-B gene [20]. In fact, the skeletal phenotype observed in this disorder was just the same as that of GC-B-KO mice, and here for the first time, the CNP/GC-B signaling was proved to be a physiological and potent stimulator of endochondral bone growth not only in mice but also in humans. Later, in 2012, Miura et al., reported a Japanese case of skeletal overgrowth phenotype and showed that it was caused by a constitutively activated mutation in the GC-B gene [21]. They also reported a Korean case of skeletal overgrowth caused by a constitutive active mutation in the GC-B gene [22]. Thus, the notion that the CNP/GC-B system is a potent stimulator of endochondral bone in human is firmly established so far.
Translational Research into Activation of CNP/GC-B Signaling for Skeletal Dysplasias
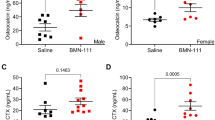
Following these results, we planned to start translational research into activation of the CNP/GC-B system for disorders with impaired bone growth in skeletal dysplasias. Achondroplasia is the most common form of skeletal dysplasias, with a birth prevalence of ~1:10,000. Patients suffering from achondroplasia exhibit short stature (final height: ~130 cm in males and ~120 cm in females) and a short-limbed dwarf phenotype owing to impaired endochondral bone growth, and it had been shown to be caused by a constitutive active mutation in the FGF receptor 3 (FGFR3) gene, followed by a decrease in the proliferation of chondrocytes in the growth plate [23, 24]. Current therapies for achondroplasia consist of distraction osteogenesis, an orthopedic procedure, and administration of growth hormone [25]. Although distraction osteogenesis gives better results, it lays a burden on patients and limits their quality of life. Growth hormone has a minimal effect. Therefore, we started translational research into activation of the CNP/GC-B system for this disease. We obtained transgenic mice with targeted overexpression of activated FGFR3 (G380R mutation in humans) in cartilage, using type II collagen promoter as a mouse model of achondroplasia (Ach mice) [26]. As shown in Fig. 4, Ach mice show short stature and an impaired bone growth phenotype, and mimic the skeletal phenotype of achondroplastic patients. We achieved targeted overexpression of CNP in the growth plate of Ach mice by crossing them with col2-CNP-Tg mice. The short stature observed in Ach mice, owing to their impaired bone growth, was recovered in the double transgenic Ach/col2-CNP-Tg mice and grew almost comparable to that of wild-type mice (Fig. 4). As was previously reported, the length of the Ach mice was significantly shorter at least at the third week after birth, and more than 10 % shorter after 6 weeks of age, than that of their wild-type litter mates. In Ach/col2-CNP-Tg mice, the decrease in the length of Ach mice was recovered, and the length became comparable to that of their wild-type litter mates. A soft x-ray picture revealed that the shortening of Ach bones was almost completely recovered in double transgenic bones. But the width of the cranium, which is made through membranous ossification, was not changed. Histological examination of the tibial growth plate showed that the width of the growth plate of Ach mice was shorter than that of wild-type mice, but was restored in the growth plate of the double transgenic mice [19].

Skeletal rescue of achondroplastic model mouse by targeted overexpression of CNP in cartilage. The gross appearance (left) and soft x-ray picture (right) of litter mates of the mice at the age of 3 months are depicted
Next, in order to test the effect of elevated plasma concentrations of CNP on endochondral bone growth, we generated transgenic mice with an elevated plasma concentration of CNP, using human serum amyloid P component promoter, which enables targeted overexpression of CNP in the liver (SAP-CNP-Tg mice) (Fig. 5). The resultant SAP-CNP-Tg mice, which have about two times the plasma CNP concentration of wild-type mice, exhibited skeletal overgrowth just like col2-CNP-Tg mice [27]. Then we mated Ach mice with the SAP-CNP-Tg mice and tried to rescue the impaired endochondral bone growth of Ach mice by increased circulating CNP. We could successfully rescue the short-stature phenotype of Ach mice in the double transgenic Ach/SAP-CNP-Tg mice (Fig. 6a). After 6 weeks of age, the short stature observed in Ach mice was completely rescued in Ach/SAP-CNP-Tg mice. Skeletal analysis by a soft x-ray picture also exhibited that the impaired bone growth of Ach mice was rescued by increased circulating CNP (Fig. 6b). The lengths of the humerus, radius, ulna, femur, and tibia of the double transgenic mice were rather longer than those of wild-type mice. Histological examination revealed that the shortened growth plate of Ach mice was rescued in Ach/SAP-CNP-Tg mice and became rather elongated in comparison with the wild-type mice [28].

