
Abstract
Circulating tumor cells (CTCs) or fetal nucleated erythrocytes in maternal peripheral blood are extremely rare cell population (~ 1/105) in circulating blood, though these cells are useful for diagnose diseases. Flow cytometry enabled us to detect such a rare cell populations. However, in order to sort those rare cell populations by fluorescent activated cell sorter (FACS), it is necessary to stain the cells with fluorescent conjugated antibody ex vivo and to obtain theoretically few-hundred milliliters of blood. Therefore, high-speed closed system for analysis and isolation of rare cell populations within the blood without cell staining is desirable. In this chapter, we described analysis of physical characteristic of hematopoietic cells by microfluidics-based devices for efficient detection of rare cell population in circulating blood.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
An organ (or tissue) that constitutes as a whole an individual is consisted itself of cells of distinct types. For example, the adult bone marrow is occupied with mature hematopoietic cells, including granulocytes, monocytes, macrophages, dendritic cells, B cells, natural killer cells, T cells, erythrocytes, and megakaryocytes, immature hematopoietic progenitor cells, and non-hematopoietic cells, such as mesenchymal cells and perivascular cells. Numerous efforts had been made to enrich and isolate distinct cell type. Density gradient centrifugation separates cells in organs based on distinct size and widely used in biological researches. Magnetic beads separation exploits antigen-antibody reactions and has been an effective method to purify cell populations of interest [1]. The MACS technology (a magnetic separation method) employed the principle that each cell types within a heterogeneous population express distinct surface markers (or surface antigens) on the cell membrane. The development of the flow cytometer, which is originally developed by Leonard Herzenberg at Stanford University in California and Marvin Van Dilla at the Los Alamos National Laboratories in New Mexico, revolutionized our approach to cell biology research, enabling isolation of cells with a particular antigen expression pattern from a wide variety of biological source. It has significantly contributed to provide new insights into our understanding of molecular cell biology, particularly in the fields of immunology, hematology, and stem cell biology. These researches also have highlighted the importance of identifications and investigations of rare populations within a heterogeneous population.
Flow cytometry is an analytical technique that measures the fluorescence intensities of a single cell stained with antibodies against different antigens tagged with different fluorophores. Another outstanding feature with the flow cytometer include isolation of viable cells expressing the proteins of interest while simultaneously analyze 10 parameters (proteins) or more from a heterogeneous population. A flow cytometer is consisted of two systems: a fluidic system and a measuring system. The fluidic system is designed to vertically align and transport cells in a sample to the flow cells by generating a laminar flow of sheath flow surrounding the sample flow from the sample loading port. A flow cytometer with the cell-sorting feature is also equipped with an electrostatic deflection system. The measuring system acquires Forward scatter (FSC) and Side scatter (SSC) as the aligned cell pass through lasers one by one. At the same time, fluorescence emitted from the fluorophores conjugated with antibodies bind to the cell as a result of excitation by the lasers is directed to a series of dichroic mirrors and optical filters to filter out noise and arrives at photomultiplier tube (PMT). The PMTs effectively amplify photons of the fluoresence by converting it to electrons, allowing measurement of the quantity of photons as a form of voltage.
With this technology, we have previously identified a population with a defined antigen expression pattern that highly purified murine hematopoietic stem cells, which represent only one in 300,000 cells within an adult bone marrow in the steady state. Hematopoietic stem cells give rise to all mature blood cells including erythrocytes, platelets, and leukocytes through step-wise differentiation processes with distinct intermediate progenitor cells while maintaining its population via self-renewal and sustain the blood cell populations throughout an adult’s life. Hematopoietic stem cells reside in bone marrow cells, spleen, and blood in adult, and fetal liver in early embryo. Although the existence of the stem cells and its importance have been widely recognized by the bone marrow transplantation, direct experimental proof was difficult to perform due to the lack of the technology to isolate a subpopulation within a heterogeneous population at that time. Furthermore, competitive dilution assay postulated hematopoietic stem cell concentration to be 0.7–1.1 and 1.0–1.6 per 105 bone marrow cells, suggesting its isolation extremely difficult [2, 3]. The development of flow cytometers made it possible for researchers to prospectively isolate any cells of interest in the bone marrow of mice and examine its long-term ability to reconstitute myeloid and lymphoid cells in recipients’ body, followed by long-term serial transplantation [4]. The first report appeared in 1988 by Spangrude et al. using specific cell surface markers (Thy-1loLineage markers−Sca-1 + ) [5]. Inspired by these seminal studies, we have demonstrated the identification of a set of surface markers that highly enrich murine hematopoietic stem cells (Lineage markers−c-Kit + Sca-1 + CD34low/−) capable of long-term reconstitution of lymphohematopoietic system in recipients when transplanted with a single donor cell [6]. Further characterization of the population by single cell transplantation assays have shown that the most primitive hematopoietic stem cells compartment present with distinct functional capabilities based on their CD150 expression levels [7]. These findings have highlighted the impact of clonal analysis on stem cell biology using flow cytometers, which is directly translatable to other fields of basic biology as well as human studies. Figure 6.1 illustrates a schematic of a flow cytometer and an example of flow diagrams in purifying murine hematopoietic stem cells.

