
Abstract
Combined pulmonary fibrosis and emphysema (CPFE) is a common but under-recognized syndrome characterized with distinct profiles of clinical, functional and radiological features from both pulmonary fibrosis and emphysema. Tobacco smoking may be situated at a major cause and differentiate prognosis of CPFE associated with PH and lung cancer from that of pulmonary fibrosis or emphysema alone. Further studies are needed to ascertain the aetiology, morbidity, mortality and management of CPFE. The establishments of definition, classification and staging of CPFE, including delineation of boundaries between IPF and CPFE, also are required. Better understanding of CPFE will be able to develop future therapeutic strategies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Combined pulmonary fibrosis and emphysema
- Idiopathic pulmonary fibrosis
- Emphysema
- Pulmonary hypertension
- Lung cancer
1 Introduction
Pulmonary emphysema and idiopathic interstitial pneumonia (IIP) are entities defined by distinct clinical, functional, radiological and pathological characteristics. Emphysema is defined as an enlargement of the distal air spaces from terminal bronchioles to alveoli due to the destruction of alveolar walls [1]. Idiopathic pulmonary fibrosis (IPF) is the most common IIP and characterized generally by not only a progressive and fatal disease but also a histopathological and/or radiological pattern of usual interstitial pneumonia (UIP) [2]. Despite traditionally considered as separate disease states, several studies have described series of combined pulmonary fibrosis and emphysema (CPFE). The combination of both disorders was described over 40 years ago by Auerbach et al. who examined a pathological study of 1824 autopsy lungs [3]. Since Cottin et al. described that CPFE exhibited emphysema at upper lobes and fibrosis at lower lobes on chest high-resolution computed tomography (HRCT) in 2005 [4], CPFE has been proposed as a distinct syndrome [5–7].
2 Epidemiology
The prevalence of CPFE in IIP has been estimated approximately from 8 to 50 % [8–17]. The variety of CPFE prevalence may be influenced by referral bias, recruit strategy and criteria definition. Nevertheless, the combination of emphysema and pulmonary fibrosis is considered as a relatively common finding on HRCT scanning (Table 13.1). Although a pattern of UIP/IPF appears to be the most common finding at lower lobes in CPFE, other patterns of fibrotic IP have been reported in the setting of CPFE [4, 9, 18, 19]. In addition, Cottin et al. described the heterogeneity of pulmonary fibrosis in CPFE associated with connective tissue diseases (CTDs) [20]. On the other hand, several patterns of emphysematous change, including paraseptal, centrilobular and bullous change, also have been reported in the setting of CPFE [4, 12, 17, 19, 20].
Most of cohort studies have demonstrated that CPFE is often observed in males over 65 years of age who are current smokers or ex-smoker of >40 pack-years [6, 7] (Table 13.2). However, despite demonstrating a similar smoking history and pulmonary function profile, CPFE associated with CTDs was observed more female dominantly and younger than “classic CPFE” [20].
3 Physical Findings
Exertional dyspnoea (functional class III or IV of the New York Heart Association) is the most common symptom despite of relatively normal spirometric values among CPFE patients [4]. Physical examination often reveals bibasilar inspiratory crackles and finger clubbing. Other signs and symptoms reported are cough, sputum production and asthenia among CPFE patients [4].
4 Radiological Features
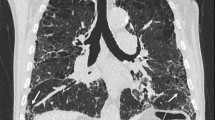
Radiological findings of CPFE are considered to be characterized by emphysema at upper lobes and fibrosis at lower lobes. On chest X-ray findings of CPFE, an interstitial pattern or reticulonodular infiltration is present at basal periphery of bilateral lungs, while a hyperlucency with thinning or reduction in pulmonary vessels is observed at bilateral apices. However, HRCT scanning is the most appropriate tool for the diagnosis of CPFE, because the estimation by chest X-ray alone does not necessarily confirm the diagnosis of this entity (Fig. 13.1).

High-resolution computed tomography (HRCT) of a male smoker aged 77 years with combined pulmonary fibrosis and emphysema. (a) Presence of paraseptal emphysema and subpleural bullae in bilateral upper lobes. (b) Images of subpleural honeycombing and traction bronchiectasis in bilateral lower lobes
Cottin et al. described the radiological criteria to determine CPFE as follows: firstly, the presence of emphysema on HRCT, defined as well-demarcated areas of decreased attenuation in comparison with contiguous normal lung and marginated by a very thin (<1 mm) wall or no wall, and/or multiple bullae (>1 cm) with upper zone predominance, and, secondly, the presence of diffuse parenchymal lung disease with significant pulmonary fibrosis on HRCT, defined as reticular opacities with peripheral and basal predominance, honeycombing, architectural distortion and/or traction bronchiectasis or bronchiolectasis; focal ground-glass opacities and/or areas of alveolar condensation may be associated but should not be prominent [4].