Generation of CNP transgenic mice with increased circulating CNP under the control of human serum amyloid P component (SAP) promoter. Gross appearance (left) and soft x-ray picture (right) of litter mates of the mice at the age of 15 weeks are depicted

Skeletal rescue of achondroplastic model mouse by increased circulating CNP. Gross appearance (left) and soft x-ray picture (right) of litter mates of the mice at the age of 3 months are depicted
Finally, we tried to administrate CNP to Ach mice. Ach mice were about 10 % shorter than wild-type mice at the beginning of administration at the age of 3 weeks. Administration of CNP at the dose of 1 μg/kg/min stimulated the growth of Ach mice, and their length was almost comparable to that of wild-type mice at the end of the 4-week administration period (Fig. 7a). A soft x-ray picture revealed that the impaired skeletal growth observed in Ach mice was recovered by administration of CNP. Administration of 0.1 μg/kg/min of CNP considerably rescued the shortness of Ach bones, and 1 μg/kg/min elongated them to an extent rather longer than those of vehicle-treated wild-type mice. Histological examination revealed that the narrowed growth plate of Ach mice was recovered and became comparable to that of wild-type mice through administration of CNP at the dose of 1 μg/kg/min (Fig. 7b) [28].

Skeletal rescue of achondroplastic model mouse by continuous intravenous injection of CNP at the dose of 1 μg/kg/min. Gross appearance (a) and histological pictures of the vertebral growth plates (b, safranin-O staining) of the mice at the end of the 4-week treatment
Conclusions and Future Prospects
We have elucidated that CNP/GC-B signaling is a physiological stimulator of endochondral bone growth and exhibited that the activation of this pathway strongly stimulates longitudinal bone growth in mice model. Furthermore, clinical reports showed that decreased signaling of this pathway causes impaired endochondral bone growth (acromesomelic dysplasia, type Maroteaux), and increased signaling causes skeletal overgrowth phenotype in humans. We started translational research into CNP/GC-B therapy for skeletal dysplasias using a mouse model of achondroplasia, and successfully rescued their impaired skeletal growth phenotype.
Nevertheless, there remain several problems to be solved to make CNP/GC-B therapy a reality for skeletal dysplasias. In the preclinical study described here, we had to administrate CNP intravenously, not subcutaneously, because CNP is easily degraded by subcutaneous endopeptidase. First, therefore, in the event that we use CNP, we must develop a way of administering CNP. Second, in relevance to this point, modification of the clearance system of CNP may be another approach for activating the CNP/GC-B system [29].
References
Nakao K, Ogawa Y, Suga S, Imura H (1992) Molecular biology and biochemistry of the natriuretic peptide system. I: natriuretic peptides. J Hypertens 10:907–912
Nakao K, Ogawa Y, Suga S, Imura H (1992) Molecular biology and biochemistry of the natriuretic peptide system. II: natriuretic peptide receptors. J Hypertens 10:1111–1114
Mukoyama M, Nakao K, Hosoda K et al (1991) Brain natriuretic peptide as a novel cardiac hormone in humans. Evidence for an exquisite dual natriuretic peptide system, atrial natriuretic peptide and brain natriuretic peptide. J Clin Invest 87:1402–1412
Mukoyama M, Nakao K, Saito Y et al (1990) Increased human brain natriuretic peptide in congestive heart failure. N Engl J Med 323:757–758
Mukoyama M, Nakao K, Saito Y et al (1990) Human brain natriuretic peptide, a novel cardiac hormone. Lancet 335:801–802
Saito Y, Nakao K, Arai H et al (1989) Augmented expression of atrial natriuretic polypeptide gene in ventricle of human failing heart. J Clin Invest 83:298–305
Saito Y, Nakao K, Nishimura K et al (1987) Clinical application of atrial natriuretic polypeptide in patients with congestive heart failure: beneficial effects on left ventricular function. Circulation 76:115–124
Sudoh T, Minamino N, Kangawa K, Matsuo H (1990) C-type natriuretic peptide (CNP): a new member of natriuretic peptide family identified in porcine brain. Biochem Biophys Res Commun 168:863–870