The workflow of a single cell transplantation of murine hematopoietic stem cells in the Nakauchi Lab. a Representative flowdiagrams showing a gating strategy to isolate a murine hematopoietic stem cell population (Lineage marker−/CD34low/Sca-1 Sc/C-kit C−). The antibodies used and its fluorescent conjugate is indicated. b A schematic illustrating the workflow of single cell transplantation assays. A bulk of whole bone marrow cells (WBMCs) is isolated from legs and pelvis of donor mice and subsequently magnetically selected against c-kit positivity. After the selection, the cells are stained with a cocktail of antibodies unique to a hematopoietic stem cell population and a single cell of the population is sorted into a well of a 96-well plate via a flow cytometer. The sorted cell is transplanted into a lethally irradiated recipient mouse together with a defined number of competitor cells. The fates of the donor cells are analyzed monthly using peripheral blood (PB) of each recipient. The self-renewal ability of the donor cells is assessed by serial transplantation, followed by monthly PB analysis at least up to 4 month after transplantation. Parts of image are adopted from [22]
2 Circulating Tumor Cells (CTCs)
Activating invasion and metastasis are one of the hallmarks of cancer and account for nearly 90 % of cancer-associated mortality [8]. However, it remains the most poorly understood aspects of cancer biology. Recent research have raised the notion that circulating tumor cells (CTCs) bridges a primary site to a distant organ and therefore understanding it would provide new therapeutic targets and diagnostic biomarkers [9]. In accordance with the hypothesis, a number of studies have shown correlations between high CTC numbers and severity of the disease, increased metastasis, and decreased time to relapse [10]. Moreover, CTCs have been detected even in cancer patients at the early stage, suggesting its diagnostic values [11]. In spite of these potential, researchers in this field have been encountering many obstacles [9]. One such obstacle includes technical issues inherited to the detection system currently employed in the clinic. Although the development of an automated immunomagnetic enrichment and staining system (CellSearchTM) have accelerated our understanding of CTCs, the use of epithelial-specific markers and enrichment processes may result in loss of subpopulations of cancer cells, particularly cancer stem cells that had undergone epithelial-to-mesenchymal transition [12, 13]. These findings pose the need for the development of a novel detection and isolation systems to further evaluate the utility and biological significance of CTCs.
3 Prenatal Diagnosis for Detection of Chromosomal Abnormality of Embryo
Prenatal diagnosis is the test for detection of congenital abnormality of embryo. There are two types of diagnosis methods including definitive diagnosis such as amniocentesis, chorionic villus sampling and non-definitive diagnosis such as ultrasonography, maternal serum screens. In order to perform definitive diagnosis, it is necessary to obtain amniotic fluid, villus or fetal blood by invasive strategy with the risk of abortion. On the other hand, ultrasonography and maternal serum screens are non-invasive technique; however, those are the inspection for the morbidity rate not for the definitive diagnosis. Recently Lo et al. reported that there are large quantities of cell free embryonic DNA in maternal circulating blood and the concentration of cell free DNA fragment derived from each chromosome are proportional to the mass of each chromosome [14, 15]. The ratio of embryonic cell free DNA fragments in maternal circulating cell free DNA fragments is about 10 ~ 15 % and this percentage increase as the week number in pregnancy increase [16]. From these observations, non-invasive prenatal testing (NIPT) which can detect abnormality in chromosomal number was developed [17]. In order to detect the chromosomal abnormality of fetus, over 107 embryonic cell free DNA fragments are sequenced by next generation sequencer and analyzed by comparing with human genome resources to determine which chromosome is the origin of fragment. Then the number of fragments in each chromosome is counted and compare with the number of fragments derived from embryo with normal karyotype to estimate the quantity of the number of each chromosome in specimen. Although the detection rate of chromosomal abnormality is high (ch13 trisomy; 91.7 %, ch18 trisomy; 100 %, ch21 trisomy; 99.1 %) by NIPT, NIPT is not definitive diagnosis because only three chromosomes (ch13, 18, 21) can be analyzed currently and chromosome abnormality other than chromosome number (chromosomal translocation or mosaicism) cannot be detected [18] (Table 6.1). Therefore non-invasive definitive diagnosis technology for detection of chromosomal abnormality is still required. It has been identified the existence of fetal nucleated erythrocytes in maternal peripheral blood. Although these cells are rare cell population (one in tens of thousands of circulating blood cells), utilization of these cells for non-invasive prenatal diagnosis has been actively studied [19–21].