UIP is the most common pattern [4, 9, 18, 19], as reporting the most frequent presence of honeycombing in the wide variety of HRCT findings of CPFE. Other patterns reported in pulmonary fibrosis include reticular opacities, ground-glass opacities, traction bronchiectasis and architectural distortion, which are compatible with non-UIP, smoking-related interstitial pneumonia (IP) or unclassifiable interstitial lung diseases (ILDs) (Table 13.3).
The various findings of emphysema also are present at upper lobes, including centrilobular, paraseptal and bullous change [4, 12, 17, 19, 20]. Centrilobular and paraseptal emphysema appear to be typical features of CPFE (Table 13.3). In the various findings of emphysema, Sugino et al. indicated a paraseptal emphysema as a predictor of poor prognosis among 46 CPFE patients [17].
5 Pathological Features
As the wide variety of radiological findings correlate closely with histopathological data, UIP is the most common pattern of pathological findings in accordance with the most frequent presence of honeycombing in HRCT findings of CPFE [4, 9]. Other patterns reported in pathological findings include nonspecific IP, desquamative IP, respiratory bronchiolitis-related ILD and unclassifiable ILD [4, 9, 18, 19]. Because of the pathological heterogeneity, a pathological criterion for diagnosis of CPFE is not mapped out [12].
6 Pulmonary Function and Gas Exchange
Pulmonary function tests show normal or subnormal findings of respiratory volume and flow, despite of severe dyspnoea on exertion and extensive radiographic findings among CPFE patients [4]. The coexistence of emphysema and fibrosis leads to an influence of pulmonary function that profiles each other in CPFE. Forced vital capacity (FVC), forced expiratory volume in the first second (FEV1) and total lung capacity (TLC) are usually within normal or subnormal range (Table 13.2). The unexpected subnormal spirometric findings in CPFE may be explained by counterbalancing effects of the restrictive disorder in pulmonary fibrosis and the propensity to hyperinflation in emphysema [5, 6]. The hyperinflation and increased pulmonary compliance in emphysema probably compensate for the loss of volume in fibrosis, resulting in the preservation of spirometric findings.
Additionally to the spurious preservation of spirometric findings, a marked impairment of gas exchange manifested as a reduction in diffusing capacity of the lung for carbon monoxide (DLco) is common [4] (Table 13.2). The severe impairment of gas exchange is likely due to not only reduction in pulmonary vascular surface and capillary blood flow but also thickness of alveolar wall.
Resting and exertional hypoxaemia also are common among CPFE patients. In the series reported by Cottin et al., CPFE patients exhibited 63 ± 14 mmHg of partial pressure of oxygen in arterial blood (PaO2) at rest on room air with 41 ± 16 mmHg of alveolar-arterial oxygen gradient and exertional decrease by 8.9 ± 5.7 % of arterial oxygen saturation (SpO2) during a 6-min walk test (6MWT) [4]. Hypercapnia does not appear to be as frequent as hypoxaemia in CPFE [4, 19]. In a consequence of exertional hypoxaemia, Jankowich M.D. et al. reported that 80 % required oxygen therapy over a 5-year period among 20 CPFE patients [12].
7 Pathogenesis
Tobacco smoking has been suggested as a major cause, as a history of smoking is a constant factor in all the cohort studies [6, 7]. In addition, possible factors also have been identified, such as exposure to agrochemical compounds or asbestos [19]. While underlying mechanisms resulted from smoking exposure have been considered, the precise and exact pathogenesis of CPFE is unclear. Although potential roles for tumour necrosis factor-α [21] and platelet-derived growth factor-β [22] have been suggested in various animal models of CPFE, it is unclear whether any of these models represent typical CPFE in human smokers.
Rogliani et al. reported an augmented expression of metalloproteinases (MMPs) in fibroblasts of lungs from CPFE patients compared with emphysema patients, suggesting the roles for MMPs in acceleration of pulmonary fibrotic process in CPFE [23]. Tasaka et al. reported an increased CXC chemokine levels in bronchoalveolar lavage fluid from CPFE patients compared with IPF patients, suggesting a relationship between the increased CXC chemokines and emphysematous change in CPFE [24]. Tzouvelekis et al. reported an elevated serum antinuclear antibodies and CD20-positive B-cell infiltration into the lungs from CPFE patients compared with IPF patients, suggesting a presence of underlying autoimmune disorders in CPFE [25]. Collectively, several mechanisms as mentioned above resulted from smoking exposure appear to underlie the pathogenesis of CPFE.