Suga S, Nakao K, Itoh H et al (1992) Endothelial production of C-type natriuretic peptide and its marked augmentation by transforming growth factor-beta. Possible existence of “vascular natriuretic peptide system”. J Clin Invest 90:1145–1149
Komatsu Y, Nakao K, Itoh H, Suga S, Ogawa Y, Imura H (1992) Vascular natriuretic peptide. Lancet 340:622
Suga S, Itoh H, Komatsu Y et al (1993) Cytokine-induced C-type natriuretic peptide (CNP) secretion from vascular endothelial cells–evidence for CNP as a novel autocrine/paracrine regulator from endothelial cells. Endocrinology 133:3038–3041
Komatsu Y, Itoh H, Suga S et al (1996) Regulation of endothelial production of C-type natriuretic peptide in coculture with vascular smooth muscle cells. Role of the vascular natriuretic peptide system in vascular growth inhibition. Circ Res 78:606–614
Kawai M, Naruse M, Yoshimoto T et al (1996) C-type natriuretic peptide as a possible local modulator of aldosterone secretion in bovine adrenal zona glomerulosa. Endocrinology 137:42–46
Zhang M, Su YQ, Sugiura K, Xia G, Eppig JJ (2010) Granulosa cell ligand NPPC and its receptor NPR2 maintain meiotic arrest in mouse oocytes. Science 330:366–369
Kiyosu C, Tsuji T, Yamada K, Kajita S, Kunieda T (2012) NPPC/NPR2 signaling is essential for oocyte meiotic arrest and cumulus oophorus formation during follicular development in the mouse ovary. Reproduction 144:187–193
Chusho H, Tamura N, Ogawa Y et al (2001) Dwarfism and early death in mice lacking C-type natriuretic peptide. Proc Natl Acad Sci U S A 98:4016–4021
Kronenberg HM (2003) Developmental regulation of the growth plate. Nature 423:332–336
Tamura N, Doolittle LK, Hammer RE, Shelton JM, Richardson JA, Garbers DL (2004) Critical roles of the guanylyl cyclase B receptor in endochondral ossification and development of female reproductive organs. Proc Natl Acad Sci U S A 101:17300–17305
Yasoda A, Komatsu Y, Chusho H et al (2004) Overexpression of CNP in chondrocytes rescues achondroplasia through a MAPK-dependent pathway. Nat Med 10:80–86
Bartels CF, Bukulmez H, Padayatti P et al (2004) Mutations in the transmembrane natriuretic peptide receptor NPR-B impair skeletal growth and cause acromesomelic dysplasia, type Maroteaux. Am J Hum Genet 75:27–34
Miura K, Namba N, Fujiwara M et al (2012) An overgrowth disorder associated with excessive production of cGMP due to a gain-of-function mutation of the natriuretic peptide receptor 2 gene. PLoS One 7:e42180
Miura K, Kim OH, Lee HR et al (2014) Overgrowth syndrome associated with a gain-of-function mutation of the natriuretic peptide receptor 2 (NPR2) gene. Am J Med Genet A 164A:156–163
Shiang R, Thompson LM, Zhu YZ et al (1994) Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell 78:335–342
Rousseau F, Bonaventure J, Legeai-Mallet L et al (1994) Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature 371:252–254
Cattaneo R, Villa A, Catagni M, Tentori L (1988) Limb lengthening in achondroplasia by Ilizarov’s method. Int Orthop 12:173–179
Naski MC, Colvin JS, Coffin JD, Ornitz DM (1998) Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development 125:4977–4988
Kake T, Kitamura H, Adachi Y et al (2009) Chronically elevated plasma C-type natriuretic peptide level stimulates skeletal growth in transgenic mice. Am J Physiol Endocrinol Metab 297:E1339–E1348
Yasoda A, Kitamura H, Fujii T et al (2009) Systemic administration of C-type natriuretic peptide as a novel therapeutic strategy for skeletal dysplasias. Endocrinology 150:3138–3144
Matsukawa N, Grzesik WJ, Takahashi N et al (1999) The natriuretic peptide clearance receptor locally modulates the physiological effects of the natriuretic peptide system. Proc Natl Acad Sci U S A 96:7403–7408
Acknowledgments
We thank Dr. D. M. Ornitz for the Ach mice used in this work.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this paper
Cite this paper
Yasoda, A., Nakao, K. (2015). Translational Research of the Activation of the C-Type Natriuretic Peptide (CNP)-Guanylyl Cyclase-B Pathway for Skeletal Dysplasia. In: Nakao, K., Minato, N., Uemoto, S. (eds) Innovative Medicine. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55651-0_15
Download citation
DOI: https://doi.org/10.1007/978-4-431-55651-0_15
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55650-3
Online ISBN: 978-4-431-55651-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)