4 Hurdles and Solutions in Search for Rare Populations in Blood
Despite its significant contribution, ex vivo cell staining with antibodies and requirement for highly skilled operators have been major hurdles with flow cytometric analysis. Furthermore, in order to obtain a sufficient number of such a rare cell population for identification as well as down-stream analysis with high robustness, precision, and accuracy, one needs to obtain theoretically few-hundred milliliters of blood from a specimen. To overcome these issues, we aim to develop a novel high-speed closed system for analysis and isolation of rare cell populations within the blood. We propose to analyze each cell in the blood at an ultra-high speed while they are in an extracorporeal circulation. The prototype of the system is illustrated in Fig. 6.2.

A conceptual design for isolating a rare cell population within blood. A schematic of the system is illustrated on the left. Detection systems that measure potential parameters are integrated in an extracorporeal circulation from a patient (e.g. a cancer patient). The cell isolation system is located after the detection system to sort out cells of interest based on the measured parameters. Potential parameters include commercially available CTC-specific probes (ALDEFLUOR kit; StemCell Technologies, Vancouver, Canada), physical properties such as cell size, viscoelasticity and weights
5 Analysis of Physical Properties of Hematopoietic Cells with a Microfluidic Device on an Ultra-High Speed Image Acquisition System
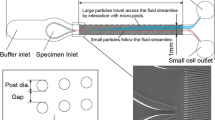
Since ex vivo antibody staining is not applicable in the proposed system, development of alternative parameters that characterize and distinguish cell types at ease is a prerequisite for this project. Although it has been long known that each cell type possesses distinct physical properties, such as weight, elasticity, dimension, and viscosity, a few studies have attempted such measurements in part due to technological hurdles. We have developed a novel microfluidic channel and an ultra high speed online image capturing system, respectively and attempted to measure physical properties of various cells, including murine bone marrow cells (mBMCs), human peripheral blood mononuclear cells (hPBMCs), a human cancer cell line, a mouse cancer cell line, and erythroblast cell lines at different stages of maturation established pluripotent stem cells, passing through the microfluidic channel while acquiring its real time images. This system allows us to measure the passing time of a cell while it travels through the region with a narrowed diameter deforming. With the passing times and diameters, we can obtain viscoelasticity of a cell. The schematic of the prototypic system is depicted in Fig. 6.3.

Prototypic design of image acquisition system. Sequential images of a cell traveling through the channel are acquired by a high-speed camera connected to a conventional microscopy and to a computer. The high-speed camera is operated by software via the computer. An illustration of the microfluidic chip is shown on the top. The software processes the acquired images and generates a 2-D plot as shown in Fig. 6.4. Images are provided by the Kaneko lab
We performed the following experiments by using the ultra high-speed image acquisition system that we have developed. At the one day before or at the day of experiments, we prepared freshly isolated bone marrow cells in a femur of a C57BL/6 mouse, human PBMCs from a healthy volunteer, a human leukemia cell line (EOL-1: chronic eosinophilic leukemia), a mouse cancer cell line (4T1: mammary gland tumor), and the erythroblast cell line. The prepared cells were diluted to a density of 1 × 107 cell/ml, if possible, using either saline or phosphate-buffered saline with 3 % Fetal bovine serum (vol/vol) immediately before running the experiment. We used human erythrocytes prepared in the lab as a reference control. Subsequently, the cell suspension was drawn through the polyvinyl tubing to which a 2.5 ml syringe is attached at the other end and the tip was inserted to the inlet of the microfluidic channel. The cells were introduced into the channel by applying a pressure with the syringe piston. When the flow rate around the region with a narrowing diameter within the channel reached to a constant value, we started acquiring images for one minute. We repeated the experiments several times switching microfluidic channels with different diameters (6–10 µm). Cell diameters and passing times were automatically extrapolated by analyzing the images via a tailor-made program. The results were exported as a excel spreadsheet containing a 2-D plot (Passing time v.s. cell diameter). Figure 6.4 shows representative data from the experiments.