Recently, individual genetic backgrounds have been considered as a predisposal in the development of CPFE. Cottin et al. reported a heterozygous mutation in SFTPC (the gene encoding surfactant protein C) in a nonsmoking young female with CPFE [26]. In addition, telomeropathy also may be on the verge of becoming a predisposal in the development of CPFE, since shorter telomeres appear to be associated with pulmonary fibrosis, emphysema and smoking exposure [27]. However, the underlying genetic predisposition in CPFE remains to be elucidated because of the limited reports.
8 Complication
8.1 Pulmonary Hypertension
Pulmonary hypertension (PH) is particularly prone to develop and a common complication during the clinical course of CPFE. Cottin et al. reported 47 % prevalence of PH (definition as an estimated systolic pulmonary arterial pressure (eSPAP) ≥ 45 mmHg by echocardiography system) among 61 CPFE patients [4]. In the study, 5-year survival rates were 25 % and 75 %, respectively, among CPFE patients with and without PH, indicating a presence of PH as a clinical determinant of prognosis in CPFE. Mejía et al. reported a complication of PH that was more frequent and severe in CPFE patients than in IPF patients [14]. The study also demonstrated a lower survival rate in CPFE patients than in IPF patients, indicating a complication of severe PH (eSPAP > 75 mmHg by echocardiography system) as a predictor of mortality in CPFE.
Cottin et al. also reported 1-year survival rate of 60 % among 40 patients with CPFE and PH, with a mean PAP of 40 ± 9 mmHg by examination of right heart catheterization (RHC), and indicated both a reduced cardiac index (<2.4 L/min/m2) and elevated pulmonary vascular resistance (>485 dyne/s/cm5) as predictors of poor prognosis [28]. In the study, while the diagnosis of PH was established at mean of 16 months after the initial diagnosis of CPFE, no significant benefit of medical therapy, including pulmonary vasodilators, corticosteroids, immunomodulators and bronchodilators, was observed.
Therefore, as no data currently supports treatment of PH in CPFE with pulmonary vasodilators, oxygen therapy and referral for lung transplantation, if appropriate, appear to be the most reasonable option for the management [29]. Randomized controlled trials are urgently required for the establishment of therapeutic strategy of PH in CPFE.
8.2 Lung Cancer
CPFE patients may be at a high risk of developing lung cancer, as both pulmonary fibrosis and emphysema possess a possibility to predispose lung cancer. Despite several limited evidence in retrospective studies, an increased prevalence of lung cancer among CPFE patients has been reported.
Kitaguchi et al. reported 46.8 % and 7.3 % prevalence of lung cancer, respectively, in 47 CPFE patients and 82 emphysema patients [19], whereas Kurashima et al. reported 33.3 % and 12.1 % prevalence, respectively, in 129 CPFE patients and 233 IPF patients [13]. The more prevalence of lung cancer among CPFE patients may be influenced by referral bias and recruit strategy. Nevertheless, it is noteworthy that CPFE appears to be prone to cause lung cancer. Smoking-related lung cancers such as squamous cell lung cancer and small cell lung cancer (SCLC) appear to predominate in pathological features of lung cancer in CPFE [19, 30, 31].
Usui et al. described clinical characteristics of 101 CPFE subjects in 1143 patients with lung cancer [30]. In the study, CPFE subjects demonstrated the worst median overall survival (10.8 months) compared with normal subjects (i.e. without lung disease) (n = 623, 53.0 months) and emphysema subjects (n = 404, 21.9 months). The poor prognosis of lung cancer may be explained by not only earlier and more frequent recurrence of lung cancer but also less tolerance to chemotherapy and performance status in association with acute exacerbation of ILD among CPFE patients than among others [30, 32].
Collectively, as the more prevalence and worse clinical course of lung cancer have been demonstrated, CPFE patients may be at a high risk of developing lung cancer. However, it remains uncertain whether CPFE is an independent risk factor of lung cancer.
9 Treatment
Therapeutic options for CPFE patients are limited and may require treatment for both IPF and emphysema [5–7]. Smoking cessation, of course, is an obvious objective and should be encouraged and supported. Oxygen therapy is appropriate for the management of hypoxaemia. Inhaled bronchodilators are more often prescribed among CPFE patients than among patients with pulmonary fibrosis alone [12, 15]. Treatment for CPFE patients with systemic corticosteroids and immunomodulator therapy (e.g. azathioprine, N-acetylcysteine or pirfenidone) have been considered similar to that for IPF, but without beneficial results in the published series.
A possibility of using pulmonary vasodilators (e.g. endothelin-1 receptor antagonist, prostanoids or phosphodiesterase type 5 inhibitors) has been raised for therapeutic strategy of PH in CPFE, but no studies have been published to date on the issue. Both hypoxic pulmonary vasoconstriction (HPV), a phenomenon to avoid worsening arterial oxygenation, and imbalance in ventilation/perfusion ratio (VA/Q) due to abnormal changes in pulmonary vascular bed and airway in CPFE may predispose PH. Pulmonary vasodilators have a possibility to worsen arterial oxygenation due to inhibition of HPV and potentiation of VA/Q mismatch by nonselective vasodilation of pulmonary vessels. Therefore, as no data currently supports treatment of PH in CPFE with pulmonary vasodilators, a referral for lung transplantation, if appropriate, appears to be the most reasonable option for the management [29].
10 Survival
The median survival of CPFE in reported series has been ranged from 1.8 to 8.5 years (Table 13.4). The various survival of CPFE may be influenced by a presence of either complication or acute exacerbation. However, it remains controversial whether CPFE patients have worse survival than patients with pulmonary fibrosis or emphysema alone.
Mejía et al. reported a worse survival in association with severe and more frequent PH in CPFE patients than in IPF patients [14]. Sugino et al. also indicated both a paraseptal emphysema and PH as predictors of worse survival in CPFE patients than in IPF patients [17]. In addition, despite the spurious preservation of spirometric findings, Schmidt et al. indicated a longitudinal decline in FEV1 as a more superior predictor of mortality than other pulmonary function parameters in CPFE patients [16]. In contrast, other studies demonstrated comparable or better survival in CPFE patients than in patients with pulmonary fibrosis or emphysema alone [11–13, 15, 24, 33]. The basis for these discrepant results is unclear and may be influenced by referral bias, recruit strategy and criteria definition.
11 Conclusion
A number of published series have demonstrated that CPFE is a common syndrome characterized with distinct profiles of clinical, functional and radiological features from both pulmonary fibrosis and emphysema. Tobacco smoking may be situated at a major cause and differentiate prognosis of CPFE associated with PH and lung cancer from that of pulmonary fibrosis or emphysema alone. Is CPFE an independent entity? CPFE is a distinct but under-recognized and common syndrome with the characteristic presentation as mentioned above [7]. However, it is clear that many aspects remain to be explored for better recognition of CPFE. Further studies are needed to ascertain the aetiology, morbidity, mortality and management of CPFE, with or without PH. The establishments of definition, classification and staging of CPFE, including delineation of boundaries between IPF and CPFE, also are required. Better understanding of CPFE will be able to develop future therapeutic strategies.
References
Celli BR, MacNee W, ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932–46.
Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–48.
Auerbach O, Garfinkel L, Hammond EC. Relation of smoking and age to findings in lung parenchyma: a microscopic study. Chest. 1974;65:29–35.
Cottin V, Nunes H, Brillet PY, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005;26:586–93.
Portillo K, Morera J. Combined pulmonary fibrosis and emphysema syndrome: a new phenotype within the spectrum of smoking-related interstitial lung disease. Pulm Med. 2012;2012:867870.
Jankowich MD, Rounds SI. Combined pulmonary fibrosis and emphysema syndrome: a review. Chest. 2012;141:222–31.
Cottin V. The impact of emphysema in pulmonary fibrosis. Eur Respir Rev. 2013;22:153–7.
Akira M, Yamamoto S, Inoue Y, et al. High-resolution CT of asbestosis and idiopathic pulmonary fibrosis. AJR Am J Roentgenol. 2003;181:163–9.
Choi SH, Lee HY, Lee KS, et al. The value of CT for disease detection and prognosis determination in combined pulmonary fibrosis and emphysema (CPFE). PLoS One. 2014;9:e107476.
Copley SJ, Wells AU, Sivakumaran P, et al. Asbestosis and idiopathic pulmonary fibrosis: comparison of thin-section CT features. Radiology. 2003;229:731–6.
Doherty MJ, Pearson MG, O’Grady EA, et al. Cryptogenic fibrosing alveolitis with preserved lung volumes. Thorax. 1997;52:998–1002.
Jankowich MD, Rounds S. Combined pulmonary fibrosis and emphysema alters physiology but has similar mortality to pulmonary fibrosis without emphysema. Lung. 2010;188:365–73.