A representative plot that can be obtained from an experiment. The horizontal axis represents the diameter of a cell at the entrance of the narrowed region in micrometer (µm), whereas the vertical axis shows the passing time of a cell in millisecond (msec). Each dot represents a cell passed through the channel
The plots obtained with mBMCs experiments clearly indicated a linear correlation between passing time and cell diameter, whereas no such trend was observed from the plots using human erythrocytes (the reference control). Moreover, we observed clusters of cells on the plot that do not fit onto the regressed line, indicating that they behave differently in the channel. Some of those cells may be dying as postulated from its morphology (Fig. 6.5). Experiments with tumor cell lines unexpectedly demonstrated heterogeneous behaviors although they are clonal cell lines. Importantly, when human PBMCs and a tumor cell line were mixed together and simultaneously investigated for their passing times and diameters, it was possible to distinguish the two from the plots, which was generated from an unbiased method (Fig. 6.6). These results suggest the utility of the proposed system and physical properties as an alternative parameter for isolating a rare population in the blood.

A representative 2-D plot obtained with mouse bone marrow cells. Combined data from five independent measurements is shown. There is a positive correlation between passing time (T) and the length (L). Note two large populations on the plot (the gray circle; the lower left and the orange circle; lower middle). Some cells showed unexpected behaviors (the red circle; top middle and the green circle; lower right). L length in micrometer (or diameter of a cell), T the passing time in millisecond

A representative 2-D plot obtained with a mixture of human peripheral blood and the 4T1 cancer cell line. The plot from one measurement is shown here. The cluster on the left represents human peripheral blood cells (hPBCs) and the right cluster designates the 4T1 cancer cells. Note the distribution of the cancer cells spreading widely between 15–25 µm through 20–80 msec., although they all derived from a clonal origin. It appears possible to roughly discriminate hPBC from the cancer cells. L length in micrometer (or diameter of a cell), T the passing time in millisecond
Furthermore, another experiments conducted with PSC-derived erythroblasts under maturation and matured erythrocytes revealed a positive correlation between nucleus sizes and the passing times (Fig. 6.7). Since high elastic deformability of erythrocytes is attributed to its nucleation during the maturation stages and is a crucial property in vivo as it travel through the blood vessel, the result further strengthen the notion that it is of importance to use enucleated and fully maturated erythrocytes for an alternative blood transfusion source. Overall, the current system may also be useful for the quality assessment of the erythrocytes generated from the PSC technology upon the clinical application.

2-D plots obtained with erythroblast cell lines at different stages of maturation. a Representative plots obtained with erythroblast cell lines at their maturation from proerythroblasts. The maturity of the cell lines is descending from 1, being erythroblast line 1 around the proethryroblast stage. The most matured line of the four (erythroblast line 4) comprised of the cells with smaller cell diameter. D length in micrometer (or diameter of a cell), T the passing time in millisecond; d the width of the narrowed region of the channel, where images are taken. b A histogram of the combined data of all four lines. Frequencies are plotted against diameters of the cells corresponding to (a). Most of the cells from line 4 appears on the left side of the histogram
6 Conclusion
The current study has demonstrated the utility of the proposed system to distinguish distinct cell types within a heterogeneous population with ease and high throughput. However, we observed some potential problems with the current system for further developmental processes. We sometimes encountered the situation where the entrance speeds of incoming cells, which are monitored at the entrance of the narrowing region, vary overtime during a data acquisition, making the data inconsistent. One possible cause of the problem may arise from the fluidic system exploited to the current system, which is generated by the potential energy. If so, we could overcome this by adopting an external sheath flow system utilized to flow cytometer. Another issue worthwhile to mention is related to surface finishing of the microfluidic chips. As we process high number of cells at a high speed, some debris from cells or media accumulate at the bottom of the channel, making a plug in between the entrance and the exit. Considering that blood contains a wide range of sticky material, this issue should be resolved. We may avoid this by coating the chip with serum, silane-coupling agents for glass bottom or by locating a filter before the chip. Although further improvement is necessary, we suppose microfluidics-based devices described here have a potential for efficient detection of rare cell population in circulating blood.