Kurashima K, Takayanagi N, Tsuchiya N, et al. The effect of emphysema on lung function and survival in patients with idiopathic pulmonary fibrosis. Respirology. 2010;15:843–8.
Mejía M, Carrillo G, Rojas-Serrano J, et al. Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest. 2009;136:10–5.
Ryerson CJ, Hartman T, Elicker BM, et al. Clinical features and outcomes in combined pulmonary fibrosis and emphysema in idiopathic pulmonary fibrosis. Chest. 2013;144:234–40.
Schmidt SL, Nambiar AM, Tayob N, et al. Pulmonary function measures predict mortality differently in IPF versus combined pulmonary fibrosis and emphysema. Eur Respir J. 2011;38:176–83.
Sugino K, Ishida F, Kikuchi N, et al. Comparison of clinical characteristics and prognostic factors of combined pulmonary fibrosis and emphysema versus idiopathic pulmonary fibrosis alone. Respirology. 2014;19:239–45.
Jankowich MD, Polsky M, Klein M, et al. Heterogeneity in combined pulmonary fibrosis and emphysema. Respiration. 2008;75:411–17.
Kitaguchi Y, Fujimoto K, Hanaoka M, et al. Clinical characteristics of combined pulmonary fibrosis and emphysema. Respirology. 2010;15:265–71.
Cottin V, Nunes H, Mouthon L, et al. Combined pulmonary fibrosis and emphysema syndrome in connective tissue disease. Arthritis Rheum. 2011;63:295–304.
Lundblad LK, Thompson-Figueroa J, Leclair T, et al. Tumor necrosis factor-alpha overexpression in lung disease: a single cause behind a complex phenotype. Am J Respir Crit Care Med. 2005;171:1363–70.
Hoyle GW, Li J, Finkelstein JB, et al. Emphysematous lesions, inflammation, and fibrosis in the lungs of transgenic mice overexpressing platelet-derived growth factor. Am J Pathol. 1999;154:1763–75.
Rogliani P, Mura M, Mattia P, Ferlosio A, et al. HRCT and histopathological evaluation of fibrosis and tissue destruction in IPF associated with pulmonary emphysema. Respir Med. 2008;102:1753–61.
Tasaka S, Mizoguchi K, Funatsu Y, et al. Cytokine profile of bronchoalveolar lavage fluid in patients with combined pulmonary fibrosis and emphysema. Respirology. 2012;17:814–20.
Tzouvelekis A, Zacharis G, Oikonomou A, et al. Increased incidence of autoimmune markers in patients with combined pulmonary fibrosis and emphysema. BMC Pulm Med. 2013;13:31.
Cottin V, Reix P, Khouatra C, et al. Combined pulmonary fibrosis and emphysema syndrome associated with familial SFTPC mutation. Thorax. 2011;66:918–19.
Nunes H, Monnet I, Kannengiesser C, et al. Is telomeropathy the explanation for combined pulmonary fibrosis and emphysema syndrome?: report of a family with TERT mutation. Am J Respir Crit Care Med. 2014;189:753–4.
Cottin V, Le Pavec J, Prévot G, et al. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur Respir J. 2010;35:105–11.
Seeger W, Adir Y, Barberà JA, et al. Pulmonary hypertension in chronic lung diseases. J Am Coll Cardiol. 2013;62:D109–16.
Usui K, Tanai C, Tanaka Y, et al. The prevalence of pulmonary fibrosis combined with emphysema in patients with lung cancer. Respirology. 2011;16:326–31.
Girard N, Marchand-Adam S, Naccache JM, et al. Lung cancer in combined pulmonary fibrosis and emphysema: a series of 47 Western patients. J Thorac Oncol. 2014;9:1162–70.
Kumagai S, Marumo S, Yamanashi K, et al. Prognostic significance of combined pulmonary fibrosis and emphysema in patients with resected non-small-cell lung cancer: a retrospective cohort study. Eur J Cardiothorac Surg. 2014;46:e113–19.
Akagi T, Matsumoto T, Harada T, et al. Coexistent emphysema delays the decrease of vital capacity in idiopathic pulmonary fibrosis. Respir Med. 2009;103:1209–15.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this chapter
Cite this chapter
Morio, Y., Takahashi, K. (2016). Combined Pulmonary Fibrosis and Emphysema (CPFE). In: Nakamura, H., Aoshiba, K. (eds) Idiopathic Pulmonary Fibrosis. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55582-7_13
Download citation
DOI: https://doi.org/10.1007/978-4-431-55582-7_13
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55581-0
Online ISBN: 978-4-431-55582-7
eBook Packages: MedicineMedicine (R0)