References
Miltenyi S, Müller W, Weichel W, Radbruch A (1990) High gradient magnetic cell separation with MACS. Cytometry 11:231–238
Harrison DE, Jordan CT, Zhong RK, Astle CM (1993) Primitive hemopoietic stem cells: direct assay of most productive populations by competitive repopulation with simple binomial, correlation and covariance calculations. Exp Hematol 21:206–219
Zhong RK, Astle CM, Harrison DE (1996) Distinct developmental patterns of short-term and long-term functioning lymphoid and myeloid precursors defined by competitive limiting dilution analysis in vivo. J Immunol 157:138–145
Morrison SJ, Weissman IL (1994) The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity 1:661–673
Spangrude GJ, Heimfeld S, Weissman IL (1988) Purification and characterization of mouse hematopoietic stem cells. Science 241:58–62
Osawa M, Hanada K, Hamada H, Nakauchi H (1996) Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science 273:242–245
Morita Y, Ema H, Nakauchi H (2010) Heterogeneity and hierarchy within the most primitive hematopoietic stem cell compartment. J Exp Med 207:1173–1182
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Plaks V, Koopman CD, Werb Z (2013) Circulating tumor cells. Science 341:1186–1188
Chaffer CL, Weinberg RA (2011) A perspective on cancer cell metastasis. Science 331:1559–1564
Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, Leach SD, Stanger BZ (2012) EMT and dissemination precede pancreatic tumor formation. Cell 148:349–361
Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133:704–715
Thiery JP, Acloque H, Huang RYJ, Nieto MA (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139:871–890
Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW, Wainscoat JS (1997) Presence of fetal DNA in maternal plasma and serum. Lancet 350:485–487.
Lo YM, Zhang J, Leung TN, Lau TK, Chang AM, Hjelm NM (1999) Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet 64:218–224
Lo YM, Tein MS, Lau TK, Haines CJ, Leung TN, Poon PM, Wainscoat JS, Johnson PJ, Chang AM, Hjelm NM (1998) Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am J Hum Genet 62:768–775
Chiu RW, Chan KA, Gao Y, Lau VY, Zheng W, Leung TY, Foo CH, Xie B, Tsui NB, Lun FM, Zee BC, Lau TK, Cantor CR, Lo YM (2008) Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl Acad Sci U S A 105:20458–20463
Palomaki GE, Deciu C, Kloza EM, Lambert-Messerlian GM, Haddow JE, Neveux LM, Ehrich M, van den Boom D, Bombard AT, Grody WW, Nelson SF, Canick JA (2012) DNA sequencing of maternal plasma reliably identifies trisomy 18 and trisomy 13 as well as Down syndrome: an international collaborative study. Genet Med 14:296–305
Huang Z, Fong C-Y, Gauthaman K, Sukumar P, Choolani M, Bongso A (2011) Novel approaches to manipulating foetal cells in the maternal circulation for non-invasive prenatal diagnosis of the unborn child. J Cell Biochem 112:1475–1485
Bianchi DW, Williams JM, Sullivan LM, Hanson FW, Klinger KW, Shuber AP (1997) PCR quantitation of fetal cells in maternal blood in normal and aneuploid pregnancies. Am J Hum Genet 61:822–829
Hamada H, Arinami T, Kubo T, Hamaguchi H, Iwasaki H (1993) Fetal nucleated cells in maternal peripheral blood: frequency and relationship to gestational age. Hum Genet 91:427–432
Yamamoto R, Morita Y, Ooehara J, Hamanaka S, Onodera M, Rudolph KL, Ema H, Nakauchi H (2013) Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 154:1112–1126
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Japan
About this chapter
Cite this chapter
Iriguchi, S., Yamaguchi, T., Nakauchi, H. (2015). Analysis of Physical Characteristic of Hematopoietic Cells. In: Arai, T., Arai, F., Yamato, M. (eds) Hyper Bio Assembler for 3D Cellular Systems. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55297-0_6
Download citation
DOI: https://doi.org/10.1007/978-4-431-55297-0_6
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55296-3
Online ISBN: 978-4-431-55297-0
eBook Packages: EngineeringEngineering (R0)